
Abstract
Bitumen is a complex mixture consisting of different hydrocarbons, which can be categorised into substance classes called SARA (saturates, aromatics, resins and asphaltenes) fractions. The saturates, aromatics and resins are further referred to as maltenes or maltene phase. To determine the SARA fractions the asphaltenes were separated from the maltenes in a first step. A standard method to further separate the maltenes is column chromatography (CC). Since column chromatography is a time-consuming and solvent-intensive process, an alternative method for separating maltenes using solid-phase extraction (SPE) was investigated. A total of nine bitumen samples of the same type in different ageing states but from different manufacturers were separated with both methods to determine the mass percentages of the fractions for investigating their comparability. The results show that both methods led to different mass percentages of the fractions. However, with the SPE, separating smaller amounts of maltenes is possible while saving a large amount of material, costs, and workload. Linear regression analyses between the colloidal indices with conventional and rheological parameters revealed that the fractions obtained from SPE led to higher coefficients of determination of the linear regression model. The obtained fractions were further structurally compared with FTIR spectroscopy, whereby the spectra of the SPE fractions and the CC fractions did not show any significant differences. The overall consideration of the results concludes that the SPE can replace the CC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In both its natural and technically produced variants, bitumen is a petroleum-based product, whereby petroleum is a conversion product from dead organic material [1]. Technically produced bitumen, a residue from crude oil processing, is mainly used for asphalt road construction. According to EN 12597, bitumen is defined as an almost non-volatile, adhesive, and waterproofing material, which is completely or almost completely soluble in toluene and highly viscous or almost solid at ambient temperatures [2]. Since the raw material of bitumen is crude oil, bitumen is an organic product with a highly complex chemical composition [1]. The detailed composition of bitumen and its properties strongly depend on the provenance and characteristics of the crude oil and the manufacturing during the distillation process [3, 4]. Bitumen mainly consists of long and cyclic aliphatic and aromatic hydrocarbons and their derivates. Furthermore, heteroatoms such as sulphur, nitrogen, and oxygen are present. The proportions of the main constituents depending on the respective literature, are carbon (80 to 88 wt.%), hydrogen (8 to 12 wt.%), sulphur (0 to 9 wt.%), nitrogen (0 to 2 wt.%), and oxygen (0 to 2 wt.%) [1, 3, 5]. Thus, bitumen consists of many diverse molecular groups, varying in their molecular weights, polarities, and functionalities, making a complete chemical characterisation difficult or nearly impossible [3, 6, 7]. To determine the complex bitumen structure, the components are generally grouped into four major substance classes called SARA fractions (saturates, aromatics, resins, asphaltenes). [1]. The SARA classification is a polarity-based separation relying on different solubilities (asphaltenes) and interactions of the different polarities between the phases and the individual fractions of the sample (saturates, aromatics, resins) [1, 3, 5,6,7].
The theory of SARA fractions and their determination originates from petroleum chemistry, as it has been used to characterise crude oils and study material changes due to ageing [8, 9]. The chemical composition of crude oil can differ due to the origin, which can also be transferred to the bitumen composition [1]. Regarding bitumen, the refining process, the subsequent processing, and the service life are further factors influencing the composition [1, 3, 10]. For petroleum, it was shown that different SARA fraction ratios also originated from different methods, as they did not lead to identical results [8, 9]. For bitumen, it was found that the chosen experimental conditions, for example, the used solvents, different dimensions of the columns, and different materials for the mobile phase, affected the proportion of the bitumen fractions [3]. Various methods are available to determine the SARA mass fractions. First, it is recommended to separate the asphaltenes as an insoluble fraction in n-heptane [5, 11]. The n-heptane soluble component is called maltenes or maltene phase. Different national standards for asphaltene separation regulate a uniform procedure based on the precipitation with n-heptane, such as DIN 51595 and ATSM D 6560 [12, 13]. However, there is no uniform standard for separating the maltene phase except for the American standard ASTM D 4124, which is barely considered in Europe [14]. Because of this situation, various methods are being used for SARA fraction determination, such as thin-layer chromatography with flame ionisation detection (TLC–FID) and column chromatography (CC) [5, 10, 11, 15]. Since there is no uniform standard for performing the maltene separation, significant differences in the experimental setup are recognisable. The most significant difference between TLC–FID and CC is that with TLC–FID, the maltenes are pulled into the stationary phase by capillary forces, while with CC, the maltenes are applied from above and flow through the stationary phase in the direction of gravity [8, 10]. The use of various non-standardised methods complicates a direct comparison of the SARA fractions emphasising the importance of specifying the detailed experimental setup [3]. However, both TLC–FID and CC have in common that they are time-consuming processes requiring high input of disposables. For this reason, there are approaches for other examination methods, such as the SARA separation with solid-phase extraction (SPE) cartridges [16,17,18,19]. The advantages of the SPE are both a relatively simple implementation and performance and considerably low material consumption, but it has not yet reached a comprehensive implementation [16,17,18,19].
