
Abstract
Depending on its composition, thermoplastic polyurethane (TPU)/polycaprolactone (PCL) blend may present interesting properties for biomedical applications. Here, we have extensively developed and characterized blends of TPU/PCL and composites with 5, 10, and 20 wt% of hydroxyapatite (HA). The thermal, rheological, and mechanical characterizations showed that the blends present an intermediate behavior between pure TPU and PCL. The 75TPU/25PCL blend exhibited the best shape memory performance at temperatures below 50 °C. Filaments of this blend presented a fixation rate of 86.6% and a recovery rate of 73.3% at 50 ℃. The addition of HA had little influence on the shape memory properties; on the other hand, it strengthened the material, reaching elastic modulus values close to 100 MPa. In vitro tests revealed a biocompatible behavior for all blends, and the composite with 10 wt% HA from the 75TPU/25PCL blend demonstrated greater cell proliferation compared to the other composites.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Shape memory polymers (SMP) are materials that can memorize a permanent shape through covalent or physical crosslinking, and a temporary shape of nonequilibrium can be achieved through fixation by crystallization, vitrification, reversible crosslinking, or other means [1,2,3]. SMP maintain their temporary shape until a triggering event, such as thermal [4,5,6], light [7,8], pH change [1], contact with specific cells [3], or exposure to other stimuli is applied, after which SMP regain their permanent shape. Among these stimuli, the most commonly used in SMP is heat, where its shape programming involves heating the material above a transition temperature (Ttrans), such as the melting point (Tm), or the glass transition temperature (Tg), followed by a deformation to the desired shape, and then cooling it below Ttrans. The polymer retains its shape due to switching units that resist permanent shape recovery but can recover its original shape after reheating above Ttrans due to permanent net points [1,2].
Recently, there is an increasing interest in the development of biocompatible SMP for diverse biomedical applications, such as in self-knotting biodegradable sutures [4,9,10], materials designed for drug delivery [3], cardiovascular implants [11,12], bone tissue engineering [7,13,14,15], among other applications detailed in the reviews of Melly et al. [2] and Delaey et al. [16]. For bone tissue engineering, the excellent shape recovery properties of SMP and easy/low-cost process capability have attracted much attention in their possible use in minimally invasive surgery for repairing critical bone defects. During the surgical process, the initial shape of the SMP implant is larger than the size of the bone defect, and the temporary shape obtained is much smaller than the defect. When the implant recovers to its initial shape in the human body, the recovered shape can perfectly adjust to the size of the bone defect, and there is a tailored force between the boundaries of the defect region and the implant, which is beneficial for osseointegration and bone ingrowth [7,13,14,17].
The development of new biocompatible SMP can be performed by chemical synthesis of polymers, but it tends to be complex and time-consuming. Alternatively, polymer blending is an easy method to prepare SMP [4]. Thermoplastic polyurethane (TPU)/polycaprolactone (PCL) blends are materials that present excellent shape memory performance [4,5,6,8,18]. In these blends, TPU is used as net points to determine the permanent shape, and PCL, which is a semicrystalline polymer with a Tm of approximately 55–60 °C, is used as switching units to control the shape fixing and recovering [4,6,19]. In addition to the excellent shape memory performance, TPU/PCL blends have a wide range of mechanical properties, from flexible to rigid, which depends on the proportion of the polymers and the TPU chemistry. Since PCL is a well-known biocompatible and biodegradable material, the biocompatibility and biodegradability of the TPU/PCL blends are also dependent on the TPU composition [11,12,20,21]. The blends formed by these polymers can also be used in 3D printing [8,18], which is interesting when considering the production of porous functional substitutes for the biomedical field.
TPU/PCL blends can be obtained by melting (internal mixer [5,19], extrusion [4,8,18], and injection molding [4]), or by solution [6,11]. The most common proportions applied in the literature range from 10 to 75 wt% of PCL [5,6,11,18,19,22,23], and the results of the shape recovery (Rr) and shape fixation ratio (Rf) vary from one study to another. This may be associated with the degree of elasticity of each TPU due to the different compositions. The shape recovery from these blends can be directly activated by heating or indirectly with light and electricity. In these cases, it is necessary to add a photoactive agent [8,22] or conductor [6], which allows the absorption of the specific stimulus and subsequent conversion into thermal energy.
Although TPU/PCL blends have attractive shape memory properties that can be explored in the biomedical field, only a few studies have been reported, focusing mainly on cardiovascular implants [11,12]. For the advancement of TPU/PCL blends in the biomedical field, the biological characteristics of these materials must be optimized due to the low bioactivity and hydrophobic character that both polymers present, which can result in weak interactions between the material and the cells [24]. One way to overcome this limitation is by adding bioactive fillers such as hydroxyapatite (HA). HA is a calcium phosphate widely used for bone repair since its similar composition to natural bone imparts osteoconductivity, bioactivity, and resorbability [25,26]. Recent studies have demonstrated the effective role of HA in the improvement of the mechanical performance, hydrophilicity, and biocompatibility of polymeric matrices [25,27,28]. Composites of TPU/HA [29,30,31] and PCL/HA [20,27,32] have demonstrated promising outcomes for bone tissue repair. However, there is still a lack of studies that report the production of TPU/PCL/HA composites and their properties.
We envisioned that the processing versatility of TPU and PCL, their mechanical properties, and the shape memory of the blend, combined with the bioactivity and osteoconductivity of HA, can result in composites with adequate properties for biomedical applications. Based on this, we produced TPU/PCL blends with different ratios and incorporated 5, 10, and 20 wt% of HA. The structural, thermal, rheological, mechanical, and biological characterization confirmed the promising properties of the TPU/PCL blend and, especially, the TPU/PCL/HA composite for bone tissue engineering and suture threads. The bioactive composite displayed a thermo-responsive trigger and high recovery ratios at temperatures below 50 °C.
Results and discussion
Mixing process and rheological characterization
The production of TPU, PCL, TPU/PCL blends, and composites with different HA contents was performed using a Haake torque rheometer. Figure 1 displays the torque as a function of time for all formulations, and Table 1 presents the torque at the end of mixing. Pure TPU [Fig. 1(a)] presented an equilibrium torque of 5.9 N m after 5 min. PCL reached a torque stability of 1N m after 1 min. The blends exhibited an intermediate behavior between that observed for TPU and PCL. The equilibrium torques for the 75T25P, 50T50P and 25T75P blends were 4.0, 2.5 and 1.4 N m, respectively. As the PCL fraction was increased, the percentage of reduction in the equilibrium torque (Table 1) of the blends compared to TPU was 30, 55 and 76%, almost proportional to the PCL mass fraction in the blend.

Torque as a function of time for (a) TPU, PCL and their blends. Torque as a function of time for the composites with 5, 10 and 20 wt% of HA in (b) TPU, (c) 75T25P, (d) 50T50P, (e) 25T75P and (f) PCL matrices.
For composites with a TPU matrix [Fig. 1(b)], the addition of HA caused a decrease in the equilibrium torque. For the TPU20HA composite, the equilibrium torque was reduced by 44% compared to pure TPU. This reduction suggests that HA might catalyze degradation reactions in the TPU matrix.
In composites with 75T25P, the effect of HA on the equilibrium torque was similar to that observed for TPU composites. For those obtained with the 50T50P matrix with 5 and 10 wt% of HA, the equilibrium torque was practically the same as in the blend. This behavior can be attributed to the higher fraction of PCL. Since PCL melts at lower temperatures, possibly a part of the HA tends to be incorporated at the beginning of the mixture by the PCL phase. With this incorporation, there is a reduction in the amount of HA directly in the TPU, and consequently, it may cause less impact on its degradation. For 20 wt% of HA, a small reduction of the equilibrium torque is observed due to the high content of HA [Fig. 1(d)]. There is also a trend in torque reduction as the mixing proceeds, which indicates thermal degradation reactions.
