
Abstract
Novel p-coumaric acid microemulsion systems were developed to circumvent its absorption and bioavailability challenges. Simplex-lattice mixture design and machine learning methods were employed for optimization. Two optimized formulations were characterized using in vitro re-dispersibility and cytotoxicity on various tumor cell lines (MCF-7, CaCO2, and HepG2). The in vivo bioavailability profiles of the drug loaded in the two microemulsion systems and in the suspension form were compared. The optimized microemulsions composed of Labrafil M1944 CS (5.67%)/Tween 80 (38.71%)/Labrasol (38.71%)/water (16.92%) and Capryol 90 (0.50%)/Transcutol P (26.67%)/Tween 80 (26.67%)/Labrasol (26.67%)/water (19.50%), respectively. They revealed uniform and stable p-coumaric acid-loaded microemulsion systems with a droplet size diameter of about 10 nm. The loaded microemulsion formulations enhanced the drug re-dispersibility in contrast to the drug suspension which exhibited 5 min lag time. The loaded formulae were significantly more cytotoxic on all cell lines by 11.98–16.56 folds on MCF-7 and CaCo2 cells and 47.82–98.79 folds on HepG2 cells higher than the pure drug. The optimized microemulsions were 1.5–1.8 times more bioavailable than the drug suspension. The developed p-coumaric acid microemulsion systems could be considered a successful remedy for diverse types of cancer.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the present context, cancer disease is among the principal causes of death globally, especially breast, liver, and colorectal cancer [1]. However, the productiveness of the current known treatment strategies is sometimes ineffective either due to high toxicity or cost [2]. Fortunately, nanotechnology offered a helping hand to enhance the cancer survival rate [3,4,5].
p-Coumaric acid (pCA) has a phenolic structure and belongs to the hydroxycinnamic acid group (Supplementary Material, Fig. 1). It is mainly consumed in our regular diet such as fruits (apples, grapes, oranges, etc.), vegetables (potatoes, onions, tomatoes, etc.), cereals (oats, wheat, and maize), and coffee [6, 7]. Recently, pCA as a bioactive compound showed great interest due to its potent anti-inflammatory, antioxidant, anti-platelet, anti-viral, anti-arthritis, and most interestingly, anti-cancer effects. Its anti-cancer property is mainly due to its anti-angiogenic and apoptotic effects [8,9,10]. Despite its numerous health benefits, pCA is a biopharmaceutical classification (BCS) class 2 drug, thus suffering from several delivery challenges summarized in its poor aqueous solubility and being a p-glycoprotein (P-gp) substrate [11, 12]. This leads to its noticed low bioavailability (BAV) after oral administration.
Up to the present, scant studies have focused on boosting pCA BAV which can be circumvented by utilizing a suitable delivery system that can overcome the poor solubility, slow dissolution rate, being a P-gp substrate, and consequently, enhancing the oral BAV of pCA [7, 13].
In this context, many solubilization approaches such as micronization, cyclodextrin inclusion complex, lipidic carriers, nanocarriers, complexation, and salt formation were thought of in order to solubilize and deliver poorly soluble drugs with higher available amounts [14, 15]. Focusing on lipid-based vehicles, these carriers have been previously specifically and successfully exploited for many poorly soluble drugs and oils such as acetazolamide, simvastatin, and methyldihydrojasmonate [16,17,18].
The efficiency of lipid-based carriers has been confirmed by a previously conducted meta-analysis study by our research group concluding that the lipid-based carriers enhance the BAV of many drugs (by evidence) in contrast to the corresponding drug suspension/solution. The microemulsion (ME) system was chosen for the delivery of pCA due to its superiority and efficiency in enhancing drug BAV in terms of the highest estimated mean when compared with other systems according to this study [19]. The microemulsion system is optically clear and thermodynamically stable composed of oil, surfactant (S), co-surfactant (CoS), and water forming nano-sized droplets spontaneously [20]. Microemulsions continue to receive considerable attention in the pharmaceutical industry due to their strong solubilization effect, long-term stability, low viscosity, compatibility, biodegradability in the biological system, and rapid absorption, making them an attractive vehicle for the delivery of bioactive molecules [21, 22]. From the pharmaceutical perspective, the resultant formulation’s droplet size after dispersion in the intestinal fluids enlarges the surface area and accordingly improves the dissolution of poorly water-soluble molecules. Moreover, the inclusion of oil in the formulation allows the lymphatic system transportation of the drug, thus reducing the pre-systemic emptying and evading the hepatic first-pass effect. Furthermore, based on the ME components, many advantageous effects may emerge, for instance, escalating the fluidity of membranes, opening of tight junctions, and inhibiting P-gp. As a result, ME can enhance the oral BAV of drugs and is predicted to elevate the pharmacokinetic properties of pCA [23,24,25].
The technique of design of experiment (DoE) [26] was adopted to provide the precise mathematical equation connecting the formulation inputs (oil, surfactants, co-surfactants, and water) with the final quality attributes of the developed ME systems (droplet size) [27,28,29]. Herein, the simplex lattice was the selected design as it is the design of choice for mixtures where the total components percentages sum up to 100%. Moreover, it is usually applied for areas comprising a triangular domain [30, 31]. In another aspect, the machine learning tools named the principal component analysis (PCA) and the hierarchical clustering assay (HCA) were exploited. They are considered as a multi-variate statistical approach [32]. They differ from univariate statistics in that they analyze the covariances of numerous variables and determine the degree of relationship between them rather than analyzing the mean and variance of only a single variable [33, 34].
To this end, the purpose of this study is to formulate a novel optimized pCA-loaded ME system, which will be characterized and assessed for its in vitro re-dispersibility, cytotoxicity, stability, and in vivo BAV.
Materials and Methods
Materials
p-Coumaric acid (HPLC grade > 98%), castor oil, Tween 80 (polysorbate 80), and dialysis membrane with 12,000–14,000 molecular weight cut-offs were purchased from Sigma-Aldrich Chemical Co., Steinheim, Germany. Labrafil M 1944 CS (oleoyl polyoxyl-6 glycerides), Labrasol (caprylocapryol polyoxyl-8 glycerides), Transcutol P (diethylene glycol monoethyl ether), Capryol 90 (propylene glycol caprylate), Maisine CC (glyceryl monolinoleate), Labrafac PG (propylene glycol dicaprylocaprate), Plurol Oleique CC 497 (polyglycerol oleate), Lauroglycol 90 (propylene glycol monolaurate), and Capryol PGMC (propylene glycol monocaprylate) were gently granted by Gattefossé, Saint-Priest, France. Sodium dihydrogen phosphate and disodium hydrogen phosphate were purchased from El Nasr Pharmaceutical Co., Cairo, Egypt. Membrane filter 0.45 µm, 13mm was obtained from Whatman, Kent, UK. PEG 400 (polyethylene glycol 400) was purchased from Nice Chemicals, Kochi, India. Finally, methanol, acetonitrile, and glacial acetic acid were all HPLC grade and were acquired from Fisher Scientific, Loughborough, UK.
Methods
Solubility of p-Coumaric Acid in Several Oils, Surfactants, and Co-surfactants
A solubility study was conducted to determine the suitable vehicles (oils, surfactants, or co-surfactants) to prepare ME systems that have a good solubilizing capacity for pCA. One gram of each vehicle was added to a stoppered vial containing an excess weight of the drug (300 mg). After then, sealed vials were warmed at 40°C ± 0.2 in a water bath for 15 min to facilitate solubility. The vials were then introduced in a shaking water bath at 25°C ± 0.3 for 48 h. Finally, after approaching equilibrium, each vial was centrifuged at 6000 rpm (8551.6 g) for 15 min and then filtered by a membrane filter (0.45 μm, 13 mm, Whatman, UK) to extract the undissolved drug [35]. The supernatant was spectrophotometrically analyzed at 310 nm after appropriate dilution of each mixture with methanol. This was performed by utilizing a prepared standard calibration curve of pCA in methanol generating a best-fitting straight line (r2 = 0.9974) and a regression equation (y = 0.1279x − 0.0289, y = Abs, and x = concentration of pCA).