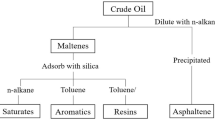
The objective of this study is to investigate the comparability of two different methods, column chromatography and solid-phase extraction, for the determination of SARA fractions. It aims to verify whether the SPE can obtain similar mass fractions of the maltenes as with column chromatography to replace column chromatography with a method that requires significantly less material. The experimental plan of the study is shown in Fig. 1. For all bitumen samples, asphaltenes and maltenes were first separated by the same method according to DIN 51595 [12]. Subsequently, CC and SPE were carried out on the maltene phases to determine the maltene mass fractions. In addition to comparing the mass fractions, a selection of virgin and aged maltene fractions were analysed using Fourier transform infrared (FTIR) spectroscopy.

Research plan
2 Materials
2.1 Bitumen and its SARA fractions
The bitumen samples used to conduct the comparison were three identical paving grade bitumen 50/70 according to EN 12591 from three different manufacturers (A, B, C) designated A 50/70, B 50/70, and C 50/70 (Fig. 1) [20]. The ageing simulation was carried out with the Rolling Thin Film Oven Test (RTFOT) according to EN 12607–1 and a double procedure of the Pressure Ageing Vessel test (PAV) according to EN 14769 combined with RTFOT [21, 22]. Previous tests have shown that a double PAV application can simulate ageing more realistically compared to recovered naturally aged bitumen samples [23]. Thus, nine bitumen samples at three different ageing stages, virgin, RTFOT and RTFOT + 2xPAV, were used for comparison [23]. Conventional and rheological bitumen parameters obtained within a previous research project were available for the bitumen samples and were therefore chosen as parameters for the correlation analyses (Table 1) [23]. The parameters are the softening point (R&B), the needle penetration (PEN), the equi-shear modulus temperatures T(|G*|= 15 kPa) and T(|G*|= 50 MPa) with their corresponding phase angles δT(|G*|=15 kPa) and δT(|G*|=50 MPa), the BBR critical temperatures T(S = 300 MPa) and T(m = 0.3) and the shear relaxation viscosity λREL [23].
The individual bituminous SARA fractions can be described as follows.
2.1.1 Saturates
The proportion of saturates in bitumen ranges from 0 to 20 wt.% acting as a diluting medium in bitumen [1, 4, 24]. The colour of the saturates fraction is primarily colourless or occasionally yellowish [1]. This fraction has the lowest molecular weight with around 600 g/mol, and its solubility parameter ranges between 15 and 17 MPa1/2 [4, 25]. Saturates are non-polar viscous oils consisting mainly of aliphatic hydrocarbons and cycloaliphatic compounds, which are straight and branched chain structures and very few polar atoms or aromatic rings [1, 4].
2.1.2 Aromatics
The aromatics fraction amounts between 30 and 65 wt.% representing the largest component of the dispersion medium for the asphaltenes [1, 3, 26]. Aromatics are dark brown viscous liquids and are the most volatile fraction in bitumen [1, 3]. Together with saturated oils, they are considered plasticisers for bitumen [1, 3]. The molecular weight of aromatics ranges around 800 g/mol, and the solubility parameter is between 17 and 18.5 MPa1/2 [4, 25]. The aromatic fraction consists mainly of non-polar aliphatic carbon chains attached to unsaturated aromatic rings [1, 3].
2.1.3 Resins
The resin content in bitumen ranges around 30 to 45 wt.% [1, 3, 24]. Resins are understood as a transition from oils to asphaltenes [4]. The primary function of the resins in bitumen is to disperse the asphaltenes [1, 3]. They are dark brown polar substances and contribute to the adhesive properties of bitumen [1, 3]. Their molecular weight ranges from 300 to 2,000 g/mol, solubility parameter lies between 18.5 and 20 MPa1/2 [4, 25]. Resins are solid or semisolid at room temperature, brittle at cold temperatures, and become fluid when heated [4]. Resins consist mainly of hydrogen and carbon atoms [1, 4]. Additionally, they can contain a small number of oxygen, sulphur, and nitrogen atoms [1].
2.1.4 Asphaltenes
The proportion of asphaltenes varies between 5 and 25 wt.% [1, 3, 24]. Asphaltenes are highly polar brown-black amorphous solids that are insoluble in n-heptane but soluble in toluene [1]. Asphaltenes are the decisive factor for bitumen properties because it is generally known that the asphaltene content of bitumen has a significant influence on the rheological properties of a bitumen [1, 25]. They offer a high molecular weight ranging between 800 and 3,500 g/mol, and their solubility parameter ranges between 17.6 and 21.7 MPa1/2 [4, 25]. In terms of composition, they are similar to the resins, mainly consisting of carbon and hydrogen atoms and some nitrogen, sulphur, and oxygen atoms [1, 3, 4].