The 25T75P blend and the 25T75P5HA composite exhibited similar behavior, with equilibrium torque occurring after 1.5 min and standing at 1.4 N m. Additionally, the early melting of the PCL, and the low HA content, may have interfered with the distributive and dispersive mixing of the TPU phase due to the low viscous stresses of the PCL and the PCL/HA mixture on the TPU. For the 25T75P10HA and 25T75P20HA formulations, the equilibrium torque increased compared to the other formulations, stabilizing at 1.7 and 1.9 N m, respectively. In these samples, the HA content may have caused enough viscous stresses to disperse the TPU fraction in the blend, as the torque between 0.5 and 2.5 min (TPU melting range) was considerably higher than that in the 25T75P blend and its composite with 5 wt% of HA.
In the composites with the PCL matrix, it was observed that there was no reduction in the equilibrium torque as a function of HA content. This suggests that there is no significant degradation in the mixing time employed. For the PCL, PCL5HA, and PCL10HA, there was no significant torque variation after 5 min, standing at 1 N m. On the other hand, the incorporation of 20 wt% of HA yielded an increase of 50% in the torque compared to pure PCL. Similar behavior was observed by Backes et al. [20] when studying the processability of PCL/HA composites by torque rheometer. Their results showed that high levels of HA significantly affect the entanglement dynamics, acting as another restriction for the movement of polymeric chains. Since PCL is the major phase in the 25T75P20HA composite, the increase in equilibrium torque observed must also be associated with this restriction of movement. It is noteworthy that, although the torque curves do not indicate degradation of PCL/HA composites, this may occur at higher HA contents [20].
Rheological properties are strongly consistent with the structural characteristics of a polymeric material. The presence and intensity of interactions between components in composites were investigated through the rheological behavior in a steady state. The curves of shear viscosity as a function of shear rate for TPU, PCL, TPU/PCL blends, and their respective HA composites are presented in Figure S1. Table 1 presents the viscosity at the shear rate of 0.01 s−1. It can be observed in Figure S.1a that PCL presents a more Newtonian behavior through the whole shear rate range analyzed [20,33,34]. TPU, on the other hand, due to its complex composition and structure with hard (HS) and soft segments (SS), which affect the rheological behavior, doesn’t present a Newtonian plateau at low shear rates. The viscosity of TPU at a shear rate of 0.01 s−1 is about three orders of magnitude higher than that of PCL. The high value of TPU’s viscosity is probably related to the molar mass and the presence of HS that form a molecular anchorage between the polymer chains, hindering their movement. Another point that must be considered is the different melting points of the polymers.
It is noteworthy that the rheological curves were obtained at 210 °C, and the low viscosity of PCL is the result of the viscosity dependence of temperature, which follows the Arrhenius equation. For higher temperatures, there is a reduction in the baseline of the rheological curve. In molecular terms, the increase in temperature increases the free volume, thus facilitating the movement of polymeric chains. Increased temperature also increases molecular vibration and the likelihood of intermolecular contacts, consequently, the entanglement and disentanglement speeds become similar for a longer time, shifting the Newtonian plateau to higher shear rates.
The TPU/PCL blends (Figure S1a) presented intermediate behavior between TPU and PCL. The 75T25P blend exhibited very similar behavior and viscosity values at a shear rate of 0.01 s−1 to pure TPU. On the other hand, the 25T75P blend was similar to pure PCL, while the 50T50P presented an intermediary curve.
Regarding the effect of the HA content on the rheological behavior of the composites obtained with TPU and with the 75T25P blend, the addition of HA caused a considerable reduction in viscosity. The composites with 5, 10 e 20 wt% of HA presented a reduction in the viscosity at a shear rate of 0.01 s−1 of 68, 84, and 92% compared to neat TPU, and 67, 91, and 95% compared to the 75T25P blend, respectively. These results suggest that HA leads to thermal degradation reactions during the processing of these composites, which is in agreement with the torque results. The degradation might occur due to the presence of hydroxyl groups (–OH) in the HA structure, which can accelerate the bond cleavage of polymer chains, mainly in urethane bonds, due to its instability above 190 °C [35,36]. In the composites from the 50T50P blend, HA also caused a reduction in viscosity compared to the blend, but this effect is much more attenuated due to the higher content of PCL.
The 25T75P blend, and the PCL composites presented a different behavior, displaying increasing values of viscosity at the shear rate of 0.01 s−1 with increasing amount of HA. A possible explanation for this is that the high HA content increases the surface area for interactions between HA particles and PCL polymer chains. Another important point that may contribute is the increase in particle–particle interactions. These considerations have already been observed in works of PCL/HA composites [20,34].
The HA increases the pseudo-plastic response for shear rates greater than 0.1 s−1 for all 25T75P composites. For shear rates greater than 1 s−1 for the 50T50P20HA composite, the viscosity decreases rapidly with increasing shear. As the shear rate increases, the disentanglement of the polymer chains and/or the cleavage of their bonds prevail over the effect of the material interaction; consequently, there is a marked reduction in viscosity. For the PCL/HA composites (Figure S1f), no significant differences were observed between the different HA concentrations in terms of viscosity for shear rates above 1 s−1. However, at low shear rates, PCL20HA presented a typical power-law behavior, associated with the large number of particles, which alter the polymer chains dynamics [20]. The greater number of particles increases the internal friction and, therefore, the viscosity at lower shear rates. However, as the rate increases, the particles and chains align and converge to the same behavior as the other samples.
The rheological properties play a critical role in understanding the results of the biomaterials proposed in this work. The data show the direction of the degradative process with the addition of HA and its possible preferential location in the PCL phase. These trends influence the final morphology of the blends and composites, such as the size of the droplets in the dispersed phase and possible agglomerates of HA, which consequently influences the biological, mechanical and shape memory responses of the studied materials.
Morphological characterization
The morphological characterization of TPU, PCL, the 75T25P blend with PCL removal in acetone, and the 50T50P and 25T75P blends without removal are shown in Fig. 2. It also presents the surface fracture and the compositional mapping of the main HA elements, which are used to analyze the distribution of HA in the composites. TPU presents a fracture with regions of large grooves and smoothness. The fracture surface of the 75T25P blend exhibits a droplet-in-matrix morphology, in which the PCL droplets are dispersed and distributed throughout the TPU matrix. For the 50T50P blend, a co-continuous structure was observed, in which the polymeric phases are continuously intertwined. Regarding the 25T75P blend, it is possible to identify the two phases without removing the TPU, and its morphology is also of droplet-in-matrix. In this case, as PCL melts easily and has a low viscosity at the process temperature, the viscous stresses were not enough to break down and disperse the TPU droplets throughout the PCL. Therefore, there was no morphology refinement in the 25T75P blend, the phases were easily identified, and large TPU domains were observed. The fracture surface of pure PCL presented small and large grooves, without smooth regions.

Cross-sectional SEM and EDS mapping (calcium (Ca) in red and phosphorus (P) in yellow) (scale bar 50 µm).
A good distribution of HA is observed in the EDS mapping of all composites based on TPU and 75T25P matrices. For the 75T25P matrix, although PCL melts first and may coat HA particles, the mapping result shows that the percentage of PCL was insufficient to induce preferential localization of HA particles, which are well dispersed in the matrix. A different trend occurred with the 50T50P composites, where specific regions with a large amount of localized HA were observed. These regions are in the PCL phase, and such behavior aligns with that observed in rheology. Nevertheless, HA is observed to be well dispersed within the PCL phase. There is a good HA distribution in the composites with the 25T75P blend, and few agglomerates are observed in the EDS mapping. In the composites with the PCL matrix, larger agglomerates are noticed, mainly with 20 wt% of HA. These may have resulted from the low viscosity of PCL at the processing temperatures used in this work. Although the internal mixer promoted relatively good dispersive and distributive mixing, the low viscosity of PCL may not have allowed sufficient viscous stresses for adequate dispersive mixing.