Construction of Pseudo-ternary Phase Diagrams (Pseudo-TPDs)
Twenty random combinations of oil, surfactant/co-surfactant (S/CoS), and water were selected in order to prepare different microemulsion combinations at room temperature by utilizing the water titration procedure in a drug-free environment [36]. The combinations corresponding pseudo-ternary phase diagrams (pseudo-TPDs) were also developed where the target was to select those possessing high ME region. Briefly, the surfactant and co-surfactant were mixed into a ratio of 1:1 (w/w). The oil was then blended with the S/CoS mixture in numerous ratios varying from 1:9 to 9:1 (w/w). The total mass percentages for the final mixture of oil, S, and CoS were maintained at 100% w/w. Then, under gentle stirring, each combination was titrated drop-wise by distilled water until turbidity was visually visible indicating the endpoint (maximum water uptake) [37]. Each component mass including the added water was then converted into gram (g) %, and pseudo-TPDs were drawn using Tri Draw (version 2.9) software (CHEMIXsoftware, Arne Standenes, Norway).
Preparation and Optimization of the Selected ME Systems Using Simplex Lattice Mixture Design and Machine Learning Methods
After selecting two novel microemulsion combinations, optimization followed, where the objective was to select the formulae having small, uniform, and stable droplet sizes, especially after dispersion in distilled water.
Simplex Lattice Mixture Design
The same equilateral triangle was selected from each pseudo-TPD for optimization employing the simplex lattice mixture design via Design-Expert® v.7.0 software. The upper and lower percentage boundaries of the independent variables (oil, S, CoS, and water) were set as shown in Supplementary Material, Table 1 keeping the total sum fixed at 100% w/w. These triangles were located above the maximum dilution line (dash lines drawn in Fig. 1) below which any formulation may change its behavior upon dilution in gastric fluids from ME to an unknown multi-phase system [38]. The simplex lattice design provided 10 runs for each ME combination to evaluate the impact of ME components on droplet size (experimental response).

Pseudo-ternary phase diagrams of a Labrafil M 1944 CS/Tween 80/Labrasol/Water (LTL ME system) and b Capryol 90/Tween80/Labrasol/Transcutol P/Water (CTTL ME system). The enclosed triangle represents the simplex lattice mixture design domain (SL), while the dashed line represents the dilution line
Microemulsion Preparation
The suggested runs (formulations) obtained from the simplex lattice design for both combinations were prepared by accurately weighing each component mass, then under continuous magnetic stirring, S and CoS were added to oil, and finally, the water was added drop-wise by utilizing a syringe to form a stable and clear system [20].
Determination of Droplet Size as an Experimental Response
Average droplet size and polydispersity index (PDI) were measured after 2 h and 24 h from preparation by using the Zetasizer Nano ZS (Malvern Instruments, UK) at a 173° scattering angle. After then, the same formulae were diluted 100-fold with distilled water in order to simulate the in vivo dilution [39] followed by the re-measurement of the droplet size and the PDI after 2 h and 24 h.
Exploiting Machine Learning Methods to Assess the Diluted Microemulsion Stability
Stability evaluation of the diluted ME formulae was performed by applying the principal component analysis (PCA) and the agglomerative hierarchical clustering (AHC) via JMP® software version 7 (SAS, CARY, NC). The evaluation was performed based on the change after 2 h and 24 h from dilution in the droplet size and PDI. Clearly, in PCA, the matrices of covariance were acquired followed by extraction of the principal components. Then, prior to analysis, all data were normalized against their standard deviation (SD) [40]. The primary objective of PCA was to decrease the dimensionality of the dataset regarding the most discriminating variables and to cluster into meaningful groups [41]. On the other hand, for AHC, the clustering was performed using the nearest neighbor/single linkage method, where the distances between data and drawn dendrograms were obtained using the Euclidean distance. The aim was to select the formulations having their 2 h and 24 h data clustered together with relatively similar Euclidean distance implying minimal changes with time and consequently indicating their stability [35].
Preparation of p-Coumaric Acid Loaded ME Formulations
After optimization, two formulae (one from each ME combination) fulfilled the previously mentioned criteria and were selected for pCA loading. This was performed by dissolving a pCA weight of 20 mg in the designated oil and surfactant and then shaking for 5 min at 25°C. The co-surfactant was then added followed by water, and the whole mixture was re-shaken for 30 min [35, 42].
Characterization of the Optimized Loaded ME Formulations
Measurement of the Droplet Size After Dilution
The drug-loaded formulations were diluted 100-fold with distilled water, and their droplet size along with PDI was measured after 2 h and 24 h of preparation to examine the physical durability of the loaded systems regarding the droplet size, PDI, drug precipitation, and dispersed phase separation [43].
In Vitro Re-dispersibility Study
The drug re-dispersibility experiment was performed in a USP basket dissolution tester (Hanson SR8 plus, Chatsworth, USA). The test started by soaking the dialysis membranes in phosphate buffer (PB) solution pH 6.8 (dialysis media) for 24 h at room temperature, where this pH was chosen as the pCA main absorption site is the intestine [43, 44]. After then, small locally fabricated tubes were occluded from the bottom with the soaked membranes. Then, an accurate volume of the drug suspension or one of the optimized ME formulae containing pCA equivalent to 20 mg was filled in the tubes. Carefully, each filled glass tube was mounted in the place of the basket of the USP dissolution tester. Each tube was immersed into a vessel containing 900 mL of PB solution pH 6.8. The study was carried out at 37 ± 0.5°C and 100 revolutions per minute (rpm) for 2 h. At specific time intervals, i.e., 5, 10, 15, 20, 30, 45, 60, and 120 min, 5 mL aliquot was taken and filtered by a 0.45 μm filter (Thermo Fisher Scientific, MA, USA), and in order to sustain the sink condition, the withdrawn volume was replaced by fresh medium 40. The concentration of pCA was examined spectrophotometrically at 286 nm after suitable dilution. The concentrations were obtained utilizing a prepared standard calibration curve of pCA in phosphate buffer (PB) solution pH 6.8 resulting in a best-fitting straight line (r2 = 0.9991) and a regression equation (y = 0.0979x + 0.0172, y = Abs, and x = concentration of pCA).
The re-dispersibility profiles of the selected stable formulations were compared with those obtained for the drug suspension.
In Vitro Cytotoxicity Assessment
p-Coumaric acid showed a reported activity on many human cancer cell lines [45]. This study was performed to examine the cytotoxic effect of pCA solution and the changes that may occur due to the drug addition to the optimized ME formulae.
Mammalian Cell Lines
Different cancer cell lines were selected, specifically, MCF-7 cells (human breast cancer cell line), Caco2 cells (human colorectal carcinoma cell lines), and HepG-2 (human hepatocellular carcinoma cell lines). All cells were grown as a monolayer on RPMI-1640 medium enriched with inactivated fetal calf serum and gentamycin at a concentration of 1 μg/mL and 50 μg/mL, respectively. The cells were then stored in a humidified room with 5% CO2 and a temperature of 37°C ± 0.2. After then, cells were sub-cultured 2–3 times a week [46].