2.1.5 Relationship between SARA fractions
The interactions between the SARA fractions can be described using models, whereby the colloidal model is most common [5, 6, 27, 28]. The colloidal model describes the bitumen structure as an oily liquid, soluble matrix (the maltenes) in which highly polar, insoluble solid particles (the asphaltenes) are dispersed [5, 29]. The most popular index, which is calculated with the SARA fractions, is the Colloidal Index (CI), also known as the Colloidal Instability Index (CII), which is based on the colloidal model explaining the behaviour of sol- and gel-type bitumen [1, 5, 6, 27]. The Colloidal Index is calculated according to Loeber et al. [27] as in Eq. (1).
In this context, higher CI values indicate a better dispersion of the micelle fractions and, thus, higher stability of the bitumen as the asphaltenes are more peptised by the resins in the bitumen system [5, 6, 27]. The solubility of the asphaltenes in a colloidal system is enhanced by intermediate polarity species such as aromatics and resins [4]. Saturates are components with the lowest polarity, while asphaltenes have the highest polarity of the SARA fractions [4].
3 Methods
To examine the mass fractions and structure of a sample, the asphaltenes, which are insoluble in n-heptane, were first separated. The maltenes were then separated into five fractions using column chromatography and solid-phase extraction. A qualitative and quantitative evaluation of the developed SPE method is based on comparing the mass fractions and spectra obtained with Fourier transform infrared spectroscopy (FTIR).
3.1 Asphaltene separation
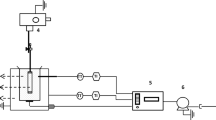
Since both the CC and the SPE were carried out on the maltene phase, separating the asphaltenes from the bitumen sample was necessary. The extraction process following DIN 51595 was used [12]. According to this method, approx. 3 g of bitumen sample was weighed into a flask and boiled under reflux in 90 ml of n-heptane for one hour (Fig. 2). The second step was the filtration of the asphaltenes using a cellulose filter type 604 ½ (particle retention of 25 μm). The filter was placed in a Soxhlet extractor, whereas the extraction was first run with 70 ml n-heptane for 1.5 h to separate the remaining maltenes from the asphaltenes. Afterwards, to regain the asphaltenes from the filter, the solvent toluene (90 ml) was used, and this step was again carried out for 1.5 h. After the second Soxhlet extraction, the solvent of the asphaltene-toluene mixture was removed using a rotary evaporator with a water bath temperature of 50 °C. Subsequently, the asphaltene fractions were oven-dried between 105 and 115 °C for 45 min. After cooling to room temperature in a desiccator for 1 h, the flasks with remaining asphaltenes were weighed, and the contents were determined by the relation to the initial weight of the sample given in mass percent [wt.%].

The maltene solutions from the filtration and n-heptane-based extraction step were combined. The solvent was evaporated at the rotary evaporator using a vacuum/pressure reduction variant. At a sample temperature of approx. 40 °C or 50 °C water bath temperature, n-heptane was aborted at approx. 120 mbar and toluene at approx. 77 mbar. To get the fractions as solvent-free as possible, the final sorting was always done at "zero", the best possible pump capacity, which was approx. 20–35 mbar. Further drying of the maltene phases at higher temperatures was not carried out to avoid a reaction of the maltene phase with the ambient atmosphere.
3.2 Maltene separation with column chromatography (CC)
The residue of the asphaltene separation, the maltenes, was further separated using the column chromatography based on Šebor et al. [8]. The separation of the maltenes was carried out in a parallel double determination. For a double determination, two glass columns with a height of 300 mm and an inner diameter of 30 mm were used. The following description and mass data refer to the procedure carried out on a single column.
Before applying the stationary phase, the column was sealed with 0.5 g cotton wool. The stationary phase was built up using two layers following Šebor et al. [8]. The first layer was created with 44 g of aluminium oxide Al2O3 (Merck no. 1.01077, particle size 0.063–0.200 mm), and the second layer with 33 g of silica gel SiO2 (Merck no. 1.07734, particle size 0.063–0.200 mm) (Fig. 3). To activate both sorbents, they were oven-dried for 1 h at 105 °C. To introduce the stationary phase into the glass column, the aluminium oxide and the silica gel were separately slurried with n-heptane. Then aluminium oxide and silica gel were filled into the column, respectively, and n-heptane was drained to just above the stationary phase in each case. The column was packed the day before the separation and sealed to prevent the stationary phase from drying out.

