
Abstract
The use of calcined clays as supplementary cementitious materials (SCMs) has been identified as a viable option to decrease the CO2 emissions related to cement production. However, while extensive data is available about kaolinitic clays in this context, other clays such as illitic clays appear to be under-studied. Therefore, in the present study, two illitic clays were compared to two low-grade kaolinitic clays in terms of transformations in the calcination temperature range 650–900 °C, and performance of the calcined clays in blended cement pastes as measured by strength evolution, heat release, hydrated phase formation and portlandite consumption. The illitic clays required a higher calcination temperature for complete dehydroxylation of their illite than what is necessary for dehydroxylation of kaolinite. These higher calcination temperatures also led to particle sintering, significantly decreasing the specific surface area of the illitic clays, particularly for the clay with the higher Fe2O3 content. Nevertheless, while the kaolinitic clays generally exhibited the best performance as SCM, the illitic clay with lower Fe2O3 content performed similar to the kaolinitic clays when calcined at optimum temperature and applied at a moderate substitution rate. These findings demonstrate that several different clays have the potential to be used as SCM and indicate possible routes to identify suitable deposits for this purpose.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The use of calcined clays from deposits with low to moderate kaolinite contents as supplementary cementitious materials (SCMs) has been identified as one of the most viable options to substantially decrease the environmental impact associated with cement production, inter alia because of their abundant availability in many parts of the world [1]. In addition, calcined clays and related geologic materials have considerable potential to be used as precursors for alkali-activated materials, which would mitigate the dependence on fly ashes and ground granulated blast furnace slags to produce these cements [1, 2]. Currently, however, the use of calcined clays with low to moderate kaolinite contents in cements is limited, partly due to insufficient knowledge of the factors that determine the reactivity of these materials and how these affect the resulting concrete properties.
The reactivity of calcined clays in cementitious systems is governed by the removal of hydroxyl groups from its clay minerals (dehydroxylation) during thermal treatment, leading to their partial amorphisation, which renders them pozzolanic. Sufficiently reactive calcined clay minerals release Si as well as Al during cement hydration, leading to formation of C–A–S–H via the pozzolanic reaction with calcium hydroxide and, because of the additional Al, potentially to more abundant formation of AFm phases. Studies of purified ‘model’ or ‘standard’ clay minerals have shown that the three principal clay mineral species or groups that occur in most of the considered clays—kaolinite, illite and smectites—differ regarding their properties relevant for their use as SCM. Generally, the highest reactivities, and the highest strengths of blended cement pastes, are obtained from calcined kaolinite (metakaolin), which also requires the lowest calcination temperatures (~ 500–800 °C, depending on its crystallinity) for optimum properties [3,4,5]. This is mainly attributed to the abundance and location of hydroxyl groups in kaolinite, which favours amorphisation on calcination, and the presence in metakaolin of significant amounts of five-coordinated Al, which is energetically unfavourable and thus highly reactive [4]. Nevertheless, also illite and smectites have been shown to exhibit pozzolanic properties after calcination at suitable temperatures, which are usually found in the range ~ 800–950 °C [3, 5,6,7,8,9]. These minerals generally show less indications of amorphisation after calcination than kaolinite, and only a limited amount of five-coordinated Al forms [5,6,7,8,9]. In addition, because of the fact that in kaolinite the octahedral sheet is connected to only one tetrahedral sheet, its dissolution rate in alkaline solution is higher than those of smectites, in which the octahedral sheet is sandwiched between two tetrahedral sheets [10]; similarly, analogous structural characteristics of the calcined minerals may contribute to the observed differences in reactivity between metakaolin and calcined 2:1 minerals (clay minerals with the octahedral sheet between two tetrahedral sheets) such as illite and smectites.
Recent research on the use of naturally occurring clays with low to moderate kaolinite content has focused either on identifying the minimum required kaolinite content when the calcination conditions are optimised for the thermal activation of the kaolinite [11, 12] (including use of these calcined clays in cement together with co-substituted limestone [12,13,14]), or on finding optimum calcination conditions with consideration not only of the kaolinite, but also of the 2:1 minerals contained in the clay, i.e. calcination temperatures substantially above 800 °C [11, 15,16,17,18]. These latter studies confirmed that reactive pozzolans can be obtained from clays with illite and/or smectites, but that these minerals require higher calcination temperatures and yield lower strengths of blended cement pastes than kaolinitic clays. However, it was also found that, due its higher specific surface area, calcined kaolinitic clays can substantially decrease the workability of concretes produced with these SCMs, requiring high amounts of superplasticisers and/or adjustment of the mix-design to mitigate this effect [16]. Taken together, the previous studies demonstrate that the occurrence of widely differing clay minerals as well as the diversity of accessory minerals in common clays does not currently allow one to draw general conclusions about the suitability of specific clay deposits regarding their use as SCM. Therefore, additional studies of a range of clays are required to establish correlations between the type of clay, its chemical and mineral composition, the calcination conditions, the fineness of the calcined material, the cement substitution rate, and the performance of blended cements.