Furthermore, the HA clusters observed in the EDS mapping are of the order of 5 to 30 μm. This result is interesting, considering that these materials can be processed in the future by 3D printing (material extrusion), where the diameter of the nozzle used for printing is generally 400 μm. Therefore, it is expected that these particles wouldn’t cause nozzle clogging during printing.
Thermal properties
The pressed samples were submitted to DSC and TGA analysis to obtain the thermal properties of the materials. TGA curves (Figure S2) were used to extract the temperature of the beginning of thermal decomposition (Tonset), the temperature(s) at which there is a maximum variation of weight (Tpeak), which were obtained by the derivative of the TGA curve (DTG, Figure S3, supplementary material), and the residue at the final temperature of analysis, which is presented in Fig. 3.

TGA and DSC results for TPU, PCL, TPU/PCL blends, and their respective composites with HA: (a) temperatures of maximum mass variation (Tpeak); (b) temperature of the onset of decomposition (Tonset); (c) residue; (d) melting temperature (Tm); (e) crystallization temperature (Tc); (f) glass transition temperature (Tg) of TPU and degree of crystallinity (Xc) of PCL.
TPU presents thermal decomposition in two stages, with Tpeak at 338 ℃ and 420 ℃ (Figure S2a and Fig. 3a). It is common for polyester-based TPUs to show a two-step decomposition behavior in the temperature ranges around 270–370 ℃ and 370–440 ℃, attributed to the thermal degradation of the urethane groups and the decomposition of the flexible segments with random scission of ester bonds, respectively [37]. TPU has lower thermal stability (Tonset = 308 ℃) when compared to PCL (Tonset = 393 ℃). In addition, PCL presents thermal degradation behavior in a single step, with Tpeak at 418 ℃. This value is very close to the second TPU Tpeak in the blends, so it was not possible to distinguish these two temperatures. The values found for PCL in this work are identical to the ones observed in the work of Beatrice et al. [33], which used the same grade of PCL, but employed a processing temperature of 140 ℃. All of the blends (75T25P, 50T50P and 25T75P) presented Tonset similar to pure TPU, and a decreasing intensity of the first degradation event with higher PCL fraction (Figure S3).
Regarding the effect of HA on the thermal properties of the matrices, the addition of 5 and 10 wt% of HA did not result in significant changes in Tonset. The variation was a maximum of 6 °C, except for 25T75P10HA (details of these values are noted in Table S1). For this specific composite, and the formulations with 20 wt% of HA, there was a more significant reduction in Tonset, as can be seen in Fig. 3b. Additionally, the greatest reduction in Tonset was observed in the composites with the 25T75P matrix when compared to the other formulations. This trend can be attributed to the viscous stresses of the PCL/HA mixture on the TPU, given that PCL melts in less than 1 min and the presence of 10 and 20 wt% of HA may have provided greater viscous stress in the low fraction of TPU (25 wt%) in the blend. Consequently, the thermo-mechanical degradation intensified. Therefore, this may be reflected in the thermal decomposition data.
For the first Tpeak of TPU, which is related to the thermal degradation of the urethane groups, there was a shift towards lower temperatures with a higher content of HA. This result comes from the hydrophilicity of HA and its hydroxyl groups (–OH), which in association with high temperature and significant level of shear imposed by the internal mixer, can accelerate the cleavage of urethane bonds, facilitating the degradation of the polymer. In the second Tpeak, no changes that exceeded a variation of 7 °C between the composites and their respective matrices were observed (Fig. 3a).
The residue at 800 °C (Fig. 3c) shows that neat TPU presents a high percentage (11%) of residue at the end of the degradation processes, which is associated with the composition of the TPU that favors the formation of carbon-rich residue during TGA under a nitrogen atmosphere [38]. On the other hand, PCL was decomposed almost completely. The residue values in the composites, except for the TPU20HA formulation, show good agreement with the nominal percentage of HA, confirming the composition and homogeneity of the samples. For TPU20HA, the formation of HA agglomerates not incorporated by the TPU matrix justifies the low value obtained.
The thermal transitions of the blends and the influence of HA addition on the characteristic temperatures (Tg, Tm, and Tc) of TPU, PCL, and TPU/PCL blends were investigated by DSC. In Figure S4, the DSC curves of the compositions during the cooling cycle and the second heating cycle are presented, and the results extracted from these are shown in Fig. 3. It should be noted that Tm for TPU was considered in the largest melting endotherm.
In the blends, Tm of both TPU and PCL was almost unaffected by the varying proportions, with values between 206–208 °C and 56–57 °C, respectively (Fig. 3d). More details of these values are noted in Table S1 of the supplementary material. The same behavior was observed for Tg (Fig. 3f) of TPU and Tc of both polymers (Fig. 3e); the maximum variation of these characteristic temperatures was 3 °C in the blends. The only exception was the 25T75P blend, in which it was not possible to determine the characteristic TPU temperatures due to the low fraction in the blend and its low melting enthalpy.
Regarding the influence of HA in the Tm of TPU and PCL, it resulted in a significant reduction for TPU, shifting from approximately 208 °C without HA to about 186 °C with 20 wt% of HA. These results indicate that HA may have acted by hindering the reassociation of the urethane group, which could have dissociated due to the instability of the urethane bonds, a phenomenon called transurethanization [35,39,40,41]. The size of the HS influences the Tm, therefore, the reduced size of the segments may have caused the shift of the melting endotherms to lower temperatures. The Tc of TPU was also influenced by HA content. Increasing amount of HA shifted Tc to higher temperatures, indicating that HA induced the crystallization of the smaller HS. Concerning the Tg of TPU, only subtle changes were observed. Differently from in TPU, the addition of HA practically did not affect Tm, Tc and Xc of PCL (Fig. 3d–f).
Mechanical characterization
The selection of materials for biomedical applications requires the knowledge of their mechanical properties. For example, biomaterials used for tissue engineering need to present similar mechanical properties to the native tissue. Therefore, the mechanical behavior of the TPU/PCL blends and TPU/PCL/HA composites were assessed by compression test. Figure 4 presents the elastic modulus of the samples, calculated from the stress × strain curves (Figure S5, supplementary material). The samples were analyzed using a 500 N load cell and did not fracture at maximum load.

Elastic modulus obtained from the compression test. Data are expressed as mean ± standard deviation. The statistical analysis was expressed relative to the matrices, unless marked with a bar (*p < 0.05; **p < 0.01; ***p < 0.001).
The pure TPU presented the lowest elastic modulus, with an average of 48.4 ± 0.8 MPa, followed by the blends 75T25P with 57.4 ± 0.7 MPa, 50T50P with 78.6 ± 2.6 MPa, 25T75P with 83.4 ± 3.8 MPa, and PCL with 92.1 ± 4.4 MPa. Similar behavior of increasing the elastic modulus with the addition of PCL was reported by other authors for TPU/PCL blends [4,5,23,42]. TPUs are generally flexible and PCL has a crystallizable structure, consequently, increasing concentration of PCL in the blend yielded increasing elastic modulus.
TPU composites with different HA contents showed a statistically significant increase in modulus compared to the TPU matrix (p < 0.001). Although the incorporation of 5 and 10 wt% of HA showed increasing modulus, the addition of 20 wt% of HA led to a slight decrease in modulus, achieving a value similar to the TPU5HA sample. This behavior might have occurred due the intensified degradation during the processing of the composite, as indicated by torque rheometer, rheological, and thermal analyses.
Regarding the composites obtained with the 75T25P, 50T50P, and 25T75P blends, the increasing addition of HA caused a crescent improvement in the elastic modulus when compared to the respective blend used as matrix (p < 0.001). The reinforcement capacity of HA in TPU and PCL matrices has already been reported in many studies [20,30,31,43]. The 75T25P20HA, 50T50P20HA and 25T75P20 composites showed an increase in the elastic modulus of 41, 25 and 36%, respectively, compared to their matrices.
For the composites with PCL, the addition of HA improves the compression modulus of the material. However, there was no significant difference between the PCL/HA composites, and this behavior has already been reported in a recent study [20]. This result may be associated with non-uniform HA dispersion. As seen in Fig. 2, the HA clusters formed in the PCL matrix. These can act as stress concentrators and affect the improvement of the elastic modulus.
