
Abstract
Neomycin B is a drug of the bacteriologic category of aminoglycosides, built by four sugar rings joined with glycosidic bonds, that recently has attracted a lot of attention because its derivatives have shown anti-carcinogenic and antiviral properties, in addition to some gene therapy applications. Although some derivatives have been synthesized, there are no theoretical studies about them. In this work, we carried out DFT calculations to predict and study stable neomycin B derivatives, obtained by substituting its first ring by Thymine, as done in previous experiments, and for Guanine as a theoretical proposal. For the two nucleobases, we obtained stable quasi-degenerated structures, pointing out that there is equal probability to synthesize their isomers. Moreover, we calculated the electrostatic potential surface to elucidate the long-range interaction mechanism.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neomycin B is a drug from the aminoglycoside family made up of four rings linked by glycosidic bonds. Its derivatives have been experimentally shown to have antiviral [1] and anticancer [2, 3] properties, in addition to being useful in inhibiting gene expression [4], at the cost of a decrease in its antibacterial effect [1,2,3]. Aminoglycosides are antibiotics that inhibit protein production by causing a misreading of the RNA message at a specific region (different for each drug) in the ribosomal A-site of the Gram-negative bacteria. It has been suggested that this behavior is the result of a very strong and specific interaction between the 2-amino-2-deoxy sugar and the ribosomal A site, and some authors have proposed that aminoglycosides work by oxidation of guanine nucleotides in the bacterial DNA [5]. However, these drugs have raised concerns due to their ototoxicity and nephrotoxicity, and their susceptibility to aminoglycoside-modifying enzymes (AMEs), a form of bacterial resistance. The AMEs work by modifying the binding capability of the drugs and therefore reducing their antibacterial activity [5]. With the rise of superbugs, the prevalence of genetic diseases and various types of cancer, some groups have been working to diversify the aminoglycosides pharmacological applications by modifying and adding to them functional groups or nucleobases, to gain specificity to certain RNA sequences.
Neomycin was discovered in 1949 as a complex mix (Neomycin A-F) produced by Streptomyces fradiae. Neomycin A is the degradation product of the remaining components, of which Neomycin B is the main component (between 85 and 95% from the total), Neomycin C is the secondary component, and the rest are scarce depending on the synthesis process [6]. Neomycin B and C are built by four rings in chair conformation; Rings I and III are an aminocyclitol, while Rings II and IV are 2-amino-2-deoxy sugar linked by O-glycosidic bonds, and both differ in the position of the aminomethyl group in Ring IV. The structural conformation of Neomycin B is shown in Fig. 1, with an Electrostatic Potential Surface (EPS) showing nucleophilic and electrophilic regions in red and blue, respectively. The frontier orbitals HOMO and LUMO have a gap of 6.9 eV, and are located in the -COOH group from Ring III and Ring I, respectively; Neomycin B is water soluble with a dipole moment magnitude of 5.13 D. It is worth mentioning that this drug is susceptible to phosphorylation in the electrophilic region around the -COOH group from Ring III (see Fig. 1) by the aminoglycoside phosphotransferase AME, that decreases the hydrogen bonding potential of the neomycin with certain RNA from the ribosomal A-Site [7].

Superimposed frontier orbitals for Neomycin B (brownish red and dark green colors for each phase), ESP magnitude at 0.001 a.u. according to the scale in the lower horizontal bar and vector of dipole moment, rings in romans. a HOMO is located on Ring I, with contributions from -COOH, b LUMO is located strongly in the -COOH from Ring III and the ring itself, with contributions from Ring I. Electrophilic regions in blue and nucleophilic regions in red
For Neomycin B, there are groups that modify Ring I by adding functional groups or nucleobases, and they have found that there is a gain in the selectivity for the bacterial ribosomal A-site [7, 8]. Other groups have linked functional groups or nucleic acids by a 1,2,3-Triazol, replacing the -COOH from Ring III, finding that their derivatives have a stronger cytotoxicity against some cancer cells, with a minor loss of their antibacterial effect [10]. Simpler modifications are those that replace the whole Ring I by a nucleobase; experiments have shown that the Neomycin-Thymine derivatives gain an important specificity against the dimerization initiation sequence (DIS) from HIV-1 and are more resistant against AMEs [1]. For the last case there is only one theoretical study carried out by Francisco [11], who modeled the Neomycin-Thymine conjugate using Density Functional Theory (DFT) with the PBE/TZP and PBE-GD3/TZP methods, to characterize the angles formed by the O-glycosidic bonds and the interatomic distances. Also, he used molecular dynamics to model the interaction between the drug and the DIS from HIV-1, finding that the affinity of the structure is due to Rings I and II, while Rings III and IV contribute to the drug selectivity.
In this work we applied DFT calculations to predict and study stable derivatives of Neomycin B, obtained by substituting Ring I for thymine and guanine, conjugating it in different spatial configurations with respect to the glycosidic bond. This kind of study is of special interest because it is in this region where the main interactions between Neomycin and the Ribosomal A-site take place, and because of the role of the nucleobases in all the interactions with the RNA fragments. Replacing the first ring should change the properties of the drug: some of the isomers could overcome the phosphorylation which can be observed as a change in the ESP surface, but without important variations in their molecular structures. Nucleobases form base pairs with hydrogen bonds, so the HOMO and LUMO are expected to be located at the nucleobases.