To contribute to these issues, two illitic clays (brick clays) and two clays with moderate kaolinite content (~ 55%) from Central Germany were compared in the present study regarding transformations on calcination and their performance as SCM. Brick clays are characterized by melting temperatures in the range 950–1200 °C, caused by significant amounts of alkalis, iron and/or calcium in the materials; usually, they contain ~ 30–60% quartz and variable amounts of different clay minerals [19]. Brick clays are available in amounts that are relevant for SCM production on an industrial scale. For example, sales of brick clays in Great Britain were ~ 9 × 106 t in 2005 [20], and 25 × 106 t of brick and special clays combined were extracted in Germany in 2009 [21]. More than 150 brick clay deposits are currently operated in Germany [22], with a medium-sized deposit typically containing 5–20 × 106 t of clay [19].
2 Materials and methods
2.1 Starting materials
Four raw clays from different deposits in Central Germany were studied. Two of the clays, designated I1 and I2, can be classified as brick clays, i.e. they fall in the compositional range of clays used for the production of bricks [19] (see below). The other two clays, designated K1 and K2, contained more kaolinite, but were used without a purification process, i.e. they can be classified as low-grade kaolinitic clays [19] (see below). All clays were preconditioned by drying at 120 °C for 48 h and grinding in an impact mill until passing through a 2-mm sieve.
Portland cement (designation PC), CEM I 42.5 R, with a sodium oxide equivalent of Na2Oeq = 0.88% was used to produce blended cement pastes and tested as a reference. Quartz powder (designation Q) was used as a filler in blended cement pastes for comparison with the calcined clays.
The chemical compositions and relevant physical properties of the starting materials are shown in Table 1.
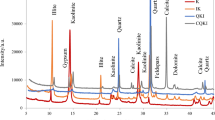
The phase assemblages of the clays, as determined by XRD (Fig. 1; for experimental conditions see Sect. 2.6), differed substantially between the illitic clays (brick clays) and the low-grade kaolinitic clays. Both illitic clays, I1 and I2, exhibited prominent reflections of illite (PDF # 00-026-0911) and kaolinite (PDF # 01-074-1784). In the diffractogram of I2, a reflection at a d-spacing of 14.3 Å (~ 6.2° 2θ) can be discerned in addition, which is assigned to smectite [23]. A small hump around d = 12.2 Å (~ 7.3° 2θ) in the same diffractogram is consistent with minor amounts of smectite with only one water layer in the interlayer space [23], possibly caused by partial dehydration during drying and grinding of the clay. Accessory minerals in both illitic clays were quartz (PDF # 00-046-1045), anatase (PDF # 00-021-1272) and feldspar (orthoclase, anorthoclase); in I2, hematite (PDF # 00-033-0664), calcite (PDF # 01-086-0174) and possibly traces cristobalite (PDF # 00-039-1425) were identified in addition.

XRD patterns (5°–75° 2θ range) of the raw clays with 10% zincite internal standard. Selected major reflections are labelled: S smectite, I illite, K kaolinite, Q quartz, Z zincite, C calcite, H hematite
The diffractograms of clays K1 and K2 exhibited a considerably higher intensity of the basal reflection of kaolinite (PDF # 01-074-1784) relative to the basal reflection of illite (PDF # 00-026-0911); indications of smectite were not found. Quartz (PDF # 00-046-1045) was an accessory mineral in both kaolinitic clays, while anatase (PDF # 00-021-1272) and minor amounts of gibbsite (PDF # 00-033-0018) were identified only in K1.
Rietveld quantitative phase analyses (RQPA) were performed using montmorillonite as the smectite mineral. The analyses showed that none of the clays contained significant amounts of amorphous phase(s); the resulting abundances of the major crystalline phases are listed in Table 2.
The TGA/DTG curves of the clays are shown in Fig. 2 (for experimental conditions and computation see Sect. 2.7). The fractions of kaolinite that were computed from the TGA curves were 17% for I1, 13% for I2, 56% for K1, and 61% for K2. These results are in good agreement with the RQPA results, though the relative order of the amounts of kaolinite in I1 and I2 was not consistent between the two methods.

a TGA curves and b DTG curves of the raw clays
Both brick clays exhibited a shoulder/peak in the DTG curves at around 600–700 °C, assigned to the dehydroxylation of illite, which was more pronounced for I2, in accord with the RQPA results. However, evaluation of the TGA curves with the tangent method yielded similar illite contents of ~ 23% for both clays.
The mass loss up to ~ 200 °C, seen in all clays, but most pronounced for I1 and I2, can be assigned to loss of adsorbed water and to loss of interlayer water from smectite-type clay minerals. However, for all clays this mass loss was low (< 1%), due to the drying of the clays at 120 °C before grinding and characterization. The similarity of the TGA/DTG curves of I1 and I2 in the temperature range < 200 °C may suggest that I1, as well, contained clay minerals with hydrated interlayers. However, smectite was identified only in I2 by XRD (Fig. 1; Table 2), and in line with this, only I2 exhibited a discernible peak in the DTG curve at ~ 800–900 °C.