To provide an even application of the maltenes into the column, 1 g maltene phase was dissolved in 10 ml n-heptane one day before. The samples were stored airtight overnight in the refrigerator. The maltenes were then applied to the glass column with a graduated pipette. The maltenes were separated into five fractions: saturates, mono-, di- and polyaromatics, and resins. Different solvents and solvent mixtures were used to separate the maltene phases, which were successively applied to the column and passed through by gravity. The solvents and solvent mixtures used to separate the different fractions are given in Fig. 3. The solvent for the first fraction, the saturates, was 290 ml n-heptane (10 ml was already used to solve the maltenes), and the throughput was collected in a flask. The flow rate was set to approx. 10 ml/min. Shortly before the solvent level reached the stationary phase, the collecting flask was changed, and the solvent or solvent mixture for the next fraction was applied. This procedure was carried out to the last fraction, again draining the solvent only to just before the stationary phase. Due to the different polarities of the components of the fractions to the respective solvent, these components absorb at different rates on the stationary phase, and the column can separate the individual fractions by the respective mobile phase. The individually collected fractions were then separated from the solvents and solvent mixtures using a rotary evaporator as described earlier. The flasks containing the individual fractions were further placed in a vacuum box and oven-dried at 45 °C until a constant mass was achieved, measured at the third decimal point. The results were referred to the weight of the initial sample.
3.3 Maltene separation with solid-phase extraction (SPE)
The SPE was conducted with a vacuum box and glass syringes from FORTUNA OPTIMA with a capacity of 24 ml connected with a Luer-lock system (Fig. 4). The work by Sakib et al. [16] and Mirwald et al. [17] served as a basis for the experimental set-up. A double determination was also carried out for the SPE, whereby the given quantities refer to a single determination. The masses of the sorbents and the solvents were adjusted from the ratio of the glass columns to the smaller syringes. The calculation was done using the volume ratios of the glass column of the CC and adjusting the volumes of the materials to the volume of the syringes. Preliminary tests to develop the method using bitumen A showed that the mass of the maltene had to be increased. To perform the SPE, 1.5 times the calculated amount of maltene was used. 0.19 g of maltenes were dissolved in 1 ml n-heptane.

a Experimental setup of the solid-phase extraction; b Schematic setup of the solid-phase extraction
Comparable to column chromatography the preparation of the SPE was carried out one day before the actual separation, and the packing of the syringe was done in the laboratory. The stationary phase was equal to the CC build with Al2O3 and SiO2, which were activated in an oven at 105 °C for 1 h. After cooling, n-heptane was added to both sorbents. The cotton wool was hand-formed into a pellet, pressed to the bottom of the syringe with the help of the plunger, and additionally compressed. The two stationary phases were then filled into the syringe. First, the aluminium oxide was filled into the syringe while the tap was closed. The tap was then opened at a pressure of approx. 200 mbar to drain off the solvent until just before the sorbent. The same procedure was conducted when filling the silica gel. The syringe was then filled with n-heptane and sealed overnight.
The next day before the actual extraction, the solvent was drained under normal pressure so that the stationary phase was slightly covered with the solvent and the maltene phase was applied using a glass pipette. After applying the maltene phase, the solvent of the first fraction was added up to the edge of the syringe. Then, the tap was opened entirely under normal pressure, and the remaining solvent was applied constantly. The resulting flow rate that occurred under normal pressure was approx. 15 ml/min. Preliminary tests have shown that with additional pressure, the solvent was drawn through the syringe too quickly, and the separation of the samples did not lead to satisfactory results. For this reason, the vacuum was only used to pack the syringe to compress the stationary phase.
The fractions were collected in glass containers inside of the vacuum box. The tap was closed when the respective solvent reached the stationary phase. The syringe was then exchanged together with the tap into the next position. The next solvent was applied, the tap was opened again under normal pressure, and the remaining solvent was refilled during the extraction. This procedure was done equally for all other fractions. The solvents and solvent mixtures were removed using a rotary evaporator, and drying was carried out as with the CC method.
3.4 Fourier transform infrared (FTIR) spectroscopy
The separated fractions were further analysed using FTIR spectroscopy with the device Spectrum Two FT-IR C 96108 from PerkinElmer. For the investigation of the samples, the attenuated total reflection (ATR) measurement with multiple reflections (zinc selenide (ZnSe) crystal, sample area approx. 575 mm2, 25 reflections) was used. Until the measurement the samples were stored airtight in the refrigerator. Therefore, the samples were dissolved in cyclohexane at a mass ratio of 1:3 (bitumen: cyclohexane). To provide representative FTIR spectra, the measurements of the samples were carried out within 24 h after dissolving using an Eppendorf pipette to apply 0.1 ml of the bitumen-cyclohexane solution to the crystal. During the waiting time after the sample was applied, the crystal was located under a fume cupboard. After a waiting time of 15 min, the solvent was allowed to evaporate, and a thin bitumen film remained (approx. 35 µm), which was measured with the following settings: 32 scans, a wavenumber range from 4,000 to 600 cm−1 and a resolution of 4 cm−1. A spectral comparison was conducted for the virgin and RTFOT + 2xPAV aged samples. SARA fractions after RTFOT ageing were not included in the analysis. After recording the reflectance spectra, they were further processed for comparison. For this purpose, the spectra were converted into absorbance spectra and then standardised. The standardisation was carried out with the Standard Normal Variate (SNV) transformation only using relevant wavenumber ranges from 3,400 to 2,500 cm−1 and 1,800 to 600 cm−1 [23, 30].