MTT Cell Viability Test
The cytotoxic effects of pCA were estimated by viability assay using microculture tetrazolium assay (MTT) as described by Ossama et al. [47] to determine the viability of cancer cells after being exposed to pCA in its pure solution form and when incorporated in the optimized formulations for 48 h. Moreover, blank ME formulations were also assessed to exclude their toxic effects. Briefly, to calculate the 50% inhibitory concentration (IC50), the optical density of each prepared concentration was examined with the microplate reader at 590 nm to discover the count of the viable cells followed by calculation of the viability percentage using the equation: cell viability % = (OD/ODc) * 100, where OD is the average optical density of treated cells (tested formulations) and ODc is that of the untreated cells. Furthermore, to draw the survival curve of each formulation in each cell line, the percentage viability against the sample concentration was plotted. Finally, the IC50 was determined from the drawn plots using GraphPad Prism software (San Diego, CA, USA) [46].
In Vivo Oral Absorption Study
The in vivo experiment was conducted for the drug suspension and the optimized loaded formulations to determine the proposed enhancement in oral BAV of pCA by utilizing the underneath-described protocol.
Oral Administration and Blood Sampling
Eighteen male Sprague-Dawley rats, weighing 200–250 g, were bought from The Nile Co., Cairo, Egypt, for pharmaceutical and chemical industries. The laboratory conditions for rats were normal. Clearly, in a 12-h light-dark cycle, rats were fed adaptively for 1 week with free access to pelleted food and water. Moreover, the rats were divided into three groups (n = 6), where 3 rats were sampled at each time interval in an alternating manner. Prior to drug administration, all animals were fasted overnight with no limitation to water. After then, each group of rats was orally administered the desired formulation where, group 1 took 100 mg/kg pCA suspension dose [48], while groups 2 and 3 got the selected formulations loaded with the same equivalent dose.
The blood samples (about 0.5 mL) were withdrawn from the retro-orbital plexus and transferred immediately to heparinized tubes at 0, 0.0833, 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h. Lactated Ringer’s solution was also used as a fluid replacement as recommended by the NIH. Consequently, the samples were centrifuged at 4000 rpm (5701.1 g) for 5 min, and the plasma was kept at −20°C until being analyzed by high-performance liquid chromatography (HPLC) [49].
The test was accepted by the Research Ethical Committee of the Faculty of Pharmacy, Ain Shams University, protocol number 273.
Development and Validation of High-Performance Liquid Chromatography (HPLC) Procedure for Precise Calculation of p-Coumaric Acid in Rat’s Plasma
A new sensitive, selective, and accurate HPLC method emerged and validated [50, 51] for pCA determination in the plasma with the aid of diclofenac sodium which was used as an internal standard (IS) as provided in the Supplementary Material.
Pharmacokinetic Assessment
The non-compartmental analysis was performed, and the pharmacokinetic parameters were estimated through the PK solver add-in program for Microsoft Excel (version 2.0, China). These parameters were the maximum plasma concentration (Cmax), time to reach maximum plasma concentration (Tmax), area under the curve (AUC0–24 h and AUC0–∞), biological half-life (t1/2), and mean residence time (MRT). In addition, the relative bioavailability was determined by dividing the AUC0–∞ of each selected formulation by that of pure drug suspension.
Statistical Analysis
All data were shown as mean ± standard deviation where n = 3. For statistical analysis, one-way ANOVA and unpaired t-test were carried out using GraphPad Prism, version 5 (GraphPad Software, San Diego, CA). Significance was reported when the p-value was ≤ 0.05.
Results and Discussion
Solubility of p-Coumaric Acid in Several Oils, Surfactants, and Co-Surfactants
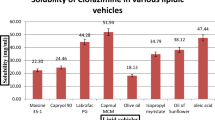
Development of stable oil in water (o/w) ME especially after dilution in the gastrointestinal tract (GIT) requires an imperative criterion which must be contemplated, that no drug precipitation should occur [52]. This can be achieved when the solubility of the drug in water is relatively lower than that in the S/CoS mixture, whereas oil solubility is also important as it will act as a drug reservoir and provide its advantages in enhancing the drug BAV [21]. As shown in Supplementary Material, Fig. 2, Labrafil M 1944 CS and Capryol 90 showed higher solubility: 16.29 ± 2.67 and 50.7 ± 5.33 mg/g, respectively. While for S and CoS, Tween 80, Transcutol P, and Labrasol were the best solubilizing agents where they revealed higher pCA water solubility (118.23 ± 2.43, 290.89 ± 10.91, and 210.57 ± 7.26 mg/g, respectively).
This may be attributed to their chemical structure and geometry where it is obvious from the chemical name that Tween 80 (polyoxyethylene (20) sorbitan monooleate), Transcutol P (diethylene glycol monoethyl ether), and Labrasol (caprylocaproyl polyoxyl-8 glycerides) are rich in hydroxyl groups (−OH) and hydrogen (−H) atoms and thus capable of forming hydrogen bonds and dipole-dipole interactions with pCA. Moreover, oils had lower drug solubility relative to S and CoS as Capryol 90 (propylene glycol caprylate) has an extremely lower number of the previously mentioned functional groups, in addition to the possibility of intramolecular hydrogen bond formation that reduces the chances for intermolecular hydrogen bonding with pCA. Furthermore, unlike Labrasol which contains caprylic (C8) and capric acid (C10), Labrafil M 1944 CS (oleoyl polyoxyl-6 glycerides) is a bulky molecule due to the presence of oleic acid (C18) which makes any interaction with pCA unfavorable and thus leads to lower solubility.
Construction of Pseudo-ternary Phase Diagrams
Pseudo-TPDs are an appropriate approach to differentiate between ME and multi-phase regions [37]. The pseudo-TPDs of the twenty random investigated combinations are provided in the Supplementary Material, Figure 3. Figure 1 depicts pseudo-TPDs of the two selected combinations: Labrafil M 1944 CS/Tween 80/Labrasol/Water (LTL ME system) and Capryol 90/Tween 80/Transcutol P/Labrasol/Water (CTTL ME system). These systems were selected due to the high solubilization capacity of their components, high ME region, and being—up to our knowledge—new carriers that were not previously utilized for the delivery of drugs including pCA. Also, the selected oils were Capryol 90 and Labrafil M 1944 CS due to their higher recorded pCA solubility relative to other oils. In addition, their chemical structure possesses small alkyl chains that will lead to the formation of small droplet sizes as was previously reported [53]. It was also mentioned that they possess surfactant properties that will lead to a reversible absorption-enhancing effect in the small intestine rather than the large intestine where other conventional absorption enhancers usually operate, hence allowing for more chances to increase pCA BAV associated with less incorporation of the S and CoS components which will consequently enhance the safety and tolerability of the system [54]. The dark (colored) area in Fig. 1 highlights the o/w ME, while the uncolored sector exhibits a turbid multi-phase region. Consequently, the same equilateral triangular area was selected for both combinations for optimization as shown in the same figure.