The elastic modulus of all composites and blends correspond to the values required for cancellous bone grafts under a compression regime (cancellous bone modulus ranges from 10 to 2000 MPa [44]). Furthermore, the elastic modulus range is also suitable for regenerating other rigid tissues such as ligaments, cartilage, and tendons [45].
Shape memory effect
Advances in SMP hold considerable promise across the biomedical field. In tissue engineering, SMP may enable minimally invasive surgeries, and the shape recovery process strengthens the contact between the material and the surrounding tissue, improving material integration [7,15]. Here, the shape memory properties in the 75T25P, 50T50P and 25T75P blends were investigated with programming carried out at the respective temperatures of 65 °C, 57 °C and 53 °C, and then cooling in an ice bath. In the 75T25P samples, it is desirable that during programming, the entire PCL fraction is molten so that during cooling, it can maintain the required shape, hence the temperature selection of 65 °C. In the 50T50P and 25T75P blends, temperatures much higher than the melting temperature cause the destruction of the original shape, so it is impossible to program the temporary shape. Therefore, the programming temperature selected for each sample ensured that the specimens could be designed to the desired state and still possess their dimensions and structure during programming.
For the shape recovery, the temperatures used were selected based on the DSC curves performed on the samples after programming (Fig. 5a). The DSC curves show a wide range for PCL melting, starting at 41 °C and ending at approximately 62.5 °C. Two melting peaks are observed within this range, one less intense at around 43 °C and the second around 55 °C, which are associated with the level and perfection of the crystals formed during the cooling. Therefore, the shape recovery was analyzed in different temperatures: 45, 50, 55 and 60 °C. Figure 5b shows the procedure of the U-bending test performed on the blends and composites in rectangular plates.

(a) DSC curves of the TPU/PCL blends after programming. (b) U-bending test performed on the blends and composites in rectangular plates. (c) Photos of the initial shape, temporary shape and shape recovery of the TPU/PCL blends at different temperatures.
In Fig. 5c, photos of the blends are displayed in their initial shape, after programming (temporary shape), and after recovery at the selected temperatures with an exposure time of 30 s. It is observed that the 75T25P sample did not show a good capacity to fix the desired temporary shape compared to the 50T50P and 25T75P blends. On the other hand, the 75T25P blend presents a higher level of shape recovery at lower temperatures, as observed in the shape recovery performed at 45 and 50 °C. For the 50T50P and 25T75P blends, temperatures below 50 °C are not sufficient to melt a sufficient portion of the PCL, and the stored energy cannot fully drive the movement of the PCL phase. It was also observed that the 75T25P blend maintains structural stability at temperatures greater than 55 ℃. That is because TPU, which is responsible for the specimen's integrity, is the major phase in this blend, which no longer occurs in the other blends. The supplementary video S1 displays detailed information about the shape recovery procedure of the 75T25P blend in the U-bending test.
Rf and Rr (obtained at 55 °C for 30 s) of the blends and the selected composites are shown in Figure S6. This recovery temperature was selected to evaluate Rr in the three blends, because it allows the best performance without destroying the plate shape of the 50T50P and 25T75P blends. As can be visualized in Fig. 5c, the 75T25P sample exhibited the lowest Rf performance (80.2%) compared to the 50T50P and 25T75P blends, which had an excellent performance, with Rf values of 97.4% and 99.7%, respectively (Figure S6a). A similar result was observed in previous works [6,19]. The low value presented by the 75T25P blend is due to the higher TPU content, which imposes a greater shape restoring force, not being completely suppressed by the low fraction of PCL (25 wt%). Although Rf of the 75T25P sample is low compared to the other blends, this is still considered a great value for SMP, and similar values for blends with proportions close to this one are presented in Table 2.
The three blends presented high values of Rr, confirming their ability as SMP. These were 74.8, 77.6 and 71.1% for 75T25P, 50T50P and 25T75P, respectively. Such values are similar to those observed in previous works [18,22]. However, the activation temperature of this work is lower than those observed in the previous ones, as shown in Table 2, and this result is interesting for the biomedical field. The values for the three blends are very close, which is unexpected, since the 25T75P sample should have a much lower Rr value than the other blends. That is because PCL is the majority fraction in this system and it has a low capacity for shape recovery. However, due to the low TPU fraction in the 25T75P blend, and poor distribution and dispersion, as observed in the micrograph of Fig. 2, TPU may not be associated with this result. A possible explanation is that Rr could be more related to the stress relief caused by deformation in the PCL crystalline regions, which did not melt in the shape programming, than the energy stored in the TPU, which provides the recovery force.
One of the requirements to apply the thermos-responsive SMP is that the sample must have mechanical resistance at the conduction temperature of the recovery process [18]. In the case of samples 50T50P and 25T75P, the recovery rate cannot reach high values at temperatures below 50 °C, but when the conduction temperature is 55 °C or higher, the shape memory recovery rate of the material increases and can reach almost 100% (for example at 60 °C, Fig. 5c). Although it was experimentally observed that the 50T50P and 25T75P samples maintained dimensional integrity (the rectangular shape of the samples was not destroyed) at the activation temperature of 55 °C, the mechanical strength was compromised. Figure S7 and the supplementary video S2 show that the 50T50P and 25T75P blends cannot displace a 4.6 g metal part with the shape recovery action activated at 55 °C. On the other hand, the 75T25P blend easily displaces it in a short time. Video S3 shows more details of the 75T25P sample mechanical resistance during the shape recovery process with weight imposition.
Regarding the effect of HA on shape memory, Rf and Rr were evaluated only in the 75T25P blend, considering that it presented a greater possibility of recovery at lower temperatures, which is important in the biomedical field. Figure S6b presents Rf and Rr results for the 75T25P blend and its composites with 5, 10 and 20 wt% HA. The presence of HA did not significantly influence the fixation and shape recovery of the specimens. Rr values of this blend and its composites were obtained at a temperature of 55 °C for 30 s of immersion. This condition was selected considering the lowest temperature with the high performance of Rr. These parameters can still be considered harsh to the human body when taking into account the maximum temperature supported by bones without causing permanent damage, which is less than 46 °C for a time of 60 s [46,47]. However, as observed in Fig. 5c, the 75T25P blend presents a certain degree of recovery at temperatures lower than 55 °C, allowing the possibility for further investigations.
Additionally, the activation temperature for recovery can be further reduced, with the decrease of the degree and perfection of PCL crystallinity by reducing molar mass and increasing the cooling rate on programming. Evidently, a balance between lower activation temperature and sufficient fixation rate must be sought for this case. In addition, the use of indirect heating, for example, with near-infrared light, is a real possibility to recover the material’s shape while preventing the surrounding tissue from being affected by the temperature. Near-infrared light is already widely used in biomedical applications due to its harmless nature and excellent tissue penetration capacity. However, it is necessary to add photothermal agents, such as magnesium particles [7] and polydopamine nanospheres [22], both already used and explored in the biomedical field. These allow the absorption of light and subsequent conversion into thermal energy, located only in the desired structure.
The shape memory behavior of the 75T25P blend was further explored at various temperatures and with samples in the filament format (Fig. 6), considering the possible use of this material for 3D printing, stent, and surgical sutures. The results showed that Rf measured 20 min after removing the material from the ice bath was 86.6%. The recovery is temperature dependent, with values of 30, 36.6, 43.3 and 73.3% with an exposure time of 10 s at temperatures of 42.5, 45, 47.5 and 50 °C, respectively (Fig. 6b). The recovery speed of the programmed filament at 50 °C is observed in more detail in the supplementary video S.4. Figure 6c shows the self-knotting ability of the filament. The application of a temperature of 50 °C was able to close the filament knot in 2 s without applying external forces, simply by the shape memory action of the material. This behavior also allows the 75T25P blend to be exploited as smart surgical sutures, as they can be applied loosely in their temporary shape. When thermally activated, the sutures will shrink and tighten into a knot due to their internal energy, thus performing their suturing function. The detailed self-knotting process is described in the supplementary video S.5.