4 Results and discussion
The results are presented in four sections. The first step of the method evaluation is to assess the measurement accuracy of the double determinations (4.1.). Subsequently, the results of the mean values of the SARA mass fractions of the two methods are compared, and the respective colloidal indices are calculated (4.2.). Furthermore, the correlations between the colloidal indices and the physical parameters are investigated using linear regression analyses (4.3.). Finally, the discussion concludes by comparing the FTIR spectra of the mass fractions (4.4.).
4.1 Measurement accuracy of the double determinations
To compare the measurement accuracies of the two methods, the absolute differences between the double determinations were calculated, respectively, for the CC and the SPE maltene separation according to Eq. (2).
where BS is the respective bitumen sample (A, B, C), m.1 is the single value of one double determination (CC-1, SPE-1), and m.2 is the single value of the other double determination (CC-2, SPE-2). As this is the calculation of the absolute differences, it is not necessary to specify the position of the single value in the equation. The aim with double determinations should be that the results are as close as possible. The absolute differences of the respective double determinations are given in Table 2. Additionally, the mean values from the absolute differences were determined for each double determination allowing a comparison of the individual samples per method. Accordingly, bitumen A virgin shows a mean deviation of 0.39 wt.% with the CC method, whereas an average difference of 0.46 wt.% was obtained with the SPE method. In total, nine comparisons can be made for nine samples. The lower value for each comparison is marked by the bold number in Table 2. Four samples showed smaller differences with the CC and four with the SPE. For one sample, B virgin, both methods showed the same mean difference of 0.39 wt.%. However, if the total values of the deviations across all samples are considered, it becomes apparent that overall, lower differences between the double determinations occurred with the SPE. In other words, if not each individual sample is considered, but the mean value is calculated over the total absolute differences per method, the mean value of all absolute differences from the CC SARA fractions is calculated as 0.52 wt.%, compared to 0.47 wt.% for the SPE SARA fractions. Thus, on average, the double determinations with the SPE produced smaller differences than the double determinations with the CC. However, as these differences are pretty small, they should not be generally accepted as significant. No definite statement can be made as to whether the accuracy of the methods depends on the age or the origin of the sample since an even distribution of samples between the two methods results and the population of nine samples with double determination seems insufficient for this assessment.
4.2 Comparison of the mean SARA mass fractions
The mean values for each separation were then determined from the two single determinations. The results of the mean SARA mass fractions are given in Fig. 5a to 5c. There are no differences between CC and SPE for the asphaltenes, as these were determined in a previous step, and the obtained maltenes were used for both CC and SPE. In addition to the determined mass fractions, the remaining residual (Rest) is calculated, which is missing to obtain 100%. A comparison of the residual proportions shows that the results with the SPE produce larger residual proportions than the determination with the CC. The rest results from material losses, which could be caused by residues remaining in the column or syringes on the one hand or by the material transfer in flasks between the individual work steps on the other hand. Apart from the residual content, the most significant differences are observed in the resins.

Mean values of the SARA mass fractions respectively for bitumen A (a), B (b) and C (c)
To investigate to what extent the results, especially the rest, influences the ratios of the SARA fractions, the colloidal indices (CI) of the SARA mass fractions were calculated based on Eq. (1) and are given in Table 3. As can be seen from the colloidal indices, the ratios of dispersed to flocculated fractions are comparatively similar. Therefore, it can be assumed that the resulting rest results from a uniform reduction of the SPE fractions and only slightly influences the calculated ratio of the SARA fractions. However, in general, it can be observed that the CI, except for the bitumen sample C RTFOT, are higher for the CC SARA fractions. This result means that when the CC is used, the calculated indices indicate higher stability of the bitumen.
4.3 Linear regression analyses
To study the relationship between the obtained SARA fractions and the given physical characteristics of the bitumen samples, linear regression analyses were performed between the two calculated colloidal indices CICC and CISPE, and the bitumen characteristics given in Table 1. The linear regression analyses were performed individually for the colloidal indices of the SARA fractions of the column chromatography and SARA fractions of the solid-phase extraction given in Table 3. In each case, the independent variable is represented by the colloidal index (x-axis) and the dependent variable is the respective parameter (y-axis). For each analysis, the values of the unaged, short-term, and long-term aged bitumen samples were plotted, which is representatively marked in Fig. 6 for bitumen B (orange). The linear regression function and the coefficient of determination R2 are given in the respective coloured boxes. 54 linear regression analyses were carried out in this way – six for each dependent variable. Figure 6 exemplarily illustrates the outcomes using the softening point. The correlations between the other properties and the colloidal indices at virgin, RTFOT and RTFOT + 2xPAV ageing conditions are given in Table 4.