2.2 Calcination of the clays
The thermal activation of the clays was carried out by batch calcination, each batch ~ 50 g of raw clay, in platinum crucibles in a muffle furnace. The calcination temperatures were 650, 850 and 900 °C. The clays were heated at a rate of 10 °C/min and then soaked at the specified temperature for 3 h. Subsequently, samples were removed from the furnace and left to cool on a lab bench for 30 min, and then stored in a desiccator at 23 °C with silica gel to avoid excessive absorption of water and potential rehydroxylation [24].
Below, calcined clays are designated by the name of the raw clay, followed by an underscore and the calcination temperature in °C.
2.3 Blended cement pastes
For the preparation of blended pastes, the Portland cement and calcined clay or quartz powder were first manually mixed and homogenised for 30 s. Two substitution rates were employed: 15 v/v% and 30 v/v%. These volume-based substitution rates are equivalent to mass-based substitution rates of ~ 13 wt% and ~ 26 wt%, respectively (the apparent densities of the calcined clays were 2.53–2.68 g/cm3).
The Portland cement and the blended cements were mixed with deionised water at a water-to-solids ratio (w/s) of 1.26 by volume (equivalent to a w/s of ~ 0.42 by weight for the blended cement pastes; w/s = 0.40 by weight for the plain Portland cement paste). The resulting pastes for strength testing and microstructural characterisation were mixed in a planetary centrifugal mixer at a rotational speed of 1250/min for 4 min. Subsequently, the pastes were cast into 20 mm × 20 mm × 20 mm cube moulds and cured for 1, 7 or 28 days at 20 °C and ~ 99% relative humidity (above water). The pastes for isothermal calorimetry were mixed for 4 min at 1600 rpm using a Janke & Kunkel IKA-WERK mechanical mixer and then ~ 15 g of the paste immediately placed in plastic ampoules and inserted in the calorimeter cell.
Superplasticizers were not applied. Thus, workability differed between pastes, depending on the calcined clays and the substitution rate, but all pastes were workable enough to be produced and cast without the occurrence of excessive air voids.
Designations of blended cement pastes below are composed of PC (for Portland cement), the substitution rate in v/v% and the supplementary material (either Q for quartz powder or the designation of the calcined clay), separated by an underscore.
2.4 Isothermal calorimetry
The heat release during hydration at 20 °C (heat of hydration) of the plain Portland cement paste and the blended cement pastes was measured up to 28 days in a TAM Air isothermal calorimeter. The time between addition of water to the cement pastes and the start of the measurements was recorded and the obtained calorimetry data adjusted accordingly.
2.5 Compressive strength testing
Compressive strength testing was performed on 16 hydrated blended cement pastes. Blends with brick clays (i.e. clays I1 and I2) were tested for calcination temperatures of 650 and 850 °C, and the low-grade kaolinitic clays (i.e. clays K1 and K2) were tested for calcination temperatures of 650 and 900 °C. Compressive strength testing of the hardened cubes was done on a 10-kN testing machine at a loading rate of 240 N/s, equivalent to 0.6 MPa for the chosen sample dimensions.
After strength testing, specimens were ground manually using mortar and pestle for subsequent XRD analyses, and the resulting powders stored in glass vials in a desiccator (flushed with argon to remove the air) until required for testing.
2.6 X-ray diffraction
Rietveld quantitative phase analysis (RQPA) was performed for the raw clays. The NIST Standard Reference Material 660c (LaB6) was used to determine the instrumental parameters of the employed XRD device; zincite (ZnO; AppliChem, grade p.a., min. 99.5%) was used as internal standard for the measurements of the clay samples. The samples were ground in a McCrone ‘micronizing’ mill with 10% zincite in propan-2-ol for 5 min, and the resulting slurries were dried at 40 °C overnight. The dry powders were filled into purpose-built sample holders by side-loading. XRD patterns were recorded in Bragg–Brentano geometry on a Rigaku Ultima IV diffractometer under the following conditions: Cu Kα radiation (λ = 1.541874 Å); tube operating at 40 kV, 40 mA; sampling interval: 0.01° 2θ; scan rate: 0.2° 2θ min−1; scanning range: 5°–150° 2θ; divergence slit: in-plane 1/6°, axial 10 mm; strip detector D/teX Ultra with 5° Soller slits. Phase identification was done with Match! version 3.5 (Crystal Impact, Germany); Rietveld analyses were performed with TOPAS version 5 (Bruker AXS, Germany).
XRD patterns for qualitative phase analysis were recorded for the calcined clays and the hydrated cement pastes. The sample powders were filled into sample holders by top-loading. Measurements were done on the same device as the measurements for RQPA, but employing the following conditions: CuKα radiation (λ = 1.541874 Å); tube operating at 40 kV, 40 mA; sampling interval: 0.02° 2θ; scan rate: 0.5° 2θ min−1; scanning range: 5°–65° 2θ; divergence slit: in-plane 0.5°, axial 10 mm; strip detector D/teX Ultra with 5° Soller slits. The sample holders were spun at 15 rpm during the measurements. Phase identification was performed with Match! version 3.5 (Crystal Impact, Germany).