Optimization of the Selected ME Combinations Using Simplex Lattice Mixture Design and Machine Learning Methods
After preparing the twenty runs (10 for each combination) suggested by the utilized simplex lattice mixture design, their droplet size (experimental response) and PDI were determined. The outcomes are reported in Supplementary Material, Table 2. Small droplet sizes were obtained from all the freshly prepared (2 h) formulations ranging from 8.615 nm to 86.02 nm for the LTL ME system and 3.105 nm to 88.33 nm for the CTTL ME system indicating that the microemulsion systems were successfully developed. This small droplet size promotes drug absorption and BAV by providing a high interfacial surface area and enhancing the rate of drug dissolution [37]. Furthermore, the small droplet size indicates the development of a better-packed layer of S and CoS at the water-oil interface, hence stabilizing the oil droplets [52, 55]. Noticeably, the droplet size measurements after 24 h either from preparation or even after dilution revealed an increase in the droplet size in almost all the investigated formulae. This may be attributed to the high surface energy of these small-diameter droplets, leading to increased collisions and aggregation by time [35]. Furthermore, the 100-fold dilution leads to a growth in the droplet size as excessive water lowers the efficiency of S/CoS in decreasing droplet size. This was more obvious in the CTTL ME system than the LTL ME system due to the latter’s lower PDI leading to better discrimination in droplet size. Interestingly, although the same runs were prepared for both systems, there was a dramatic difference in droplet size; this may be attributed to the incorporation of another CoS in the CTTL ME system “Transcutol P” which is responsible for the increased droplet size because it is familiar that inclusion of S causes condensation of the interfacial film while CoS addition expands the film [36, 56]. Regarding the PDI, it is normal to be high in the undiluted ME formulations (> 0.2) [35, 56], owing to the extremely low interfacial tension of the surfactant monolayer even approaching zero in some cases. Therefore, there are more chances to obtain non-spherical shape particles such as worm-like or irregular shapes. In contrary to normal emulsions, it forms spherical structures as high interfacial tension favors the minimization of the droplet interfacial areas, as it is well-established that the sphere is characterized by the lowest interfacial area for a given volume [57]. Non-spherical structures usually score high PDI values in dynamic light scattering particle size measuring techniques as they are mainly light intensity-based techniques. On the other hand, the 100-fold diluted formulations are more uniform in size with lower PDI, and thus it is more important to consider the droplet size, PDI, and stability of the diluted formulations as it simulates the in vivo dilution [39].
Figure 2 depicts the contour and the 3D plots of models which explains the differences in the droplet sizes between the prepared formulations of both systems. The apexes of the triangles represent the limit of the ME components, while each red dot denotes the droplet size of the prepared formulation. The droplet size value increases moving from the blue to red region. It is obvious in the 2 h CTTL ME system that by increasing the oil percentage—moving from the apex to the bottom—the droplet size increased. On the other hand, as S/CoS and water percentages increased, the droplet size decreased which was also reported in many articles [43, 46, 58]. While for the LTL ME system, this was not the trend. By increasing the water percentage, the droplet size decreased, while the oil and S/CoS percentage had no major effect on the droplet size. This could be obvious by observing the magnitude of the water coefficient in the generated model in the Supplementary Material, Table 3 that describes the contribution of independent variables to the respective response, where the coefficient of water had the highest magnitude, implying that water was critical for determining the globule size [58]. Finally, the effect of dilution and storage for 24 h are obvious on the plots and coincide with the aforementioned explanation.



Contour (left) and 3D plots (right) of the generated models, i.e., a–d for the LTL ME system and e–h for the CTTL ME system, where a, e; b, f; c, g; and d, h corresponds to 2 h after preparation, 24 h after preparation, 2 h after dilution, and 24 h after dilution, respectively. The response (globule size) value increases moving from the blue to red colors
Accordingly, many formulae showed small and uniform droplet sizes, but not all of them were stable for 24 h after dilution, as shown in Supplementary Material, Table 2, which is an essential criterion to guarantee stable droplet size until the drug is absorbed and reaches systemic circulation. Based on this, multivariate statistics were conducted using HCA and PCA to assess the stability of formulations after 24 h of dilution and select the most stable formulation in each system. In HCA, data were organized according to the similarity between formulations (no significant change in droplet size after 24 h dilution) and represented in a tree-like diagram, while in PCA, the most stable formulae were chosen by locating the similar formulations in the same quadrant near each other. Results are shown in Fig. 3, where the 2 h after dilution formulations were coded from F1 to F10, while the same formulations 24 h after dilution were numbered from F11 to F20. The microemulsion formulations were depicted as points (scores) and the measurements as vectors (loadings), respectively. The first principal component accounted for 55.6 and 55.8% of the variation between the formulations for the investigated first and second systems, respectively, while the second principal component accounted for 44.8 and 44.6%, respectively. It is also worth noting that the DS (droplet size) and the PDI were correlated (both positive) with respect to the first (main) principal component which implies that increasing the DS is associated with an increase in PDI may be due to the presence of aggregations which distort the readings [41].

a Scores and loading plots of PCA and b HCA dendrograms of the prepared formulations for both systems, i.e., (i) LTL ME system and (ii) CTTL ME system. The 2 h after dilution formulations’ codes are 1 to 10, and the 24 h after dilution codes are 11 to 20 for the same formulations. DS, droplet size; PDI, polydispersity index
Results showed that F1 LTL ME and F5 CTTL ME systems possessed superior stability as 1 and 11 in the LTL ME system and 5 and 15 in the CTTL ME system were clustered together in HCA and located close to each other in PCA, and thus were selected for further characterization. This stability may be due to the presence of lower oil content and optimum percentage of S and CoS compared to less stable formulations. The S and CoS were responsible for the ultra-low value of the interfacial tension which imparts stability to ME. Moreover, the optimum concentration of S and CoS allowed the formation of a coat around the oil globules protecting them and avoiding coalescence. Any further increase in the S and CoS concentration may lead to flocculation and consequently disrupt the ME stability [59].
Characterization of Optimized ME Formulations
Preparation of p-Coumaric Acid Loaded ME Formulations and Re-measurement of Droplet Size After Dilution
It is important that the chosen ME systems remain stable upon addition of drug and dilution in terms of droplet size and PDI since it is assumed that adding a drug may affect the droplet size, and hence different characteristics may evolve [39]. After preparation and dilution 100 times of the loaded F1 LTL and F5 CTTL ME systems, droplet size and PDI were measured. The results are displayed in Table 1, concluding that the amount of the solubilized drug (20 mg of pCA) was not sufficient to cause any remarkable difference in droplet size or PDI, i.e., p-value > 0.05, which coincides with Moghimipour et al. results [42]. Also, no drug precipitation or phase separation was observed in any formulae.
In Vitro Re-dispersibility Study of p-Coumaric Acid
Figure 4 illustrates the comparison between the drug dispersion from F1 LTL and F5 CTTL formulations versus the pure drug suspension. Results displayed that the optimized formulae enhanced the dispersion rate of pCA significantly (p-value less than 0.05) at the first 15 min after which 100% of the drug was re-dispersed in all three samples with no sign of drug precipitation occurred along the 120 min indicating that the whole dose will be available for absorption. Obvious lag time for the first 5 min in the dispersion profile of the pure drug suspension was shown after which the drug started to re-disperse in the dissolution medium. This was a result of poor pCA aqueous solubility. The lag time was also reported in the re-dispersibility study of many drugs [60,61,62]. On the other hand, the optimized ME systems enhanced the rate of drug dispersion due to the formed nano-sized droplets that increased the surface area, in addition to surfactants and co-surfactants that kept the drug in the solubilized state [56, 62]. By comparing the loaded F1 LTL and F5 CTTL ME formulations, results displayed that F1 was better in escalating the dispersion rate of pCA at the first 5 min, while F5 was better at 10 min. By reaching 15 min, both formulae reached 100% of drug dispersion. This closeness in enhancing the dispersion rate may be due to the similar droplet size of both formulae, in addition to the convergence of the S and CoS percentages used in F1 (80% w/w) and F5 (77.42% w/w).