Linear regression analyses between the softening points R&B (y-axis) and colloidal indices (x-axis) at virgin, RTFOT and RTFOT + 2xPAV ageing conditions, respectively, for CC SARA fractions (a) and SPE SARA fractions (b)
The results per independent variable per bitumen sample show that for 27 comparisons, in 23 cases, higher coefficients of determination are found for the colloidal indices calculated from the SPE SARA fractions (Fig. 6 and Table 4). In each case, the higher coefficient of determination is marked in bold (Table 4). Only for four comparisons higher R2 were found for the CC SARA fractions. These four analyses involve the needle penetration of bitumen A and C, the phase angle δT(|G*|=15 kPa) of bitumen C and the equi-shear modulus temperatures T(|G*|= 50 MPa) of bitumen C. For all other dependent variables of the bitumen samples, higher coefficients of determination were found with the CI of the SPE SARA fractions. This result is a strong indication that the colloidal indices of the SPE SARA fractions show a higher correlation with the conventional and rheological parameters than the CC SARA fractions. This means that even though it has been shown that there are differences between the masses of the SARA fractions of the CC and SPE, which initially made it seem that the SPE might not be suitable for use, the linear regression analyses with consideration of the ageing development have shown that when related to physical parameters, the SPE SARA fractions show higher correlations. Although the trend favours SPE SARA fractions, the correlations with CC SARA fractions were cumulative higher for bitumen C in three out of nine comparisons. Thus, it can be concluded that the respective bitumen sample is also decisive for the result. This result further indicates that even within same graded bitumen samples with similar physical characteristics, results from chemical tests cannot be directly transferred from one bitumen sample to another.
4.4 FTIR spectra of the SARA fractions
In addition to comparing the mass fractions, FTIR spectroscopy was used to detect possible structural differences between the maltene fractions (SAR: saturates, aromatics, resins) of the two methods as well as general differences in the SAR fractions. Therefore, the differences between the spectra of the fractions from the two methods were compared first. For this purpose, the standardised wavenumbers of the fingerprint region between 1,800 and 600 cm−1 were used for calculating difference spectra. The calculation was done by subtracting the standardised absorbance values of the spectra of the SPE from the CC given as absolute numbers according to Eq. (2). Then the mean values of the differences were calculated from the difference spectra. If there are no differences between the CC and SPE spectra, the differences and, thus, the mean values would be close to zero. Table 5 shows the calculated mean values of the absolute differences between the spectra of the CC and SPE fractions. As can be seen, the mean values of the differences between the spectra result as marginal. Thus, it can be concluded that the fractions obtained with both methods structurally do not significantly differ from each other. The result supports the objective to replace the CC method with the SPE method as similar spectra are produced after separation.
However, the mean values confirm that the maltene separation methods have different effects on the three bitumen samples A, B, and C. While the mean values of fractions F1 and F2 are comparably higher for bitumen A, for bitumen C the mean values of the difference of the fractions F4 and F5 spectra are comparably higher. Therefore, it can be concluded that no uniform deviation can be observed across all fractions, but the deviation is somewhat dependent on the bitumen composition.
Since the difference spectra have shown no significant structural differences between the CC and SPE fractions, the general differences between spectra of the SPE SAR fractions are analysed in a second step. The most significant differences between the SAR factions are mainly visible in the fingerprint region between 1800 and 600 cm−1. The standardised FTIR spectra of the CC separation (dashed line) and the SPE separation (continuous line) were given representatively based on bitumen B in Fig. 7. Because bitumen B showed the lowest mean values of the absolute differences and thus the deviation between the CC spectrum and SPE spectrum was the smallest, bitumen B was selected to represent the differences in the individual fractions. In Fig. 7a to 7e, each fraction is shown individually, whereby the grey spectra are the standardised FTIR spectra of the total B 50/70, virgin (dashed), and long-term aged (solid) to display the differences between the individual fractions and the total bitumen. These figures visualise what the mean values of Table 4 have already shown: the deviations of the CC and SPE spectra are relatively small and almost unrecognisable using the graphical representations.

Standardised FTIR spectra of (a) saturates (F1), b monoaromatics (F2), c diaromatics (F3), d polyaromatics (F4), e resins (F5)
Moreover, Fig. 7 illustrates the increase in the bands representing the oxygen compounds, carbonyls at 1,700 cm−1 and sulfoxides at 1,030 cm−1, from F1 to F5. For fractions F4 and F5, the bands of the sulfoxides even show a higher curve than the sulfoxides of the total bitumen. For both the F5 virgin fraction and the F5 long-term aged fraction, the bands of the oxygen compounds are higher than those of the total bitumen. The polarities of the fractions increase from F1 (saturates) to F5 (resins). At the same time, both the carbonyls and the sulfoxides are polar oxygen compounds. In general, the solubility of polar and non-polar substances is that similar dissolves in similar [16]. The oxygen compounds are thus primarily addressed by the solvents used for F4 (polyaromatics) and F5 (resins) in terms of their polarity. This observation explains why no differences between virgin and long-term aged are detectable in fractions F1 to F3. The differences between virgin and aged fractions only become apparent for fraction F4 regarding the carbonyls and fraction F5 for both carbonyls and sulfoxides. It can be concluded that although the mass fractions of fractions F1 to F3 change with ageing, this change does not affect the structural composition of the fraction. Only in the case of the polyaromatics and the resins, there are structural changes in the FTIR spectra in addition to the mass changes due to ageing.