2.7 Thermogravimetric analysis
Thermogravimetric analysis (TGA) measurements were performed on raw clays, calcined clays as well as hydrated cement pastes using a Mettler Toledo TGA/DSC3+STARe. Sample masses of ~ 10 mg were heated in Al2O3 crucibles first to 40 °C and kept at that temperature for one hour, then the samples were heated to 1000 °C (clays) or 900 °C (cement pastes) at a heating rate of 5 °C/min; the nitrogen flux during the measurements was 80 ml/min. From the TGA results, the derivative thermogravimetry (DTG) curves were calculated.
The dehydroxylation of the kaolinite, illite and smectite occurs at temperature intervals that partially overlap, but may allow their differentiation in DTG curves (see e.g., Refs. [4, 25]). Under the present experimental conditions, the mass losses due to dehydroxylation of kaolinite, illite and smectite were found at approximately 400–600, 600–700 and 800–900 °C, respectively. Where a distinctive feature could be identified in the DTG curves, the corresponding mass loss (\(m_{{{\text{H}}_{2} {\text{O}}}}\)) was determined by the tangent method from the TGA curve.
The mass fractions of the individual clay minerals (fclaymin) in the clays was calculated from the determined mass losses according to
where Mclaymin and \(M_{{{\text{H}}_{2} {\text{O}}}}\) are the molar masses of the clay mineral and water, respectively, and \(n_{{{\text{H}}_{{2}} {\text{O}}}}\) is the number of moles of water released on dehydroxylation of the clay mineral. For the numerical values of \(n_{{{\text{H}}_{{2}} {\text{O}}}}\) and Mclaymin, the following idealised stoichiometries were assumed for the dehydroxylation of kaolinite, illite and smectite (montmorillonite), respectively:
The degree of dehydroxylation (DTG) of kaolinite and the 2:1 minerals (illite and smectite, considered together) in the calcined clays was done by comparing the respective mass losses in the TGA/DTG curves of the raw clays and the calcined clays:
where fclaymin,calcined is the mass fraction of clay mineral remaining after calcination, obtained from Eq. (1) applied to the TGA curve of the calcined clay.
The amount of bound water in the hydrated cement pastes was defined as the mass loss in the temperature interval 50–500 °C (thus excluding the weight loss due to decomposition of carbonates). The portlandite content of the pastes was determined by the tangent method from the step in the TGA curves at about 400–450 °C.
3 Results and discussion
3.1 Transformations of the clays during calcination
The diffractograms of the clays after calcination at 650, 850 and 900 °C (Electronic Supplementary Material Figs. S1–S4) reveal that already after calcination at 650 °C, the kaolinite had become virtually completely amorphous, i.e. its peaks were absent, while the 002 reflection of illite at a Bragg angle of 8.8° 2θ was diminished at this temperature, but remained present up to 900 °C. These observations are in line with previous studies on purified clay minerals [4,5,6]. The main reflection of the smectite in I2 at d = 14.3 Å (~ 6.2° 2θ) remained present, but is shifted to higher Bragg angles and broadened, after calcination at 650 °C, while it is no longer detectable after calcination at 850 °C, again in accord with studies on purified montmorillonite [5, 7].
Figure 3 demonstrates that the alteration of the illite on calcination can be followed by the shift of its 004 reflection from ~ 5.00 Å (~17.8° 2θ) to 5.04 Å (17.6° 2θ). In all clays, this shift was only partial after calcination at 650 °C (viz. peaks were present at ~ 5.00 Å and 5.04 Å), while calcination at ≥ 850 °C left only the peak at 5.04 Å (Fig. 3, Figs. S1–S4). A close inspection of the diffractograms published by He et al. [6] and Hollanders et al. [5] shows that the same peak shift occurred in the temperature range ~ 650–800 °C for the purified illites in these studies as well. The XRD patterns of the calcined illite in the present samples were matched by that of dehydroxylated muscovite (PDF # 00-046-0741) [26]. As muscovite has a structure that is closely related to that of illite, this strongly suggests that the shift of the 004 reflection of illite is caused by rearrangement of its structure due to dehydroxylation. Consequently, the incomplete shift of that peak after calcination at 650 °C indicates that this temperature was not sufficient to fully dehydroxylate and transform the illite.

004 reflection in the XRD patterns of raw and calcined clay I2, showing the transition of the illite to dehydroxylated illite
A confirmation of these conclusions is provided by the degrees of dehydroxylation, as determined by TGA (Table 3; Fig. S5). While the dehydroxylation of the kaolinite was virtually complete already after calcination at 650 °C, the degree of dehydroxylation was determined to be only ~ 60–70% for the 2:1 minerals (i.e. mainly illite) at this calcination temperature. Only after treatment at ≥ 850 °C, illite and smectite in I1 and I2 were virtually completely dehydroxylated. In line with these results, a wide dehydroxylation range of ~ 500–800 °C was previously inferred by Garg and Skibsted [9] from 29Si MAS NMR spectroscopy data for an illite-dominated clay (with about 30% interstratified smectite layers).