In vitro re-dispersibility profiles of the pure drug suspension, F1 LTL and F5 CTTL, formulations through a 120 min and b zooming on the first 20 min to discriminate between different profiles
In Vitro Cytotoxicity Assessment
The cell viability using MCF-7, HepG2, and Caco2 cell lines was exploited to examine the potential cytotoxicity of pCA-loaded ME systems. The drug in the ME systems is speculated to reach the tumor cells in vivo owing to the small globule size of the microemulsions. The globules are expected to evade the immunity system elements due to the presence of the surfactant “Labrasol” in its composition. This surfactant contains PEG moieties [3] that will form a stealth effect, thus allowing the formulations to approach the tumor area and to be uptaken by the cells by the enhanced permeation and retention (EPR) effect. The results in Fig. 5 revealed that the cytotoxicity of the loaded and unloaded (blank) ME systems increased in a dose-dependent manner, unlike the pure drug solution. Moreover, the pure drug solution showed cell toxicity with IC50 values of 104 ± 4.20, 89.90 ± 3.10, and 29 ± 1.30 µg/mL for MCF-7, HepG2, and Caco2 cells, respectively. This sensitivity difference of the cell lines against any chemical compound such as pCA could be due to the unique features of each type of cell, hence leading to different defense processes towards pCA [63].

Cell viability of a pure p-coumaric acid solution (medicated ME systems) and b plain ME systems (blank formulations)
Furthermore, the addition of the drug to the optimized ME systems increased the cytotoxic effect of pCA compared to the pure drug alone showing a very significant reduction in the IC50 values: 6.28 ± 0.46, 0.91 ± 0.08, and1.77 ± 0.16 µg/mL for loaded F1 and 7.22 ± 0.67, 1.88 ± 0.19, and 2.42 ± 0.51 µg/mL for loaded F5 on MCF-7, HepG2, and Caco2 cells, respectively. Since the cytotoxicity of an anti-cancer drug relies on its accumulation in tumor cells, thus the reduced IC50 confirmed the internalization of the investigated drug-loaded nano-carrier inside the tumor cells, the release of the drug from formulae, and being accumulated inside tumor cells [64]. This accumulation is owed to the small droplet size of the carriers, around 10 nm, that enhanced the solubility of pCA as proven by the in vitro experiment discussed earlier, and leads to better interaction with cancer cells and thus higher drug uptake by endocytosis and pinocytosis bypassing the general diffusion pathway of the free drug [22, 65,66,67,68]. Further, it is proclaimed that the ME improves molecule absorption either by enhancing permeability or by tight junction modulation. This is ascribed to the inclusion of S and CoS in the ME systems (Tween 80, Transcutol P, and Labrasol). These components serve as penetration enhancers via disrupting cell membranes and opening tight junctions especially Labrasol by interacting with F-actin and actin-anchoring protein (ZO-1) leading to improved transcellular transport of drugs [69,70,71]. Nevertheless, the ME is responsible for preventing drug efflux from cells by inhibiting the efflux-mediated transporters (P-gp) which is mainly responsible for anti-cancer drug resistance [72]. This inhibition is due to the incorporated Tween 80 and Labrasol where the latter one when combined with Transcutol P as in F5 CTTL augments the inhibition [3, 63, 73].
Comparing the loaded F1 LTL with the F5 CTTL ME systems, the differences in the recorded IC50 values were insignificant, proving that the shown reduced cytotoxicity was mainly due to the drug effect and not due to the formulae components. Furthermore, due to concerns regarding the safety of formulae towards non-cancerous cells, the blank (unloaded) ME systems—F1 and F5—were evaluated, and they showed extremely lower cytotoxic effects with F1 scoring an IC50 values of 1714 ± 226, 244 ± 46, and 672 ± 96 µg/mL and F5 scoring an IC50 values of 786 ± 78, 588 ± 108, and 960 ± 178 µg/mL for MCF-7, HepG2, and Caco2 cells, respectively. These results confirmed the systems’ safety. However, their cytotoxic effects increased in a concentration-dependent manner due to the continuous cells’ exposure to S and CoS (48 h) making the formulae cytotoxic at higher concentrations 71. Fortunately, this effect is reversible as reported in many articles [69, 74]. Moreover, this cytotoxicity effect will not be observed in vivo, as these formulae will be diluted in gastric fluids reaching a safe extent. Sha et al. reported that when SMEDDS (self-emulsifying drug delivery system) was diluted (1:50–1:2000) to form ME, no toxic effects were reported [69]. Furthermore, a high volume of distribution and high metabolic rates of ME components will contribute to the lower toxicity of the ME systems [75]. To this end, incorporating pCA into ME systems leads to a synergistic cytotoxic effect making it possible to decrease the treatment dose due to the significantly higher inhibitory effect at a lower drug concentration.
In Vivo Oral Absorption Study
Figure 6 represents the F1 LTL and F5 CTTL ME systems plasma profiles versus the pure drug suspension. The oral administration of the ME systems exhibited a significantly higher absorption and relative BAV as revealed in Table 2 (p < 0.05), in addition to a greater Cmax (8.31 ± 0.19 and 9.07 ± 0.14 μg/mL) and AUCtotal values (38.34 ± 3.15 and 46.82 ± 2.79 μg/mL.h) for F1 and F5, respectively, compared to that of pure drug suspension having a Cmax at 6.28 ± 0.18 μg/mL and AUCtotal of 26.32 ± 0.42 μg/mL.h. According to these results and with respect to the number of sampled animals in each group, the statistical power of analysis was calculated as 50% (calculated using Giga calculator, https://www.gigacalculator.com/). It is recommended that the number of sampled animals should be increased in future similar experiments in order to increase the power of analysis to 80% or more. Also, the recorded Tmax in F1 and F5 ME systems were less than the pure drug suspension. These obtained results conformed with the in vitro results, where the pure drug suspension showed a lag time of 5 min and a lower re-dispersibility rate than the optimized ME system formulae.

Plasma concentration time profile of pCA in rat’s plasma
The significant differences in the parameters resulting in higher drug absorption and BAV of the optimized ME systems relative to the pure drug suspension were probably attributed to the larger surface area obtained from the oral administration of the ME systems due to droplet size reduction to about 10 nm, which improved diffusion across intestinal membranes and increased mucosal permeability [46]. Additionally, the promotion of lymphatic transportation of the loaded ME systems occurs due to the oil in the formulae and the small globules, thus bypassing the portal vein route and the first-pass effect [49, 76, 77]. Moreover, high concentrations of the S and the CoS in the ME, as discussed earlier, were able to disturb the cell membranes and open the tight junctions of the intestinal epithelium, thus increasing the permeability of the intestinal barrier. Furthermore, P-gp inhibition contributed to this higher BAV.
On the other hand, pure pCA suspension could not dissolute rapidly in the GIT because of its insufficient solubility, in addition to the absence of all the previously mentioned ME system advantages. Consequently, low drug concentrations and poor oral absorption were achieved.
Furthermore, by comparing F1 LTL and F5 CTTL ME systems, the latter showed superior pharmacokinetic parameters. This superiority may be due to the difference in the constituents where Capryol 90—the oil component—in F5 assisted in enhancing the drug absorption more than Labrafil M 1944 CS—the oil component—in F1 as reported by Hu et al. [78]. Also, F5 contains higher concentrations of surfactants and co-surfactants and consequently augmented the penetration and the absorption boosting effects [71, 72, 79].
Conclusion
In the current study, an effort was performed to augment the poor aqueous solubility and low BAV of p-coumaric acid by developing two novel optimized pCA-ME formulations. The optimized formulations provoked a notable enhancement in the drug dispersion rate contrary to the pure drug suspension. The prepared pCA-ME formulations were proven safe and revealed a pronounced improvement in the antiproliferative activity of pCA against MCF-7, Cacao2, and HepG2 cell lines. Moreover, the results of the in vivo pharmacokinetic study showed notably improved pharmacokinetic parameters with approximately 1.5–2-fold enhancement in bioavailability. To sum up, the prepared p-coumaric acid microemulsion systems could be used as a potential orally administered remedy for cancer disease.