5 Conclusion
The main objective of this study was to develop and evaluate a method for the fractionation of maltenes based on the principles of chromatographic separation. The advantage of the new method should be that material consumption, time and workload are reduced, which simultaneously means cost savings. For this purpose, the maltene phases of nine bitumen samples in different ageing stages were separated by standard column chromatography to obtain reference values. Subsequently, with the help of solid-phase extraction, a method was developed with which these nine maltene phases were also separated. For both methods, the asphaltenes were separated in a preliminary step following existing standards. With column chromatography and solid-phase extraction, the maltenes were separated using five different solvents or solvent mixtures to separate the maltenes into five fractions: saturates, monoaromatic, diaromatics, polyaromatics and resins. Both methods use gravity, as the samples are applied to the devices from above in each case. No additional vacuum was used during SPE, as tests showed that the sample was only pulled through the syringe without proper separation when additional vacuum was applied.
The results have revealed that slightly lower deviations between the double determinations of the individual fractions occurred with the SPE. Both methods lead to different mass fractions of the maltene fractions whereas the calculated CI displays that based on the chosen calculation method CC leads to slightly higher colloidal indices than SPE. According to the chosen calculation method, higher indices imply a better micelles distribution and thus higher stability of the colloidal material [5, 6, 27]. Linear regressions of the colloidal indices with conventional and rheological parameters have revealed that SPE colloidal indices, on average, lead to higher coefficients of determination R2 of the linear regression models than CC colloidal indices. Furthermore, the fractions are structurally compared by FTIR spectroscopy. The spectra of the SPE fractions and the CC fractions do not show any significant differences, which in turn further confirms the intention to replace CC with SPE.
In general, CC and SPE are sensitive methods with a high workload, whereby the results strongly depend on the respective person conducting the separations. Both methods, therefore, require good training. The preparation of the columns and the syringes must be done one day before the actual measurement. However, it has been shown that the time required for the pure SPE separation is significantly shorter than for the CC, as less solvent is used for the SPE, which flows through the significantly smaller stationary phase. This lower material consumption also reduces the time required for subsequent rotation of the solvent and drying of the fractions. These findings provide positive feedback for introducing SPE as an economical and rapid method for maltene separation.
References
Hunter RN, Self A, Read J (2015) The Shell Bitumen handbook. Thomas Telford Publishing, London
European Committee for Standardization (CEN)–EN 12597 (2014) Bitumen and bituminous binders–Terminology. CEN, Brussels
Porto M, Caputo P, Loise V, Eskandarsefat S, Teltayev B, Rossi CO (2019) Bitumen and bitumen modification: a review on latest advances. Appl Sci. https://doi.org/10.3390/app9040742
Paliukaitė M, Vaitkus A, Zofka A (2014) Evaluation of bitumen fractional composition depending on the crude oil type and production technology. In: International conference on environmental engineering, Vilnius Gediminas Technical University Press Technika. Enviro. Doi: https://doi.org/10.3846/enviro.2014.162
Lesueur D (2009) The colloidal structure of bitumen: consequences on the rheology and on the mechanisms of bitumen modification. Adv Coll Interface Sci 145:42–82. https://doi.org/10.1016/j.cis.2008.08.011
Tabatabaee HA, Kurth TL (2017) Analytical investigation of the impact of a novel bio-based recycling agent on the colloidal stability of aged bitumen. Road Mater Pavement Des 18:131–140. https://doi.org/10.1080/14680629.2017.1304257
Dhasmana H, Hossain K, Karakas AS (2019) Effect of long-term ageing on the rheological properties of rejuvenated asphalt binder. Road Mater Pavement Des 22(6):1268–1286. https://doi.org/10.1080/14680629.2019.1686051
Šebor G, Blažek J, Nemer MF (1999) Optimization of the preparative separation of petroleum maltenes by liquid adsorption chromatography. J Chromatogr A 847:323–330. https://doi.org/10.1016/S0021-9673(99)00329-5
Kharrat AM, Zacharia J, Cherian VJ, Anyatonwu A (2007) Issues with comparing SARA methodologies. Energy Fuels 21:3618–3621. https://doi.org/10.1021/ef700393a