The effect of calcination on the median particle size and the specific surface area of the clays is shown in Fig. 4. Calcination at 650 °C caused slight to moderate increases of the median particle size with no significant reduction of the specific surface area; this indicates agglomeration of the clay particles without sintering during calcination. At calcination temperatures of ≥ 850 °C, a substantial decrease of the specific surface areas of the clays was observed (except for K2 at 850 °C), which was most pronounced for the illitic clays, particularly for I2. This behaviour of the illitic clays can be explained by the fact that brick clays generally contain comparatively high amounts of elements that act as flux (Na, K, Fe and Ca), which lowers their melting temperature to ~ 950–1200 °C [19]; accordingly, they require comparatively low temperatures for sintering and melt formation. That the most significant decrease of the specific surface area at ≥ 850 °C was observed here for clay I2 is explained by the fact that this clay had the highest Fe2O3 content (Table 1). Melting and glass formation of illitic clays on calcination was also previously inferred by Trümer et al. [16]; however, complete melting did not occur for the clays in the present study, as evidenced by the remaining reflections of dehydroxylated illite in the XRD patterns of I1 and I2 (Figs. S1 and S2).

a Median particle size and b specific surface area of the raw and calcined clays. Error bars represent one standard deviation in each direction
Despite partial sintering, the densities of the calcined clays, measured by helium pycnometry, were not significantly different from the raw clays, viz. in the range 2.62–2.68 g/cm3 for the illitic clays, and 2.53–2.63 g/cm3 for the kaolinitic clays.
3.2 Performance of blended cement pastes
3.2.1 Compressive strength
Figure 5 shows the compressive strengths of the cement pastes after 1, 7 and 28 days of hydration, relative to the compressive strength of the plain Portland cement paste (PC) after 28 days of hydration; the compressive strength of the latter was 74.8 MPa.

Compressive strength of the blended cement pastes with a 15 v/v% and b 30 v/v% cement replacement, relative to the plain Portland cement paste, after 1, 7 and 28 days of hydration. The compressive strength of the plain Portland cement paste was 74.8 MPa. Error bars represent one standard deviation in each direction
At early ages (i.e. 1 and 7 days), all blended pastes with 15 v/v% substitution rate exhibited significantly lower strengths than the plain PC paste. Most of the pastes blended with calcined clays performed similar to the paste blended with quartz powder at these hydration times, but the pastes blended with I1_850, K2_650 and K2_900 possessed slightly higher strengths. After 28 days of hydration, the highest compressive strengths were obtained with pastes blended with I1_850, K1_900, K2_650 and K2_900, all of which exhibited at least 94% of the compressive strength of the plain PC paste, i.e. considerably higher than the strength of PC_15_Q.
For a substitution rate of 30 v/v%, the highest compressive strengths were again exhibited by the pastes blended with I1_850, K1_900, K2_650 and K2_900, though in this case, the difference between I1_850 and the calcined kaolinitic clays was more pronounced. However, the strengths of the pastes with I1_650 as well as with I1_850 were higher than that of PC_30_Q.
Pastes blended with calcined clay I2, at both substitution rates, possessed compressive strength that were similar to that of the paste blended with quartz powder, i.e. this clay did not contribute to the strength of the cement pastes significantly more than the quartz powder (viz., only through the filler effect [27,28,29]).
For I1, the compressive strength at both substitution rates was higher for the higher calcination temperature (850 °C) than for calcination at 650 °C. A similar tendency was also observed for K1 and K2, though exceptions from this general trend occurred.
3.2.2 Isothermal calorimetry
The heat release during hydration of the cement pastes was obtained from isothermal calorimetry up to 28 days (Figs. S6–S9). For all clays except I2, for all calcination temperatures and at both substitution rates, the cumulative heat release of the pastes blended with calcined clays until about 4 days of hydration was similar to the cumulative heat release of the paste blended with quartz powder. For I2, the cumulative heat release at 4 days was lower than the heat released by the corresponding quartz-blended paste, particularly at a calcination temperature of 650 °C and a substitution rate of 15 v/v%. At later ages, the heat released from the pastes blended with calcined clays increased over that of the paste blended with quartz powder, and at ages between approximately 10 and 25 days, some of the clay-blended pastes outperformed even the plain Portland cement paste in terms of the cumulative heat release.
A comparison of the cumulative heat release of the blended cements after 28 days of hydration is provided in Fig. 6. All pastes with calcined clays except PC_15_I1_650 and PC_15_I2_650 exhibited cumulative heat releases that were substantially higher than that of the corresponding quartz-powder blended paste and similar to each other at the same substitution rate, respectively. PC_15_I1_650 and PC_15_I2_650 released approximately the same heat as the corresponding quartz powder-blended paste, i.e. substantially less heat than the other pastes blended with 15 v/v% calcined clay. Though these results are in general agreement with the compressive strength results (PC_15_I1_650 and PC_15_I2_650 had comparatively low strengths, Fig. 5), the overall correlation between the cumulative heat release and the compressive strengths was poor. For example, the good strength performance of PC_15_I1_850, PC_15_K2_650 and PC_15_K2_900 is not mirrored by a particularly high heat release from these pastes (Fig. 6a). The same lack of agreement between strength and heat release after 28 days was observed for the pastes with a substitution rate of 30 v/v% (Fig. 6b). Equally poor relationships were obtained when comparing the cumulative heat release of the pastes at other durations of hydration, e.g. at 6 days (Fig. S10), with their compressive strengths.