(a) Shape memory test in a filament of the 75T25P blend. (b) Photos of the initial shape, temporary shape with average Rf, and shape recovery with average Rr at 42.5, 45, 47.5 and 50 °C. (c) Demonstration of the knot tightening process in a 75T25P blend filament, after only 2 s at 50 °C, the knot is firmly tightened by itself.
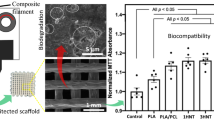
Culture cellular assay
The resazurin assay was carried out to evaluate the proliferation of osteoblasts during 1 and 7 days of culture in the samples of TPU, PCL, TPU/PCL blends and composites with 5, 10 and 20 wt% of HA from the 75T25P blend (Fig. 7). After 1 day, the 75T25P/HA composites showed no significant difference compared to the 75T25P blend. The 50T50P and 25T75P blends showed similar behavior to PCL, achieving a greater cell proliferation when compared to TPU. After 7 days, all samples reached a substantial increase in reduction percentage compared to the first day, resulting from a higher number of viable osteoblasts cells that metabolized resazurin into resuforin. The 75T25P blend exhibited a percentage reduction close to that of TPU, the 25T75P blend close to PCL, and the 50T50P blend an intermediary result between the two polymers. Similar to the first day, PCL and the 50T50P and 25T75P blends showed a significant difference compared to TPU.

Cytotoxicity assay after 1 and 7 days of osteoblast culture (MC3T3-E1) on TPU, PCL, TPU/PCL blends and 75T25P/HA composites with HA at 5, 10 and 20 wt%. The statistical analysis was expressed relative to the TPU at the same time point, unless marked with bars (*p < 0.05; **p < 0.01).
The 75T25P/HA composites with higher HA contents improve the cellular response compared to the pure 75T25P blend, evidencing the positive effect of the HA addition. During cell culture, extracellular matrix proteins are adsorbed on the biomaterial’s surface, assisting in subsequent cell attachment. For osteoblast culture, fibronectin and vitronectin are two important proteins that affect cell attachment and spread. It is known that calcium phosphate additives can aid in the adhesion of these two proteins, thus promoting the fixation and proliferation of osteoblasts [48]. In the 75T25P5HA composite, the low HA content does not significantly affect osteoblast proliferation, and this result may be associated with the encapsulation of HA particles by the matrix, which ends up not impacting cell proliferation. For the 75T25P10HA composite, the content of 10 wt% of HA was effective to increase the proliferation of osteoblasts (p < 0.05) in relation to the pure blend. For 75T25P20HA, although there is an increase in the reduction percentage, there is no significant difference compared to the 75T25P blend; this might be related to the thermal degradation of the matrix by HA at a content of 20 wt%. As raised by the thermal analysis data, the high HA content may have acted by preventing the reassociation of the urethane groups, which might affect the proliferation of osteoblasts.
Conclusions
Shape memory bioactive materials based on TPU/PCL blends and TPU/PCL/HA composites were developed by melt blending for applications in the biomedical field. The rheological and thermal characterization of the composites indicated that the addition of HA intensified thermal degradation reactions, mainly in the TPU matrix, in which lower viscosities and thermal stability were observed. Despite of that, the composites are still stable under physiological conditions. The increasing HA concentration (5, 10 and 20 wt%) favored the mechanical properties of the blends (75T25P, 50T50P, and 25T75P), achieving elastic moduli up to 113.2 MPa, i.e., 134% higher than pure TPU and 23% higher than pure PCL.
Regarding shape memory capability, the 75T25P blend presented the best performance at temperatures below 50 °C. The addition of HA did not change the fixation or shape recovery rate. In 1.4 mm filaments, the recovery achieved was of 30% of its initial size at 42.5 °C, and 73.3% at 50 °C after 10 s. The biocompatibility results showed that TPU, PCL, TPU/PCL blends, and the 75T25P composites with HA allow cell proliferation. Overall, the 75T25P10HA composite presented the most promising results, combining mechanical strength, bioactivity and shape memory ability.
Ultimately, the results indicate that the developed composites can provide a promising strategy in the biomedical field, exploring shape memory to enhance the fixation of the material with the tissues and in the development of minimally invasive surgeries for tissue repair. Future works will be focused on reducing recovery temperature and the development of 4D printed materials.
Experimental
Materials
TPU (Elastollan 590 A 13000, density of 1.25 g cm−3 and hardness of 92 shore A) was kindly donated by BASF Co., Ltd. PCL (CAPA 6500) was kindly donated by Ingevity/Perstorp. HA [(Ca5(OH)(PO4)3, Ca/P ratio of 1.62, d50 < 4 μm, #21,223, CAS#: 1306-06-5] was purchased from Sigma Aldrich. TPU and PCL were cryogenically milled using a Mikro-Bantam® hammer mill (Hosokawa Micron Powder Systems) and dried under vacuum for 48 h at 60 °C for TPU and 40 °C for PCL.
Preparation of TPU/PCL blends and TPU/PCL/HA composites
The processing of TPU, PCL, TPU/PCL blends with different ratios of TPU (25, 50, and 75 wt%), and composites with 5, 10 and 20 wt% of HA were performed employing an internal mixer coupled to a Haake torque rheometer (Rheomix 600p, roller rotors, semi-interpenetrating, and counter-rotating) with the following conditions: 5 min of mixture at 60 rpm, and 190 °C. Before their use, TPU and PCL were dried for 24 h in a vacuum oven, while HA was dried for 24 h at 110 °C in an air-circulating oven. Afterward, the different compositions presented in Table S2. The blends were named with the first letter of the polymers (T = TPU and P = PCL) and with the mass ratio between the polymers, e.g., 75T25P indicates that this blend is composed of 75 wt% TPU and 25 wt% PCL. The composites have the name of the matrix plus the percentage of HA, e.g., 75T25P20HA is the composite obtained from the 75T25P blend with 20 wt% of HA. The TPU/HA and PCL/HA composites were produced to understand the influence of HA on each polymer separately and to compare with the composites from the blends in different proportions.
Scheme 1 summarizes the experimental procedure. The materials collected after blending were granulated in small pellets and hot-pressed in a hydraulic press (Marconi, MA098) for mechanical, biological, rheological, microstructural, thermal, and shape memory analyses. For this purpose, a metallic mold was used, and the following pressing conditions were applied: (i) pressure of 10 bar at 210 °C with a setting time of 5 min; (ii) cooling under pressure down to 160 °C; (iii) releasing the pressure and cooling to room temperature. This condition was determined after several attempts so that the final material was free of air bubbles. The same equipment was used to stamp circular disks (8 mm in diameter and 3 mm in thickness) out of the compressed samples using a biopsy punch. The disks were used for mechanical testing and cell culture.

Experimental setup and characterizations.
Characterizations
Rheological measurements
The rheological behavior of the compressed samples was evaluated in steady state using a compact modular rheometer (MCR 702 MultiDrive, Anton Paar) coupled with a parallel-plate geometry of 25 mm diameter, gap between plates of 1.0 mm, and nitrogen atmosphere. The rheological characterization was assessed from 0.01 to 100 s−1 at 210 °C.
Microstructural analysis
The morphology of TPU, PCL, TPU/PCL blends and composites with different HA contents was investigated by scanning electron microscopy (SEM), using a Philips XL-30 FEG. In parallel, compositional mapping via energy dispersive spectroscopy (EDS) was used in the composites to evaluate the distribution of HA within the matrices. All compressed samples were cryo-fractured in liquid nitrogen, and after fracture, only the 75T25P blend was subjected to the PCL extraction process with acetone at 40 °C for 40 min. This procedure was performed due to the difficult visualization of the phases in this composition. Prior to analysis, all samples were fixed on supports and covered with a thin layer of gold.