Methods
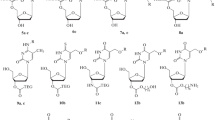
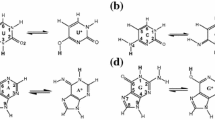
We first search for the optimal configuration for thymine and guanine with the DFT B3LYP/6-31G** method that has shown a good agreement between theoretical and experimental results [11, 12], implemented in the Gaussian-09 software [14]. We found bond length relative differences lower than 1.18% (at the amine from guanine) [15]. Afterwards, we built the conjugates by replacing Ring I of Neomycin B with thymine/guanine [16] in two different sites for each nucleobase: 1’N and 9’N in guanine, 1’N and 3’N in thymine (see Fig. 2). In order to get a general overview of the behavior of the three remaining rings and the nucleobase, geometry optimization calculations were done with different initial dihedral angles α and β (see Fig. 2). This was carried out using the B3LYP/6-21G** method and all atoms were left without constrictions. From this procedure, a set of local minima was obtained, and the subset with relative energies lower than 1 meV was reoptimized using the B3LYP/6-31G** method to get the complex conformations closer to the global minima. With this energetic criterion, using 6-21G** base, for Neomycin-Guanine at site 1N (NeomG1) two sets of local minima configurations were predicted, while for site 9’N (NeomG9) only one. For Neomycin-Thymine at site 1’N (NeomT1) and 3’N (NeomT3) we found three and two sets, respectively, with 6-31G**.

Neomycin-Thymine showing the O-glycosidic bonds (θ) and the dihedral angles α and β between Ring II and the nucleobase. For thymine, the conjugates were built by binding atoms 1’N and 3’N with the O-glycosidic bond from Ring II, while for guanine the conjugates were linked with atoms 1’N and 9’N (Fig. S1)
Results
According to our results, when employing the larger base 6-31G**, we did not get any significant structural difference. For the case of Neomycin-Guanine systems, the NeomG9a configuration is the lowest energy isomer, while the NeomG1b is the highest with a ΔE of 446.9 meV between them (see Table 1). For Neomycin-Thymine, in general the NeomT1 configurations are more stable than NeomT3, being NeomT1a the lowest energy isomer and NeomT3a the highest with only 126.3 meV between them. We used the frontier orbitals HOMO and LUMO along with the ESP and dipole moment magnitude to get an overview of the reactivity of systems predicted to be stable, looking to understand what the experiments have shown. In Table 1 there is a compendium of our results. For the Neomycin-Guanine isomers there are two stable configurations with dihedral angles β around 80° (conf. 1a and conf. 9a), while all the configurations take different dihedral angles α. We can see that the distances d between the nucleobase and Ring II centroids are larger than the corresponding distance at Neomycin B (4.94 Å). In general, the O-glycosidic angles, do not vary considerably with respect to their amplitudes in Neomycin B, although it is in the glycosidic bond to the nucleobase where the largest variation occurs, within ranges of (− 3.4 ± 0.4)° for the purine, and (− 2.2 ± 0.6)° for the pyrimidine (see Tables S1-S3 for a comparison with some structural parameters in [11]).
The HOMO–LUMO gaps for Neomycin-Guanine are around 5.40 eV, compared with the 6.9 eV for Neomycin, which is indicative that those molecules are chemically softer. As shown in Fig. 3, frontier orbitals can be found on the Nucleobase with or without a small contribution due to the -COOH group from Ring III. In all cases, the main contribution to the frontier orbitals is on the guanine. It is remarkable that NeomG1a and NeomG9a have greater dipole moment magnitudes than Neomycin B, but the magnitude for NeomG1b is the lowest (Table 1). Although this conjugate and their isomers would interact preferably through the nucleobases, they would do it with different intensities. Contrary to what is seen in Neomycin B, the ESP (Fig. 3a and b) shows larger nucleophilic regions around Ring III for NeomG1a (the isomer with the largest dipole moment) compared with NeomG1b, also, the electrophilic regions are shown as less intense in the nucleobase for NeomG1b. The ESP for the NeomG9a is an intermediate case. The most polar configuration, NeomG1a, is the best candidate to resist phosphorylation of the resistant bacteria. This conjugate has not yet been synthesized, but we expect it leads to the formation of guanine-cytosine base pairs in experiments interacting with some RNA fragments.

HOMO, LUMO, ESP surface and dipole moment vectors for (a, b) NeomG1a, (c, d) NeomG1b, (e, f) NeomT1a and (g, h) NeomT1b. The selected conjugates for this figure have the larger (a, b, e & f) and lower (c, d, g & h) ESP magnitude contrast, respectively. For surfaces corresponding to additional conjugates see Fig. S2
The dihedral angles scanning using 6-21G** suggests that there are potential barriers between local minima, indicating that up to 5 different Neomycin-Thymine isomers can coexist in a racemic mixture after a synthesis process. We will try to explain what differences arise from these structural changes in the next paragraphs.