Cumulative heat release of the blended cement pastes with a 15 v/v% and b 30 v/v% cement replacement after 28 days of hydration
These findings are seemingly in contrast to the results of Avet et al. [12], who found a good correlation between the compressive strengths after 28 days of blended cement mortars with low-grade kaolinitic clays with varying kaolinite contents, all calcined at 800 °C, and the cumulative heat release after 6 days at 20 °C of blended pastes with these calcined clays. However, the apparent discrepancy between the present results and the previous study is likely related to the fact that in the latter study the calcined clays were not blended with Portland cement for the calorimetric measurements, but with a mix of portlandite, K2SO4 and KOH, and the employed w/s ratio was 1.20. Additional possible reasons for the lack of correlation between the cumulative heat release and the strength of the blended cements in the present study will be discussed in the next section.
3.2.3 Bound water and portlandite consumption
Figures 7 and 8 show plots of the bound water content and the portlandite content of the cement pastes, respectively, versus their relative compressive strength after 28 days of hydration. The pastes with quartz powder are marked in red in these plots. (The individual values of all pastes are listed in Tables S1–S4; the individual TGA and DTG curves are shown in Figs. S11 and S12.) Though the scatter of the data is significant, some general trends are clearly discernible.

Bound water content versus relative compressive strength after 28 days of hydration of the plain Portland cement paste (square symbol) and the blended cement pastes with 15 v/v% (circles) and 30 v/v% (triangles) cement replacement. The dashed line serves as a guide to the eye

Portlandite content (%CH) versus relative compressive strength after 28 days of hydration of the plain Portland cement paste (square symbol) and the blended cement pastes with 15 v/v% (circles) and 30 v/v% (triangles) cement replacement. Dashed lines serve as guides to the eye
First, the bound water content as well as the portlandite content were lowest for pastes with 30 v/v% cement replacement, intermediate for pastes with 15 v/v% cement replacement, and highest for the plain Portland cement paste (with one exception for the bound water content, PC_15_K2_650, discussed below). This is due to dilution of the cement by addition of calcined clays or quartz powder to the blends. As the cement is the main reactant to form portlandite and the other cementitious phases such as C–(A–)S–H and AFm phases—all of which are hydrates—, its dilution leads to decreased portlandite and bound water contents to an extent that is dependent on the ability of the replacement material to form hydrates too. With decreasing bound water content, the compressive strength of the pastes decreased (Fig. 7), because the volume of hydrated phases formed on reaction of the cement or the blends with water determines the strength of the hardened paste by filling the capillary pore space [30, 31].
Second, within each group of blended pastes (15 v/v% or 30 v/v% substitution rate), the compressive strengths increased with decreasing portlandite content (Fig. 8); the quartz-blended pastes exhibited the lowest strengths and the highest, or almost the highest, portlandite content within each group. This observation is attributed to the differing extents of the pozzolanic reaction of the calcined clays: The higher the portlandite consumption of a calcined clay, i.e. the lower the portlandite paste of the hardened paste was, the higher was the amount of C–(A–)S–H and AFm phases formed through its pozzolanic reaction, which contributed to the strength of the pastes. An increased formation of AFm phases in blended cement pastes with calcined clays, compared to the plain Portland cement paste, was also evident in the XRD patterns of the pastes (Fig. S13): An increased fraction of hemicarbonate (PDF # 00-041-0221) was detected in the blended pastes, particularly in those with comparatively high compressive strength. The particularly high bound water content of PC_15_K2_650, higher than that of the plain Portland cement paste, may be caused by this or related differences of the relative abundances of the various hydrates in these pastes.
Slight variations can be expected between the phase assemblages of the blended pastes with calcined clays too, caused by different relative amounts of Si and Al released from calcined illite, kaolinite and smectite, and also due to additional elements (e.g., Mg and Fe) released by the clays. This provides a possible explanation for the poor relationship between the heat release and the compressive strength of the pastes that was observed here (Sect. 3.2.2): Different amounts of dissolved elements may induce precipitation of phases with considerably differing enthalpies of formation, particularly through formation of phases with different Fe contents [32], without exhibiting an equivalent effect on the strength of the pastes.
An additional factor that could have contributed to the poor correlation of strengths with heat release is the degree of reaction of the clinker in blends with the same substitution rate but different calcined clays. The specific surface area and the surface chemistry of the calcined clays influence the magnitude of their accelerating effect on the reaction of the clinker [27,28,29], which determines the heat release from the clinker reactions. However, any increase of the extent of reaction of the clinker would also be expected to cause an increase of the amounts of hydrates formed in the pastes and, thus, of their bound water contents. Though the pertinent data exhibited considerable scatter, the compressive strengths were clearly related to bound water content (Fig. 7), while no such correlation was found for strength versus heat of hydration. The scatter of the bound water-versus-strength data may be related to the extent of clinker reaction, but the fact that there was a correlation between bound water and compressive strength, while this was not the case for the heat release, indicates that differences between the degree of reaction of the clinker in blends with different calcined clays cannot fully account for the lack of correlation between strength and heat of hydration.