Thermal characterization
The characteristic glass transition (Tg) and crystalline melting (Tm) temperatures of TPU, PCL, blends and composites, and the degree of crystallinity (Xc) of PCL were evaluated by differential scanning calorimetry (DSC). The DSC curves were obtained in an equipment from TA Instruments, model Q2000, with the nitrogen gas flow set to 50 mL min−1. The method consisted of: (i) heating the sample from 30 to 240 °C at 10 °C min−1; (ii) 3 min isotherm; (iii) cooling the sample at a rate of 10 °C min−1 to − 70 °C; (iv) heating at 10 °C min−1 from − 70 to 240 °C. The Xc was calculated using the following equation:
where N is the mass fraction of the HA and/or TPU in the blend or composite, ∆Hm is the melting enthalpy, and ∆Hmº is the melting enthalpy considering a 100% crystalline PCL (139.0 J g−1 [33]). Previous to the measurements, the compressed samples were cut to obtain 7 mg of material and dried under a vacuum oven for 48 h at 40 °C.
The thermal stability of TPU, PCL, blends and all composites was evaluated by thermogravimetric analysis (TGA) using an equipment from TA Instruments, model Q50. The analyses comprised of heating the samples from 25 to 800 °C, using a heating rate of 20 °C min−1 under a nitrogen atmosphere with flow rate of 100 mL min−1. Previous to the measurements, the compressed samples were cut to obtain 15 mg of material and dried under a vacuum oven for 48 h at 40 °C. TGA analysis was also used to estimate the HA content incorporated in the matrices.
Compression testing
The stamped disks were tested in a universal testing machine Instron, model 5569, using a cell load of 500 N, a pre-load of 15 N, and a deformation rate of 1.3 mm min−1. The data were acquired and analyzed with the Bluehill 2.15 software. Five samples were tested for each composition, and the statistical analyses between sample groups were performed using one-way ANOVA followed by the Tukey test considering p < 0.05.
Shape memory test
The shape memory test was first performed on the 75T25P, 50T50P and 25T75P matrices using rectangular samples with 60 × 5 × 1 mm dimensions. Based on the results, the blend with the best performance was selected to evaluate the influence of HA on the shape recovery rate (Rr) and the shape fixation rate (Rf) of the composites, which are important indicators of an SMP performance. Rr is the capacity of the material to keep the memory of its permanent shape, while Rf assesses the extent to which the switching units fix the temporary shape [2]. For the blends and the selected composites, the shape memory was evaluated by the U-bending test [13,14,17]. In the original state, the angle of the specimen was noted as θ0, and then the following steps were performed: (I) heating the samples for 3 min at temperatures of 65, 57 and 53 ºC for 75T25P, 50T50P and 25T75P blends, respectively (temperatures were chosen based on DSC results, as explained further in the manuscript); (II) still under heating, the plates were deformed in a U shape (θP); (III) with the deformation still applied, the samples were rapidly cooled in an ice bath for 3 min; (IV) the fixed angle θf was measured after keeping the samples at room temperature for 20 min; (V) the samples were reheated at different temperatures (45, 50, 55 and 60 ℃) for 30 s; (VI) Finally, the recovery angle θr was measured after 20 min at room temperature. The heating of the samples was carried out in a water bath heated by heating plates, with the temperature controlled by thermometers. Rf and Rr were defined from Eqs. 2 and 3, respectively [13,17].
where θ0 = 0° and θp = 180° in this case.
The shape memory of the 75T25P blend was also evaluated using a stretched filament procedure. For this purpose, the sample was obtained as a 1.4 mm filament in diameter and 30 mm in length, using a heated metal syringe and mechanical pressure to extrude the material through the nozzle. Briefly, the samples were heated at 65 ℃ for 3 min, followed by deformation (Ɛp) to 100% of their initial length. Subsequently, they were quickly cooled in an ice bath for 3 min, with force still applied. After 20 min at room temperature, the fixed strain (Ɛf) of the filaments was measured using a caliper, and they were reheated at different temperatures (42.2, 45, 47.5, and 50 ℃) for 10 s. Finally, the residual strain (Ɛr) was measured. Rr and Rf were calculated according to Eqs. 4 and 5, respectively [4,6,13].
Osteoblast proliferation
The disks of TPU, PCL, TPU/PCL blends and composites of 75T25P with 5, 10 and 20 wt%, were sterilized with 70% alcohol under UV light on both sides for 15 min. After sterilization, the samples were rinsed with phosphate buffer solution (PBS), placed in a 48 well-plate and pre-incubated with 500 µL of the medium containing 89% v/v of α-MEM (Gibco) supplemented with 10% fetal bovine serum (FBS, Vitrocell) and 1% of antibiotic–antimycotic (Vitrocell) for 24 h in an incubator (series II 3110, Thermo Fisher Scientific) at 37 ℃, humidified and containing 5% CO2. After pre-incubation, pre-osteoblastic mouse cells (MC3T3-E1), previously cultivated, were seeded on the samples using a drop of 500 µL with a cell density of 50,000 cells. After the first day of cultivation, the samples were transferred to a new culture plate, and the culture medium was changed every 2–3 days. Following the supplier protocol, resazurin assay (#R7017, Millipore-Sigma) was used to assess cell proliferation. After 1 and 7 days of culture, the medium was removed, 500 µL of a medium/resazurin solution (90/10) was added per well, and the plate was incubated in the dark for 4 h at 37 °C. Three samples per composition were analyzed, including three negative controls containing cell-free resazurin solution. Subsequently, 100 µL of the solution of all samples (in triplicate) was transferred to a black plate where the fluorescence endpoint measurement was performed in a plate reader SpectraMax® M5 (560 nm/590 nm). An autoclaved resazurin solution was used as positive control to estimate the cell viability. Statistical analyses between sample groups were performed using analysis of variance (ANOVA) and Tukey’s post-test with a significance level of p < 0.05.
Supporting Information
Stress versus strain curves for TPU, PCL, and all polymer blends and composites; DSC curves for TPU, PCL, and all polymer blends and composites; TGA and DTG curves for TPU, PCL, and all polymer blends and composites; and table summary with results of the DSC and TGA (PDF).
Data availability
The data generated during this study might be available upon request.