Data Availability
The datasets produced and/or analyzed during the present work are available from the corresponding author on reasonable request.
References
Nagai H, Kim YH. Cancer prevention from the perspective of global cancer burden patterns. J Thorac Dis. 2017;9(3):448–51. https://doi.org/10.21037/jtd.2017.02.75.
Heredia JFV, Gironés SR, Munarriz BJ, Guillén JSC. Management of the toxicity of chemotherapy and targeted therapies in elderly cancer patients. Clin Transl Oncol. 2020;22(4):457–67. https://doi.org/10.1007/s12094-019-02167-y.
Panda R, Kuotsu K. Fabrication, characterization, and in vitro evaluation of pegylated glyceride Labrasol® nanostructured lipid carrier composites of methotrexate: the pathway to effective cancer Therapy. Asian J Pharm Clin Res. 2019;12(6):229–37. https://doi.org/10.22159/ajpcr.2019.v12i6.33377.
Moorthi C, Manavalan R, Kathiresan K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Sci. 2011;14(1):67–77.
Nakamura Y, Mochida A, Choyke PL, Kobayashi H. Nanodrug delivery : is the enhanced permeability and retention effect sufficient for curing cancer ? Bioconjug Chem. 2016;27(10):2225–38. https://doi.org/10.1021/acs.bioconjchem.6b00437.
Zhao Z, Moghadasian MH. Bioavailability of hydroxycinnamates : a brief review of in vivo and in vitro studies. Phytochem Rev J. 2010;9:133–45. https://doi.org/10.1007/s11101-009-9145-5.
Ferreira PS, Victorelli FD, Fonseca-Santos B, Chorilli M. A review of analytical methods for p-coumaric acid in plant-based products, beverages, and biological matrices. Crit Rev Anal Chem. 2018;49(1):21–31. https://doi.org/10.1080/10408347.2018.1459173.
Ou J, Huang J, Ou S. p -Coumaric acid and its conjugates : dietary sources, pharmacokinetic properties and biological activities. J Sci Food Agric. 2015;96:2952–62. https://doi.org/10.1002/jsfa.7578.
Biswick T, Park DH, Shul YG, Choy JH. p-Coumaric acid-zinc basic salt nanohybrid for controlled release and sustained antioxidant activity. J Phys Chem Solids. 2010;71(4):647–9. https://doi.org/10.1016/j.jpcs.2009.12.058.
Ouimet MA, Stebbins ND, Uhrich KE. Biodegradable coumaric acid-based poly(anhydride-ester) synthesis and subsequent controlled release. Macromol Rapid Commun. 2013;34(15):1231–6. https://doi.org/10.1002/marc.201300323.
Salameh D, Brandam C, Medawar W, Lteif R, Strehaiano P. Highlight on the problems generated by p-coumaric acid analysis in wine fermentations. Food Chem. 2008;107(4):1661–7. https://doi.org/10.1016/j.foodchem.2007.09.052.
Zhang X, Song J, Shi X, Miao S, Li Y, Wen A. Absorption and metabolism characteristics of rutin in Caco-2 cells. Sci World J. 2013;2013:1–8.
Parul J, Geeta A, SL H, Amanpreet K. Bioavailability enhancdement of poorly soluble drugs by SMEDDS: a review. J Drug Deliv Ther. 2013;3(1):98-109. https://doi.org/10.22270/jddt.v3i1.360.
Marasini N, Yan YD, Poudel BK, Choi H, Yong CO, Kim JO. Development and optimization of self-nanoemulsifying drug delivery system with enhanced bioavailability by Box-Behnken design and desirability function. J Pharm Sci. 2012;101(12):4584–96. https://doi.org/10.1002/jps.23333.
Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012;2012:1–10. https://doi.org/10.5402/2012/195727.
Yehia R, Hathout RM, Attia DA, Elmazar MM, Mortada ND. Anti-tumor efficacy of an integrated methyl dihydrojasmonate transdermal microemulsion system targeting breast cancer cells: in vitro and in vivo studies. Colloids Surfaces B Biointerfaces. 2017;155:512–21. https://doi.org/10.1016/j.colsurfb.2017.04.031.
Safwat S, Ishak RAH, Hathout RM, Mortada ND. Nanostructured lipid carriers loaded with simvastatin: effect of PEG/glycerides on characterization, stability, cellular uptake efficiency and in vitro cytotoxicity. Drug Dev Ind Pharm. 2017;43(7):1112–25. https://doi.org/10.1080/03639045.2017.1293681.
Guinedi AS, Mortada ND, Mansour S, Hathout RM. Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm. 2005;306:71–82. https://doi.org/10.1016/j.ijpharm.2005.09.023.
Nasser N, Hathout RM, Abd-Allah H, Sammour OA. Enhancement of oral bioavailability of drugs using lipid-based carriers: a meta-analysis study. Drug Dev Ind Pharm. 2020;46(12):2105–10. https://doi.org/10.1080/03639045.2020.1851245.
Subongkot T, Ngawhirunpat T. Development of a novel microemulsion for oral absorption enhancement of all-trans retinoic acid. Int J Nanomedicine. 2017;12:5585–99. https://doi.org/10.2147/IJN.S142503.
Shah N, Seth A, Balaraman R, Sailor G, Javia A, Gohil D. Oral bioavailability enhancement of raloxifene by developing microemulsion using D-optimal mixture design: optimization and in-vivo pharmacokinetic study. Drug Dev Ind Pharm. 2017;44(4):687–96. https://doi.org/10.1080/03639045.2017.1408643.
Guo R, Fu X, Chen J, Zhou L, Chen G. Preparation and characterization of microemulsions of myricetin for improving its antiproliferative and antioxidative activities and oral bioavailability. J Agric Food Chem. 2016;64(32):6286–94. https://doi.org/10.1021/acs.jafc.6b02184.
Hathout RM, Mansour S, Geneidi AS, Mortada ND. Visualization, dermatopharmacokinetic analysis and monitoring the conformational effects of a microemulsion formulation in the skin stratum corneum. J Colloid Interface Sci. 2011;354(1):124–30. https://doi.org/10.1016/j.jcis.2010.10.025.
Singh PK, Iqubal MK, Shukla VK, Shuaib M. Microemulsions: current trends in novel drug delivery systems. J Pharm Chem Biol Sci. 2014;1(1):39-51. http://www.jpcbs.info.
Hathout RM, Mansour S, Mortada ND, Geneidi AS, Guy RH. Uptake of microemulsion components into the stratum corneum and their molecular effects on skin barrier function. Mol Pharm. 2010;7(4):1266–73. https://doi.org/10.1021/mp100068s.
Bowden GD, Pichler BJ, Maurer A. A design of experiments (DoE) approach accelerates the optimization of copper-mediated F-fluorination reactions of arylstannanes. Sci Rep. 2019;9(1):1–10. https://doi.org/10.1038/s41598-019-47846-6.
Zhang T, Youan BC. Analysis of process parameters affecting spray-dried oily core nanocapsules using factorial design. Am Assoc Pharm Sci. 2010;11(3):1422–31. https://doi.org/10.1208/s12249-010-9516-7.
Abdel-hafez SM, Hathout RM, Sammour OA. Towards better modeling of chitosan nanoparticles production : screening different factors and comparing two experimental designs. Int J Biol Macromol. 2014;64:334–40. https://doi.org/10.1016/j.ijbiomac.2013.11.041.
Hathout RM, Gad HA, Abdel-Hafez SM, et al. Gelatinized core liposomes: a new Trojan horse for the development of a novel timolol maleate glaucoma medication. Int J Pharm. 2018;556:192–9. https://doi.org/10.1016/j.ijpharm.2018.12.015.