Tauste R, Moreno-Navarro F, Sol-Sánchez M, Rubio-Gámez MC (2018) Understanding the bitumen ageing phenomenon: a review. Constr Build Mater 192:593–609. https://doi.org/10.1016/j.conbuildmat.2018.10.169
Haghshenas HF, Rea R, Reinke G, Zaumanis M, Fini E (2022) Relationship between colloidal index and chemo-rheological properties of asphalt binders modified by various recycling agents. Constr Build Mater 318:126161. https://doi.org/10.1016/j.conbuildmat.2021.126161
German Institute for Standardization (DIN)–DIN 51595 (2000) Testing of petroleum products–determination of the content of asphaltenes–Precipitation with heptane. Beuth Verlag, Berlin
American Society for Testing and Materials (ASTM)–ASTM D6560 (2012) Standard test method for determination of asphaltenes (heptane insolubles) in crude petroleum and petroleum products. ASTM, Pennsylvania
American Society for Testing and Materials (ASTM)–ASTM D4124-09 (2018) Standard Test Method for Separation of Asphalt into Four Fractions. ASTM, Pennsylvania
Weigel S, Stephan D (2018) Relationships between the chemistry and the physical properties of bitumen. Road Mater Pavement Des 19(7):1636–1650. https://doi.org/10.1080/14680629.2017.1338189
Sakib N, Bhasin A (2018) Measuring polarity-based distributions (SARA) of bitumen using simplified chromatographic techniques. Int J Pavement Eng 20(12):1371–1384. https://doi.org/10.1080/10298436.2018.1428972
Mirwald J, Werkovits S, Camargo I, Maschauer D, Hofko B, Grothe H (2020) Understanding bitumen ageing by investigation of its polarity fractions. Constr Build Mater 250:118809. https://doi.org/10.1016/j.conbuildmat.2020.118809
Mirwald J, Werkovits S, Camargo I, Maschauer D, Hofko B, Grothe H (2020) Investigating bitumen long-term-ageing in the laboratory by spectroscopic analysis of the SARA fractions. Constr Build Mater 258:119577. https://doi.org/10.1016/j.conbuildmat.2020.119577
Guo M, Liang M, Fu Y, Sreeram A, Bhasin A (2021) Average molecular structure models of unaged asphalt binder fractions. Mater Struct 54:173. https://doi.org/10.1617/s11527-021-01754-2
European Committee for Standardization (CEN)–EN 12591 (2009) Bitumen and bituminous binders–specification of paving grade bitumens. CEN, Brussels
European Committee for Standardization (CEN)–EN 12607 (2013) Bitumen and bituminous binders–determination of the resistance to hardening under the influence of heat and air—part 1: RTFOT method. CEN, Brussels
European Committee for Standardization (CEN)–EN 14769 (2012) Bitumen and bituminous binders–accelerated long-term ageing conditioning by a pressure ageing vessel (PAV). CEN, Brussels
Schwettmann K, Nytus N, Radenberg M, Stephan D (2022) Bitumen reuse: physical and chemical approach to investigate the effectiveness of rejuvenators. Road Mater Pavement Des. https://doi.org/10.1080/14680629.2022.2060129
Corbett LW (1969) Composition of asphalt based on generic fractionation, using solvent deasphaltening, elution-adsorption chromatography and densimetric characterization. Anal Chem 41(4):576–579. https://doi.org/10.1021/ac60273a004
Speight JG (2014) The chemistry and technology of petroleum. CRC Press, Boca Raton
Lu X, Isacsson U (2002) Effect of ageing on bitumen chemistry and rheology. Constr Build Mater 16(1):15–22. https://doi.org/10.1016/S0950-0618(01)00033-2
Loeber L, Muller G, Morel J, Sutton O (1998) Bitumen in colloid science: a chemical, structural and rheological approach. Fuel 77(13):1443–1450. https://doi.org/10.1016/S0016-2361(98)00054-4
Petersen JC, Glaser R (2011) Asphalt oxidation mechanisms and the role of oxidation products on age hardening revisited. Road Mater Pavement Des 12(4):795–819. https://doi.org/10.1080/14680629.2011.9713895
Petersen JC (2009) A review of the fundamentals of asphalt oxidation: chemical, physicochemical, physical property, and durability relationships. Transportation Research Board https://www.trb.org/Publications/Blurbs/162366.aspx. Accessed 29 May 2022
Weigel S (2018) Bitumen and chemometrics: innovative characterisation of bitumen based on FTIR spectroscopy and multivariate analysis methods. Dissertation, Technische Universität Berlin
Acknowledgements
We would like to thank the German Research Foundation (DFG), whose financial support within the framework of the project "Post carbon road – The endless cyclic reuse of bitumen" (project number 392670763) made it possible to obtain the results presented.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwettmann, K., Höhne, P. & Stephan, D. Comparison of column chromatography and solid-phase extraction on virgin and aged bituminous maltene phases. Mater Struct 55, 242 (2022). https://doi.org/10.1617/s11527-022-02079-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-022-02079-4