References
C.C. Hornat, M.W. Urban, Shape memory effects in self-healing polymers. Prog. Polym. Sci. 102, 101208 (2020). https://doi.org/10.1016/j.progpolymsci.2020.101208
S.K. Melly, L. Liu, Y. Liu, J. Leng, Active composites based on shape memory polymers: overview, fabrication methods, applications, and future prospects. J. Mater. Sci. 55, 10975–11051 (2020). https://doi.org/10.1007/s10853-020-04761-w
J. Chen, L.E. Hamilton, P.T. Mather, J.H. Henderson, Cell-responsive shape memory polymers. ACS Biomater. Sci. Eng. 8, 2960–2969 (2022). https://doi.org/10.1021/acsbiomaterials.2c00405
X. Jing, H.Y. Mi, H.X. Huang, L.S. Turng, Shape memory thermoplastic polyurethane (TPU)/poly(ε-caprolactone) (PCL) blends as self-knotting sutures. J. Mech. Behav. Biomed. Mater. 64, 94–103 (2016). https://doi.org/10.1016/j.jmbbm.2016.07.023
S. Bhattacharya, R. Hailstone, C.L. Lewis, Thermoplastic blend exhibiting shape memory-assisted self-healing functionality. ACS Appl. Mater. Interfaces 12, 46733–46742 (2020). https://doi.org/10.1021/acsami.0c13645
M. Qu, H. Wang, Q. Chen, L. Wu, P. Tang, M. Fan, Y. Guo, H. Fan, Y. Bin, A thermally-electrically double-responsive polycaprolactone—thermoplastic polyurethane/multi-walled carbon nanotube fiber assisted with highly effective shape memory and strain sensing performance. Chem. Eng. J. 427, 131648 (2022). https://doi.org/10.1016/j.cej.2021.131648
Y. Zhang, C. Li, W. Zhang, J. Deng, Y. Nie, X. Du, L. Qin, Y. Lai, 3D-printed NIR-responsive shape memory polyurethane/magnesium scaffolds with tight-contact for robust bone regeneration. Bioact. Mater. 16, 218–231 (2022). https://doi.org/10.1016/j.bioactmat.2021.12.032
H. Bi, G. Ye, H. Yang, H. Sun, Z. Ren, R. Guo, M. Xu, L. Cai, Z. Huang, Near infrared-induced shape memory polymer composites with dopamine-modified multiwall carbon nanotubes via 3D-printing. Eur. Polym. J. 136, 109920 (2020). https://doi.org/10.1016/j.eurpolymj.2020.109920
A. Lendlein, R. Langer, Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 296, 1673–1676 (2002). https://doi.org/10.1126/science.1066102
S. Afewerki, S.V. Harb, T.D. Stocco, G.U. Ruiz-Esparza, A.O. Lobo, Polymers for surgical sutures, in: Advanced Technologies and Polymer Materials for Surgical Sutures, Elsevier, 2023: pp. 95–128. Doi: https://doi.org/10.1016/B978-0-12-819750-9.00004-8
M. Ansari, M. Golzar, M. Baghani, M. Soleimani, Shape memory characterization of poly(ε-caprolactone) (PCL)/polyurethane (PU) in combined torsion-tension loading with potential applications in cardiovascular stent. Polym. Test. 68, 424–432 (2018). https://doi.org/10.1016/j.polymertesting.2018.04.032
S.H. Ajili, N.G. Ebrahimi, M. Soleimani, Polyurethane/polycaprolactane blend with shape memory effect as a proposed material for cardiovascular implants. Acta Biomater. 5, 1519–1530 (2009). https://doi.org/10.1016/j.actbio.2008.12.014
C. Shuai, Z. Wang, S. Peng, Y. Shuai, Y. Chen, D. Zeng, P. Feng, Water-responsive shape memory thermoplastic polyurethane scaffolds triggered at body temperature for bone defect repair. Mater. Chem. Front. 6, 1456–1469 (2022). https://doi.org/10.1039/d1qm01635k
Y.J. Wang, U.S. Jeng, S.H. Hsu, Biodegradable water-based polyurethane shape memory elastomers for bone tissue engineering. Acs Biomater. Sci. 4, 1397–1406 (2018). https://doi.org/10.1021/acsbiomaterials.8b00091
Y. Zhang, J. Hu, R. Xie, Y. Yang, J. Cao, Y. Tu, Y. Zhang, T. Qin, X. Zhao, A programmable, fast-fixing, osteo-regenerative, biomechanically robust bone screw. Acta Biomater. 103, 293–305 (2020). https://doi.org/10.1016/j.actbio.2019.12.017
J. Delaey, P. Dubruel, S. Van Vlierberghe, Shape-memory polymers for biomedical applications. Adv. Funct. Mater. 30, 1909047 (2020). https://doi.org/10.1002/adfm.201909047
Y. Zhang, J. Hu, X. Zhao, R. Xie, T. Qin, F. Ji, Mechanically robust shape memory polyurethane nanocomposites for minimally invasive bone repair. ACS Appl. Bio Mater. 2, 1056–1065 (2019). https://doi.org/10.1021/acsabm.8b00655
Y. Wang, J. Zhang, M. Li, M. Lei, Y. Wang, Q. Wei, 3D printing thermo-responsive shape memory polymer composite based on PCL/TPU blends. J. Polym. Res. 29, 243 (2022). https://doi.org/10.1007/s10965-022-03095-2
X. Xu, P. Fan, J. Ren, Y. Cheng, J. Ren, J. Zhao, R. Song, Self-healing thermoplastic polyurethane (TPU)/polycaprolactone (PCL)/multi-wall carbon nanotubes (MWCNTs) blend as shape-memory composites. Compos. Sci. Technol. 168, 255–262 (2018). https://doi.org/10.1016/j.compscitech.2018.10.003
E.H. Backes, C.A.G. Beatrice, K.M.B. Shimomura, S.V. Harb, B.C. Pachane, H.S. Selistre-de-Araujo, L.C. Costa, F.R. Passador, L.A. Pessan, Development of poly(Ɛ-polycaprolactone)/hydroxyapatite composites for bone tissue regeneration. J. Mater. Res. 36, 3050–3062 (2021). https://doi.org/10.1557/s43578-021-00316-0
Y.-S. Joo, J.-R. Cha, M.-S. Gong, Biodegradable shape-memory polymers using polycaprolactone and isosorbide based polyurethane blends. Mater. Sci. Eng. C. 91, 426–435 (2018). https://doi.org/10.1016/j.msec.2018.05.063
Y. Chen, X. Zhao, C. Luo, Y. Shao, M.-B. Yang, B. Yin, A facile fabrication of shape memory polymer nanocomposites with fast light-response and self-healing performance. Compos. Part A Appl. Sci. Manuf. 135, 105931 (2020). https://doi.org/10.1016/j.compositesa.2020.105931
S. Qi, J. Fu, Y. Xie, Y. Li, R. Gan, M. Yu, Versatile magnetorheological plastomer with 3D printability, switchable mechanics, shape memory, and self-healing capacity. Compos. Sci. Technol. 183, 107817 (2019). https://doi.org/10.1016/j.compscitech.2019.107817
J. Lin, W. Wang, J. Cheng, Z. Cui, J. Si, Q. Wang, W. Chen, Modification of thermoplastic polyurethane nanofiber membranes by in situ polydopamine coating for tissue engineering. J. Appl. Polym. Sci. 137, 1–12 (2020). https://doi.org/10.1002/app.49252
N. Ramesh, S.C. Moratti, G.J. Dias, Hydroxyapatite–polymer biocomposites for bone regeneration: a review of current trends. J. Biomed. Mater. Res.—Part B Appl. Biomater. 106, 2046–2057 (2018). https://doi.org/10.1002/jbm.b.33950
Y. Han, Q. Wei, P. Chang, K. Hu, O.V. Okoro, A. Shavandi, L. Nie, Three-dimensional printing of hydroxyapatite composites for biomedical application. Crystals (2021). https://doi.org/10.3390/cryst11040353
S.A. Park, S.H. Lee, W.D. Kim, Fabrication of porous polycaprolactone/hydroxyapatite (PCL/HA) blend scaffolds using a 3D plotting system for bone tissue engineering. Bioprocess Biosyst. Eng. 34, 505–513 (2011). https://doi.org/10.1007/s00449-010-0499-2
S.V. Harb, N.J. Bassous, T.A.C. de Souza, A. Trentin, S.H. Pulcinelli, C.V. Santilli, T.J. Webster, A.O. Lobo, P. Hammer, Hydroxyapatite and β-TCP modified PMMA-TiO2 and PMMA-ZrO2 coatings for bioactive corrosion protection of Ti6Al4V implants. Mater. Sci. Eng. C 116, 111149 (2020). https://doi.org/10.1016/j.msec.2020.111149
H.Y. Mi, S.M. Palumbo, X. Jing, L.S. Turng, W.J. Li, X.F. Peng, Thermoplastic polyurethane/hydroxyapatite electrospun scaffolds for bone tissue engineering: effects of polymer properties and particle size. J. Biomed. Mater. Res.—Part B Appl. Biomater. 102, 1434–1444 (2014). https://doi.org/10.1002/jbm.b.33122
H.Y. Mi, X. Jing, M.R. Salick, T.M. Cordie, X.F. Peng, L.S. Turng, Morphology, mechanical properties, and mineralization of rigid thermoplastic polyurethane/hydroxyapatite scaffolds for bone tissue applications: effects of fabrication approaches and hydroxyapatite size. J. Mater. Sci. 49, 2324–2337 (2014). https://doi.org/10.1007/s10853-013-7931-3