4 Conclusions
In all of the four clays investigated in the present study—two illitic clays (I1 and I2) and two low-grade kaolinitic clays (K1 and K2)—, calcination caused complete transformation of the kaolinite to metakaolin already at 650 °C, while dehydroxylation of the illite in the illitic clays occurred more gradually from 650 to 850 °C. The shift of the 004 reflection of illite appears to be a good indicator of the extent of its dehydroxylation. Alongside with dehydroxylation, particle sintering occurred at ≥ 850 °C, leading to increased particle sizes and decreased specific surface areas of the clays. This latter decrease was much more significant in the illitic clays, and most pronounced in the I2, which had the highest Fe2O3 content of the clays.
Nevertheless, three of the four calcined clays exhibited pozzolanic activity, i.e. they contributed substantially more to the compressive strength of blended cement pastes than quartz powder (which affects cement hydration only through the filler effect) at equal substitution rates, at all studied calcination temperatures between 650 and 900 °C. The observed correlation between the compressive strength of the blended cement pastes and the bound water content as well as portlandite consumption after 28 days of hydration indicates that it was mainly the amount of hydrates formed that determined the contribution of the calcined clays to the cement strength, as expected. The two calcined low-grade kaolinitic clays exhibited the best performance, with the compressive strength of blended pastes with both calcined clays observed to be close to that of a plain Portland cement paste at 15 v/v% substitution rate. For both clays, a calcination temperature of 900 °C usually caused slightly higher strengths than calcination at 650 °C, though exceptions from this trend occurred.
Importantly, it was found that the illitic clay (brick clay) with a lower Fe2O3 content, calcined at 850 °C, performed very similar to the kaolinitic clays—in terms of strength evolution as well as reactivity as measured by bound water content and portlandite consumption of blended cement pastes after 28 days of hydration—, when employed at a substitution rate of 15 v/v%. However, at a substitution rate of 30 v/v%, the same calcined clay exhibited a performance that was inferior to the kaolinitic clays (except K1 calcined at 650 °C), though its performance was still significantly better than that of the quartz powder. The calcined illitic clay I2 with a higher Fe2O3 content (5.9%) performed not as good as the other illitic clay at both substitution rates, most likely because of a substantially lower specific surface area after calcination, caused by increased sintering and/or melt formation at ≥ 850 °C. Future studies should attempt to elucidate which other mechanisms play a role for the effect of Fe2O3 content on the performance of calcined clays as SCMs and their optimum calcination temperature.
The heat released in isothermal calorimetry measurements was not a good measure of the performance of the calcined clays in blended cements in terms of strength development. The poor correlation between the evolved heat of reaction and the compressive strength of cement pastes is possibly related to different relative amounts of Si, Al, Fe and other elements—that can precipitate as hydrates with different enthalpies of formation—released by the calcined clays, and to different degrees of reaction of the clinker in the blends with calcined clays. Additional studies are required to quantify the relative contributions of these two, and possibly other, causes for the observed variations of the heat of hydration of the blended cement pastes. However, it has previously been shown [12] that a good correlation between calorimetry results and the compressive strength of blended cements with low-grade kaolinitic clays is obtained when adjusted mixes that approximate the pore solution composition of hydrating cements are used for calorimetry, enabling prediction of calcined clay performance already at this stage.