The HOMO–LUMO gaps for the five stable isomers of Neomycin-Thymine are in the range from 4.83 to 5.43 eV; therefore, these molecules presumably present lower chemical stability than Neomycin B. In all cases (with the exception of NeomT1b) we identified a nucleophilic region around Ring III which can be associated with the presence of the HOMO located in the same ring and small contributions on the glycosidic bonds of the adjacent rings (see Fig. 3e y g). This behavior would lead to isomers with different resistance against phosphorylation and could explain the selective targeting towards the HIV-1-DIS reported in [1,2,3,4]. See SI for a brief discussion on the population analysis of the different isomers and Mulliken charges (final paragraph and Figs. S3–S5).
The dipole moments for the Neomycin-Thymine isomers change as a function of the dihedral angle α in a range from 3.90 to 7.22 D, making those molecules more polar (1a, 1c and 3a) or less polar (1b and 3b) than Neomycin B (5.13 D). In fact, isomers NeomG9a and NeomT1a, which are the most stable, are also the most water soluble, except for NeomG1a (see Table S4). Our results explain that the previous measurements [1] of Isothermal Titration Calorimetry (ITC) which showed binding with the DIS of the HIV-1 RNA, are related to the frontier orbitals and ESP distribution; while the low affinity towards the ribosomal A site can be due to the contributions of the different isomers present in the mixture resulting from the synthesis process.
Discussion
The dihedral angle influences the properties of the conjugates, so it is relevant to differentiate between the various isomers of the same conjugate. Since the energy differences between isomers are small, they can coexist in a racemic mixture. The question now is: How high is the energy barrier between the different local minima of a conjugate?
Varying the dihedral angles change the HOMO-LUMO gaps, hence isomers equally stable may have different interactions with different compounds.
It is remarkable how the nucleophilic regions are larger in the compounds with greater dipole moments; furthermore, they are above ring III of the conjugate. This can result in that all predicted structures could be more resistant to phosphorylation than Neomycin B. This can have relevance in the treatment against resistant bacteria.
The different conjugates have a wide range for the dipole moment magnitude, suggesting different solubility and interactions (dipole–dipole) in an aqueous media, that can result in different mobility through cellular and bacterial membranes; however, molecular dynamics simulations could help to elucidate this behavior.
For future work, it would be interesting to study the differences between the isomers of the same conjugate, and contributing to the design of corresponding in vitro or ITC experiments. We hope this study motivates further research of this kind of systems using DFT methods or inspire more experimental research on aminoglycoside-nucleobase conjugates.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
E. Ennifar et al., ACS Chem. Biol. (2013). https://doi.org/10.1021/cb400498n
W. Wang et al., Med. Chem. Res. (2018). https://doi.org/10.1007/s00044-018-2169-x
D.D. Vo et al., Org. Biomol. Chem. 16(1), 6262–6274 (2018)
S.A. Aggag et al., Anima. Gene (2022). https://doi.org/10.1016/j.angen.2022.200130
B. Willis, D.P. Arya, Adv. Carbohydr. Chem. Biochem. 60, 251–302 (2006)
R.S. Vardanyan, V.J. Hruby, Synthesis of essential drugs (Elsevier, Amsterdam, 2006), pp.275–478
K.M. Krause et al., Cold Spring Harb. Perspect. Med. (2016). https://doi.org/10.1101/cshperspect.a027029
G.C. Sati et al., ACS Infect. Dis. (2017). https://doi.org/10.1021/acsinfecdis.6b00214
K.F. Blount et al., Chem. Bio. Chem. (2006). https://doi.org/10.1002/cbic.200600109
S. Wang et al., Med. Chem. Res. (2018). https://doi.org/10.1007/s00044-018-2169-x
P. Francisco in Results and Analysis, Theoretical study of the interaction of aminoglycosides and gold complex with HIV DIS sequences, (PhD. Thesis UNAM 2020), pp. 39–95, at https://tesiunam.dgb.unam.mx/
V.T. Sabe et al., Eur. J. Med. Chem. (2021). https://doi.org/10.1016/j.ejmech.2021.113705
N. Ye et al., Drug Discov. Today (2022). https://doi.org/10.1016/j.drudis.2021.12.017
M. J. Frisch et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009
M. Preuss et al., J. Comput. Chem. (2003). https://doi.org/10.1002/jcc.10372
For 3D models of those structures see by example http://www.chemspider.com and https://www.ncbi.nlm.nih.gov/
Acknowledgments
Authors acknowledge the DGTIC-UNAM for providing supercomputing resources on “Miztli” through the project LANCAD-UNAM-DGTIC-298.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hernández Castillo, J.M., Molina Brito, B., Soto Mercado, J.R. et al. Design and theoretical characterization of neomycin-guanine and neomycin-thymine using DFT methods. MRS Advances 8, 125–130 (2023). https://doi.org/10.1557/s43580-023-00540-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43580-023-00540-2