Mandlik SK, Adhikari S, Deshpande AA. Application of simplex lattice design in formulation and development of buoyant matrices of dipyridamole. J Appl Pharm Sci. 2012;2(12):107–11. https://doi.org/10.7324/JAPS.2012.21221.
Patel DM, Patel NM, Pandya NN, Jogani PD. Gastroretentive drug delivery system of carbamazepine: Formulation optimization using simplex lattice design: a technical note. Am Assoc Pharm Sci. 2007;8(1):1–5. https://doi.org/10.1208/pt0801011.
Gabr S, Nikles S, Pferschy Wenzig EM, rdjomand-Woelkart K, Hathout RM, El-Ahmady S, Motaal AA, Singab A, Bauer R. Characterization and optimization of phenolics extracts from Acacia species in relevance to their anti-inflammatory activity. Biochem Syst Ecol. 2018;78:21–30. https://doi.org/10.1016/j.bse.2018.03.001.
Todorov H, Searle-white E, Gerber S. Applying univariate vs. multivariate statistics to investigate therapeutic efficacy in (pre) clinical trials : a Monte Carlo simulation study on the example of a controlled preclinical neurotrauma trial. PLoS ONE J. 2020;15:1–20. https://doi.org/10.1371/journal.pone.0230798.
Hardy JG, Sdepanian S, Stowell AF, Aljohani AD, Allen MJ, Anwar A, Barton D, Baum JV, Bird D, Blaney A, et al. Potential for chemistry in multidisciplinary interdisciplinary and transdisciplinary teaching activities in higher education. J Chem Educ. 2021;98(4):1124–45. https://doi.org/10.1021/acs.jchemed.0c01363.
Fagir W, Hathout RM, Sammour OA, ElShafeey AH. Self-microemulsifying systems of finasteride with enhanced oral bioavailability: multivariate statistical evaluation, characterization, spray-drying and in vivo studies in human volunteers. Nanomedicine. 2015;10(22):3373–89.
Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. Am Assoc Pharm Sci. 2007;9(3):344–52. https://doi.org/10.1208/aapsj0903041.
Wang L, Yan W, Tian Y, Xue H, Tang J, Zhang L. Self-microemulsifying drug delivery system of phillygenin: formulation development, characterization and pharmacokinetic evaluation. Pharmaceutics. 2020;12(2):1–17. https://doi.org/10.3390/pharmaceutics12020130.
Ramli S, Norhman N, Zainuddin N, Ja’afar SM, Rahman IA. Nanoemulsion based palm olein as vitamin E carrier. Malaysian J Anal Sci. 2017;21(6):1399–408. https://doi.org/10.17576/mjas-2017-2106-22.
Akula S, Gurram AK, Devireddy SR. Self-microemulsifying drug delivery systems: an attractive strategy for enhanced therapeutic profile. Int Sch Res Not. 2014;2014:1–11. https://doi.org/10.1155/2014/964051.
Hathout RM. Using principal component analysis in studying the transdermal delivery of a lipophilic drug from soft nano-colloidal carriers to develop a quantitative composition effect permeability relationship. Pharm Dev Technol. 2013;19(5):598–604. https://doi.org/10.3109/10837450.2013.813544.
Mendes M, Basso J, Silva J, Cova T, Sousa J, Pais A, Vitorino C. Biomimeting ultra- small lipid nanoconstructs for glioblastoma treatment: a computationally guided experimental approach. Int J Pharm. 2020;587:119661. https://doi.org/10.1016/j.ijpharm.2020.119661.
Moghimipour E, Salimi A, Eftekhari S. Design and characterization of microemulsion systems for naproxen. Adv Pharm Bull. 2013;3(1):63–71. https://doi.org/10.20959/wjpps20177-9193.
Bindhani S, Mohapatra S, Kar RK. Preparation, characterization and stability studies of solid self emulsifying drug delivery system of nifedipine. Int J Appl Pharm. 2020;12(2):94–102. https://doi.org/10.22159/ijap.2020v12i2.36406.
Garrait G, Jarrige JF, Blanquet S, Beyssac E, Cardot JM, Alric M. Gastrointestinal absorption and urinary excretion of trans-cinnamic and p-coumaric acids in rats. J Agric Food Chem. 2006;54(8):2944–50. https://doi.org/10.1021/jf053169a.
Pei K, Ou J, Huang J, Ou S. p-Coumaric acid and its conjugates: dietary sources, pharmacokinetic properties and biological activities. J Sci Food Agric. 2016;96(9):2952–62. https://doi.org/10.1002/jsfa.7578.
Li F, Song S, Guo Y, et al. Preparation and pharmacokinetics evaluation of oral self-emulsifying system for poorly water-soluble drug lornoxicam. Drug Deliv. 2015;22(4):487–98. https://doi.org/10.3109/10717544.2014.885615.
Ossama M, Hathout RM, Attia DA, Mortada ND. Enhanced allicin cytotoxicity on HEPG-2 cells using glycyrrhetinic acid surface-decorated gelatin nanoparticles. ACS Omega. 2019;4(6):11293–300. https://doi.org/10.1021/acsomega.9b01580.
Sharma SH, Chellappan DR, Chinnaswamy P, Nagarajan S. Protective effect of p-coumaric acid against 1,2 dimethylhydrazine induced colonic preneoplastic lesions in experimental rats. Biomed Pharmacother. 2017;94:577–88. https://doi.org/10.1016/j.biopha.2017.07.146.
Gui SY, Wu L, Peng DY, Liu QY, Yin BP, Shen JZ. Preparation and evaluation of a microemulsion for oral delivery of berberine. Pharm J. 2008;63(7):516–9. https://doi.org/10.1691/ph.2008.8018.
Marquele FD, Oliveira ARM, Bonato PS, Lara MG, Fonseca MJV. Propolis extract release evaluation from topical formulations by chemiluminescence and HPLC. J Pharm Biomed Anal. 2006;41(2):461–8. https://doi.org/10.1016/j.jpba.2005.12.022.
Paithanka HV. HPLC method validation for pharmaceuticals: a review. Int J Univers Pharm Bio Sci. 2013;2(4):229–40.
Goyal U, Arora R, Aggarwal G. Formulation design and evaluation of a self-microemulsifying drug delivery system of lovastatin. Acta Pharm. 2012;62(3):357–70. https://doi.org/10.2478/v10007-012-0022-1.
Ukai H, Iwasa K, Deguchi T, Morishita M, Katsumi H, Yamamoto A. Enhanced intestinal absorption of insulin by capryol 90, a novel absorption enhancer in rats: implications in oral insulin delivery. Pharmaceutics. 2020;12(5):1–16. https://doi.org/10.3390/pharmaceutics12050462.
Nasr AM, Qushawy MK, Elkhoudary MM, Gawish AY, Elhady SS, Swidan SA. Quality by design for the development and analysis of enhanced in-situ forming vesicles for the improvement of the bioavailability of fexofenadine hcl in vitro and in vivo. Pharmaceutics. 2020;12(5):1–22. https://doi.org/10.3390/pharmaceutics12050409.
Venkatesh G, Majid MIA, Mansor SM, Nair NK, Croft SL, Navaratnam V. In vitro and in vivo evaluation of self-microemulsifying drug delivery system of buparvaquone. Drug Dev Ind Pharm. 2010;36(6):735–45. https://doi.org/10.3109/03639040903460446.
Jaiswal P, Aggarwal G, Harikumar SL, Singh K. Development of self-microemulsifying drug delivery system and solid-self-microemulsifying drug delivery system of telmisartan. Int J Pharm Investig. 2014;4(4):195–206. https://doi.org/10.4103/2230-973x.143123.