References
Scrivener KL, John VM, Gartner EM, UN Environment (2018) Eco-efficient cements: potential economically viable solutions for a low-CO2 cement-based industry. Cem Concr Res 114:2–26
Provis JL (2018) Alkali-activated materials. Cem Concr Res 114:40–48
He C, Osbæck B, Makovicky E (1995) Pozzolanic reactions of six principal clay minerals: activation, reactivity assessments and technological effects. Cem Concr Res 25:1691–1702
Fernandez R, Martirena F, Scrivener KL (2011) The origin of the pozzolanic activity of calcined clay minerals: a comparison between kaolinite, illite and montmorillonite. Cem Concr Res 41:113–122
Hollanders S, Adriaens R, Skibsted J, Cizer Ö, Elsen J (2016) Pozzolanic reactivity of pure calcined clays. Appl Clay Sci 132–133:552–560
He C, Makovicky E, Osbæck B (1995) Thermal stability and pozzolanic activity of calcined illite. Appl Clay Sci 9:337–354
He C, Makovicky E, Osbæck B (1996) Thermal treatment and pozzolanic activity of Na- and Ca-montmorillonite. Appl Clay Sci 10:351–368
Garg N, Skibsted J (2014) Thermal activation of a pure montmorillonite clay and its reactivity in cementitious systems. J Phys Chem C 118:11464–11477
Garg N, Skibsted J (2016) Pozzolanic reactivity of a calcined interstratified illite/smectite (70/30) clay. Cem Concr Res 79:101–111
Bauer A, Berger G (1998) Kaolinite and smectite dissolution rate in high molar KOH solutions at 35° and 80°C. Appl Geochem 13:905–916
Taylor-Lange SC, Lamon EL, Riding KA, Juenger MCG (2015) Calcined kaolinite–bentonite clay blends as supplementary cementitious materials. Appl Clay Sci 108:84–93
Avet F, Snellings R, Alujas Diaz A, Ben Haha M, Scrivener K (2016) Development of a new rapid, relevant and reliable (R3) test method to evaluate the pozzolanic reactivity of calcined kaolinitic clays. Cem Concr Res 85:1–11
Avet F, Scrivener K (2018) Investigation of the calcined kaolinite content on the hydration of Limestone Calcined Clay Cement (LC3). Cem Concr Res 107:124–135
Maraghechi H, Avet F, Wong H, Kamyab H, Scrivener K (2018) Performance of Limestone Calcined Clay Cement (LC3) with various kaolinite contents with respect to chloride transport. Mater Struct 51:125
Buchwald A, Hohmann M, Posern K, Brendler E (2009) The suitability of thermally activated illite/smectite clay as raw material for geopolymer binders. Appl Clay Sci 46:300–304
Trümer A, Ludwig H-M, Rohloff K (2014) Investigation into the application of calcined clays as composite material in cement. ZKG Int 67(9):52–57
Alujas A, Fernández R, Quintana R, Scrivener KL, Martirena F (2015) Pozzolanic reactivity of low grade kaolinitic clays: influence of calcination temperature and impact of calcination products on OPC hydration. Appl Clay Sci 108:94–101
Schulze SE, Rickert J (2019) Suitability of natural calcined clays as supplementary cementitious material. Cem Concr Compos 95:92–97
Lorenz W, Gwosdz W (2003) Handbuch zur geologisch-technischen Bewertung von mineralischen Baurohstoffen (Geologisches Jahrbuch, Sonderhefte, Reihe H, Heft SH 16). Schweizerbart, Stuttgart
Bloodworth A, Highley D, Cowley J (2007) Brick clay—mineral planning factsheet. British Geological Survey. https://www.bgs.ac.uk/downloads/start.cfm?id=1350. Accessed 29 Aug 2016
Krakow L, Schunke F (2015) Resource efficiency in brick and tile industry (Ziegelindustrie International Sonderdruck). Bauverlag, Gütersloh
Krakow L, Schunke F (2016) Current clay potential in Germany. Part 1: general introduction. Ziegelind Int 2:18–25
Meunier A (2005) Clays. Springer, Berlin
Emmerich K (2000) Spontaneous rehydroxylation of a dehydroxylated cis-vacant montmorillonite. Clays Clay Miner 48:405–408
Bernal SA, Juenger MCG, Ke X, Matthes W, Lothenbach B, De Belie N, Provis JL (2017) Characterization of supplementary cementitious materials by thermal analysis. Mater Struct 50:26
Keppler H (1990) Ion exchange reactions between dehydroxylated micas and salt melts and the crystal chemistry of the interlayer cation in micas. Am Mineral 75:529–538
Berodier E, Scrivener K (2014) Understanding the filler effect on the nucleation and growth of C–S–H. J Am Ceram Soc 97:3764–3773
Oey T, Kumar A, Bullard JW, Neithalath N, Sant G (2013) The filler effect: the influence of filler content and surface area on cementitious reaction rates. J Am Ceram Soc 96:1978–1990
Kumar A, Oey T, Falzone G, Huang J, Bauchy M, Balonis M, Neithalath N, Bullard J, Sant G (2017) The filler effect: the influence of filler content and type on the hydration rate of tricalcium silicate. J Am Ceram Soc 100:3316–3328
Odler I (1991) Strength of cement (final report). Mater Struct 24:143–157
Lothenbach B, Matschei T, Möschner G, Glasser FP (2008) Thermodynamic modelling of the effect of temperature on the hydration and porosity of Portland cement. Cem Concr Res 38:1–18
Lothenbach B, Kulik DA, Matschei T, Balonis M, Baquerizo L, Dilnesa B, Miron GD, Myers RJ (2019) Cemdata18: a chemical thermodynamic database for hydrated Portland cements and alkali-activated materials. Cem Concr Res 115:472–506
Acknowledgements
This work was supported through the programme KMU innovativ of the German Federal Ministry of Education and Research (BMBF) (Förderkennzeichen 01LY1610B). The authors are grateful to H.-J. Schröder, K. Schröder-Grahle (both at KERATON Kies- und Tongruben GmbH, Plessa) and M. Neubert (Technische Universität Bergakademie Freiberg) for valuable discussions. The authors would also like to thank S. Simon (BAM) for his continuous support with extending the XRD lab and equipment for RQPA of clays.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Msinjili, N.S., Gluth, G.J.G., Sturm, P. et al. Comparison of calcined illitic clays (brick clays) and low-grade kaolinitic clays as supplementary cementitious materials. Mater Struct 52, 94 (2019). https://doi.org/10.1617/s11527-019-1393-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-019-1393-2