Z. Cui, Z. Zheng, C. Su, J. Si, Q. Wang, W. Chen, Porous 3-D thermoplastic polyurethane (TPU) scaffold modified with hydroxyapatite (HA) nanoparticles using an ultrasonic method. J. Mater. Sci. 54, 11231–11242 (2019). https://doi.org/10.1007/s10853-019-03683-6
M.H. Kim, C. Yun, E.P. Chalisserry, Y.W. Lee, H.W. Kang, S.H. Park, W.K. Jung, J. Oh, S.Y. Nam, Quantitative analysis of the role of nanohydroxyapatite (nHA) on 3D-printed PCL/nHA composite scaffolds. Mater. Lett. 220, 112–115 (2018). https://doi.org/10.1016/j.matlet.2018.03.025
C.A.G. Beatrice, K.M.B. Shimomura, E.H. Backes, S.V. Harb, L.C. Costa, F.R. Passador, L.A. Pessan, Engineering printable composites of poly (ε-polycaprolactone)/β-tricalcium phosphate for biomedical applications. Polym. Compos. 42, 1198–1213 (2021). https://doi.org/10.1002/pc.25893
B. Huang, P.J. Bártolo, Rheological characterization of polymer/ceramic blends for 3D printing of bone scaffolds. Polym. Test. 68, 365–378 (2018). https://doi.org/10.1016/j.polymertesting.2018.04.033
N.J. Hossieny, M.R. Barzegari, M. Nofar, S.H. Mahmood, C.B. Park, Crystallization of hard segment domains with the presence of butane for microcellular thermoplastic polyurethane foams. Polymer (Guildf) 55, 651–662 (2014). https://doi.org/10.1016/j.polymer.2013.12.028
R.J. Gaymans, Segmented copolymers with monodisperse crystallizable hard segments: novel semi-crystalline materials. Prog. Polym. Sci. 36, 713–748 (2011). https://doi.org/10.1016/j.progpolymsci.2010.07.012
J. Datta, P. Kasprzyk, Thermoplastic polyurethanes derived from petrochemical or renewable resources: a comprehensive review. Polym. Eng. Sci. 58, E14–E35 (2018). https://doi.org/10.1002/pen.24633
M.M. Aslzadeh, M. Abdouss, G.M.M. Sadeghi, Preparation and characterization of new flame retardant polyurethane composite and nanocomposite. J. Appl. Polym. Sci. 127, 1683–1690 (2013). https://doi.org/10.1002/app.37809
P.R. Nair, C.P.R. Nair, D.J. Francis, Phosphazene-modified polyurethanes: synthesis, mechanical and thermal characteristics. Eur. Polym. J. 32, 1415–1420 (1996). https://doi.org/10.1016/S0014-3057(96)00079-1
B. Finnigan, D. Martin, P. Halley, R. Truss, K. Campbell, Morphology and properties of thermoplastic polyurethane nanocomposites incorporating hydrophilic layered silicates. Polymer (Guildf) 45, 2249–2260 (2004). https://doi.org/10.1016/j.polymer.2004.01.049
W.P. Yang, C.W. Macosko, S.T. Wellinghoff, Thermal degradation of urethanes based on 4,4′-diphenylmethane diisocyanate and 1,4-butanediol (MDI/BDO). Polymer (Guildf) 27, 1235–1240 (1986). https://doi.org/10.1016/0032-3861(86)90012-1
H.-Y. Mi, X. Jing, E. Yu, X. Wang, Q. Li, L.-S. Turng, Manipulating the structure and mechanical properties of thermoplastic polyurethane/polycaprolactone hybrid small diameter vascular scaffolds fabricated via electrospinning using an assembled rotating collector. J. Mech. Behav. Biomed. Mater. 78, 433–441 (2018). https://doi.org/10.1016/j.jmbbm.2017.11.046
Y.S. Cho, S. Choi, S.H. Lee, K.K. Kim, Y.S. Cho, Assessments of polycaprolactone/hydroxyapatite composite scaffold with enhanced biomimetic mineralization by exposure to hydroxyapatite via a 3D-printing system and alkaline erosion. Eur. Polym. J. 113, 340–348 (2019). https://doi.org/10.1016/j.eurpolymj.2019.02.006
T. Almela, I.M. Brook, K. Khoshroo, M. Rasoulianboroujeni, F. Fahimipour, M. Tahriri, E. Dashtimoghadam, A. El-Awa, L. Tayebi, K. Moharamzadeh, Simulation of cortico-cancellous bone structure by 3D printing of bilayer calcium phosphate-based scaffolds. Bioprinting 6, 1–7 (2017). https://doi.org/10.1016/j.bprint.2017.04.001
C.F. Guimarães, L. Gasperini, A.P. Marques, R.L. Reis, The stiffness of living tissues and its implications for tissue engineering. Nat. Rev. Mater. 5, 351–370 (2020). https://doi.org/10.1038/s41578-019-0169-1
G.C.V. Rhoon, T. Samaras, P.S. Yarmolenko, M.W. Dewhirst, E. Neufeld, N. Kuster, CEM43°C thermal dose thresholds: a potential guide for magnetic resonance radiofrequency exposure levels? Eur. Radiol. 23, 2215–2227 (2013). https://doi.org/10.1007/s00330-013-2825-y
K. Kniha, N. Heussen, E. Weber, S.C. Möhlhenrich, F. Hölzle, A. Modabber, Temperature threshold values of bone necrosis for thermo-explantation of dental implants—a systematic review on preclinical in vivo research. Materials (Basel) 13, 3461 (2020). https://doi.org/10.3390/ma13163461
N. Eliaz, N. Metoki, Calcium phosphate bioceramics: a review of their history, structure, properties, coating technologies and biomedical applications. Materials (Basel) 10, 334 (2017). https://doi.org/10.3390/ma10040334
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. The authors are also grateful to Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grant numbers 2018/26060-3, 2021/11538-8 and 2020/11496-0), and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant numbers 306835/2017-7, 307665/2018-6, 314093/2021-4 and 305109/2022-7), and National Institute of Science and Technology for Rheology of Complex Materials Applied to Advanced Technologies (INCT-Rhe9) grant 406765/2022-7.
Author information
Authors and Affiliations
Contributions
LAP (Pinto) conceptualized the study, performed the experiments and the analyses, wrote the original draft, and reviewed and edited the document, table, figures, and videos. EHB conceptualized the study, advised the execution of the experiments and analyses, reviewed, and edited the document, table, figures, and videos. SVH performed the experiments and the analyses, wrote the original draft, and reviewed and edited the document, table, figures, and videos. GMP performed rheological analysis, discussed rheological results, reviewed, and edited the final document. DAVC performed SEM analysis, reviewed, and edited the final document. RJEA supervised the rheological analysis, discussed rheological results, reviewed, and edited the final document. GJMF supervised the rheological analysis, discussed rheological results, reviewed, and edited the final document. HSSA supervised the cell viability assay, discussed the cell viability results, reviewed, and edited the final document. LCC supervised the findings of the work, reviewed, and edited the final document, table, figures, and videos. LAP (Pessan) responsible for funding and supervision of the findings of the work, conceptualized the study, advised the execution of the experiments and analyses reviewed and edited the final document.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 Shape recovery process in U-bending test (MP4). (MP4 12434 KB)
Supplementary file3 Shape recovery process with weight imposition for the blends (MP4). (MP4 9383 KB)
Supplementary file4 Shape recovery process with weight imposition for 75T25P blend (MP4). (MP4 9657 KB)
Supplementary file5 Recovery speed of the programmed filament (MP4). (MP4 5519 KB)
Supplementary file6 Self-knotting process in a 75T25P blend filament (MP4). (MP4 9758 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pinto, L.A., Backes, E.H., Harb, S.V. et al. Shape memory thermoplastic polyurethane/polycaprolactone blend and composite with hydroxyapatite for biomedical application. Journal of Materials Research 39, 90–106 (2024). https://doi.org/10.1557/s43578-023-01172-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43578-023-01172-w