Mcclements DJ. Nanoemulsions versus microemulsions : terminology, differences, and similarities. Soft Matter J. 2012;8:1719–29. https://doi.org/10.1039/c2sm06903b.
Mundada VP, Sawant KK. Nanomedicine & nanotechnology enhanced oral bioavailability and anticoagulant activity of dabigatran etexilate by self-micro emulsifying drug delivery system : systematic development, in vitro, ex vivo and in vivo evaluation. J Nanomedicine Nanotechnol. 2018;9(1):1–13. https://doi.org/10.4172/2157-7439.1000480.
Sarheed O, Dibi M, Ramesh KVRNS. Studies on the effect of oil and surfactant on the formation of alginate-based O / W lidocaine nanocarriers using nanoemulsion template. Pharmaceutics. 2020;12(1223):1-21. https://doi.org/10.3390/pharmaceutics12121223.
Hong EP, Kim JY, Kim SH, et al. Formulation and evaluation of a self-microemulsifying drug delivery system containing bortezomib. Chem Pharm Bull. 2016;64(8):1108–17. https://doi.org/10.1248/cpb.c16-00035.
Hwang KM, Park SA, Kim JY, Park CW, Rhee YS, Park ES. Formulation and in vitro evaluation of self-microemulsifying drug delivery system containing fixed-dose combination of atorvastatin and ezetimibe. Chem Pharm Bull. 2015;63(6):423–30. https://doi.org/10.1248/cpb.c14-00814.
Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system ( SMEDDS ) of tacrolimus. Am Assoc Pharm Sci. 2008;9(1):13–21. https://doi.org/10.1208/s12249-007-9014-8.
Forouz F, Dabbaghi M, Namjoshi S, Mohammed Y, Roberts MS, Grice JE. Development of an oil-in-water self-emulsifying microemulsion for cutaneous delivery of rose bengal: investigation of anti-melanoma properties. Pharmaceutics. 2020;12(10):1–16. https://doi.org/10.3390/pharmaceutics12100947.
Abdelrady H, Hathout RM, Osman R, Saleem I, Mortada ND. Exploiting gelatin nanocarriers in the pulmonary delivery of methotrexate for lung cancer therapy. Eur J Pharm Sci. 2019;133:115–26. https://doi.org/10.1016/j.ejps.2019.03.016.
Desai P, Thakkar A, Ann D, Wang J, Prabhu S. Loratadine self-microemulsifying drug delivery systems (SMEDDS) in combination with sulforaphane for the synergistic chemoprevention of pancreatic cancer. Drug Deliv Transl Res. 2019;9(3):641–51. https://doi.org/10.1007/s13346-019-00619-0.
Chen YC, Chen BH. Preparation of curcuminoid microemulsions from: Curcuma longa L to enhance inhibition effects on growth of colon cancer cells HT-29. R Soc Chem Adv. 2018;8(5):2323–37. https://doi.org/10.1039/c7ra12297g.
Pham DT, Saelim N, Tiyaboonchai W. Paclitaxel loaded EDC-crosslinked fibroin nanoparticles: a potential approach for colon cancer treatment. Drug Deliv Transl Res. 2020;10(2):413–24. https://doi.org/10.1007/s13346-019-00682-7.
Bilia AR, Isacchi B, Righeschi C, Guccione C, Bergonzi MC. Flavonoids loaded in nanocarriers: an opportunity to increase oral bioavailability and bioefficacy. Food Nutr Sci. 2014;5(13):1212–27. https://doi.org/10.4236/fns.2014.513132.
Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–86. https://doi.org/10.1016/j.ejps.2005.01.001.
Ujhelyi Z, Kalantari A, Vecsernyés M, et al. The enhanced inhibitory effect of different antitumor agents in self-microemulsifying drug delivery systems on human cervical cancer HeLa cells. Molecules. 2015;20(7):13226–39. https://doi.org/10.3390/molecules200713226.
Li X, Yuan Q, Huang Y, Zhou Y, Liu Y. Development of silymarin self-microemulsifying drug delivery system with enhanced oral bioavailability. Am Assoc Pharm Sci. 2010;11(2):672–8. https://doi.org/10.1208/s12249-010-9432-x.
Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted inhibitors of P-glycoprotein increase chemotherapeutic-induced mortality of multidrug resistant tumor cells. Sci Rep. 2018;8(1):1–18. https://doi.org/10.1038/s41598-018-19325-x.
Pollard J, Rajabi-Siahboomi A, Badhan RKS, Mohammed AR, Perrie Y. High-throughput screening of excipients with a biological effect: a kinetic study on the effects of surfactants on efflux-mediated transport. J Pharm Pharmacol. 2019;71(6):889–97. https://doi.org/10.1111/jphp.13072.
Ujhelyi Z, Fenyvesi F, Váradi J, et al. Evaluation of cytotoxicity of surfactants used in self-micro emulsifying drug delivery systems and their effects on paracellular transport in Caco-2 cell monolayer. Eur J Pharm Sci. 2012;47(3):564–73. https://doi.org/10.1016/j.ejps.2012.07.005.
Patel K, Pati A, Mehta M, Gota V, Vavia P. Medium chain triglyceride (MCT) rich, paclitaxel loaded self nanoemulsifying preconcentrate (PSNP): a safe and efficacious alternative to taxol®. J Biomed Nanotechnol. 2013;9:1–11. https://doi.org/10.1166/jbn.2013.1710.
Wei L, Sun P, Nie S, Pan W. Preparation and evaluation of SEDDS and SMEDDS containing carvedilol. Drug Dev Ind Pharm. 2005;31(8):785–94. https://doi.org/10.1080/03639040500216428.
Li Y, Song J, Tian N, et al. Improving oral bioavailability of metformin hydrochloride using water-in-oil microemulsions and analysis of phase behavior after dilution. Int J Pharm. 2014;473(1–2):316–25. https://doi.org/10.1016/j.ijpharm.2014.07.011.
Hu L, Wu H, Niu F, Yan C, Yang X, Jia Y. Design of fenofibrate microemulsion for improved bioavailability. Int J Pharm. 2011;420(2):251–5. https://doi.org/10.1016/j.ijpharm.2011.08.043.
Hill M, Cunningham RN, Hathout RM, Johnston C, Hardy JG, Migaud ME. Formulation of antimicrobial tobramycin loaded PLGA nanoparticles via complexation with AOT. J Funct Biomater. 2019;10(2):26. https://doi.org/10.3390/jfb10020026.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The authors asseverate that no assets, grants, or other support were gained during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Nayera Nasser: methodology, software, formal analysis, data curation, investigation, and writing—original draft preparation. Rania M. Hathout: conceptualization, methodology, software, formal analysis, investigation, data curation, writing—reviewing and editing, and supervision. Hend Abd-Allah: formal analysis, visualization, investigation, writing—reviewing and editing, and supervision. Omaima A. Sammour: formal analysis, visualization, investigation, writing—reviewing and editing, and supervision.
Corresponding author
Ethics declarations
Ethical Approval
This study was executed in agreement with the principles of the Declaration of Helsinki. Approval was gained by the Research Ethical Committee of the Faculty of Pharmacy, Ain Shams University, protocol number 273.
Consent to Participate
Not relevant as the study includes no human subjects.
Consent for Publication
Not relevant as the work has no human subjects.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nasser, N., Hathout, R.M., Abd-Allah, H. et al. Simplex Lattice Design and Machine Learning Methods for the Optimization of Novel Microemulsion Systems to Enhance p-Coumaric Acid Oral Bioavailability: In Vitro and In Vivo Studies. AAPS PharmSciTech 25, 56 (2024). https://doi.org/10.1208/s12249-024-02766-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-024-02766-1