
Abstract
Bioavailability/bioequivalence studies supporting clinical drug development or commercial supply of drug formulations are often time, cost, and resource intensive. The drug’s pharmacokinetic (PK) variability, systemic half-life, and safety issues may pose additional challenges. The stable isotope label (SIL) approach provides a useful tool to significantly reduce the study size in clinical PK studies. Tafenoquine (TQ) is an 8-aminoquinoline under development for preventing Plasmodium vivax malaria relapse. This SIL study assessed the impact of differences in the in vitro dissolution profiles on in vivo exposure of TQ tablets. Fourteen healthy volunteers received a single dose of 300 mg TQ Intermediate Aged or 300 mg TQ Control formulations in this single-center, two-arm, randomized, open-label, parallel-group study. Endpoints included the geometric means ratio of the area under the concentration-time curve (AUC(0-t) and AUC(0-∞); primary endpoint) and maximum plasma concentration (Cmax) for Intermediate Aged versus Control TQ; correlation of PK parameters for venous versus peripheral (via microsample) blood samples; and safety and tolerability endpoints. Geometric mean ratios for PK parameters (AUC and Cmax) and their 90% confidence intervals fell well within standard bioequivalence limits (0.80–1.25). Only one mild adverse event (skin abrasion) was reported. In summary, this SIL methodology-based study demonstrates that the observed differences in the in vitro dissolution profiles between the Control and Intermediate Aged TQ tablets have no clinically relevant effect on systemic TQ exposure. The SIL approach was successfully implemented to enable the setting of a clinically relevant dissolution specification.
Clinical trial: This study (GSK study number 201780) is registered at clinicaltrials.gov with identifier NCT02751294.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Systemic drug exposures can be impacted by minor changes in formulation characteristics. In vitro dissolution testing plays a key role in understanding and optimizing a drug’s formulation, absorption profile, and release characteristics [1]. It is a regulatory expectation to employ a discriminating dissolution method for the commercial testing of oral solid dosage forms. Such methods should detect variations in the manufacturing process parameters and are used for quality control testing. The in vivo data can be used to characterize the biorelevance of the dissolution method which can then be used to support a clinically meaningful specification, thus establishing a shelf-life for the medicine.
Relative bioavailability or bioequivalence (relBA/BE) studies are often needed to compare exposures when optimizing formulations in a drug’s clinical development program or for commercial supply. These comparisons are generally performed in Phase 1 studies of healthy volunteers. The drug’s pharmacokinetic (PK) variability and systemic half-life play a key role in defining the number of subjects and study duration and design. Consequently, conducting relBA/BE studies with drugs that have high PK variability and long half-lives can pose considerable challenges.
A method or study design that addresses these challenges can be valuable in the drug development process [2]. The stable isotope label (SIL) method provides an effective approach for comparing qualitatively and quantitatively similar formulations and can generate in vivo (PK) data to support in vitro tests [2]. Using a stable isotope, the plasma concentrations of an enriched and non-enriched pharmaceutical agent can be measured from the same plasma sample (i.e., subject); thus, the SIL compound operates as internal references [3]. Co-administration of an enriched and non-enriched drug and the resulting correlation in the PK parameters derived from the same sample have been shown to reduce variability in the statistical test used to compare formulations, thus significantly decreasing the required number of subjects in both crossover and parallel group designs [3, 4].
The present study applies the SIL approach to assess the impact of different in vitro dissolution profiles caused by storage of tablets on the in vivo exposure of antimalarial drug tafenoquine (TQ). TQ is an 8-aminoquinoline antimalarial drug with a long half-life of approximately 15 days and is being co-developed by GlaxoSmithKline and Medicines for Malaria Venture (MMV) for the radical cure of Plasmodium vivax relapse. TQ has been assessed in Phase 2 [5] and Phase 3 clinical studies [6]. The current investigational regimen is TQ 300 mg single dose (administered as two 150 mg capsules) plus standard 3-day chloroquine [5]. While the long half-life of TQ offers the potential for a convenient dosing regimen [6, 7], it also brings its own challenges to drug development as discussed above.
Using the SIL method, this study aimed to determine whether the differences in the dissolution profiles of two TQ formulations led to any changes in in vivo drug exposure. The data could underpin the setting of clinically relevant dissolution specification for commercial tablet testing.
MATERIALS AND METHODS
Materials
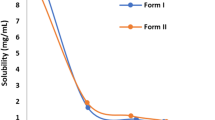
Two batches of TQ 150 mg tablets were investigated in the present study: tablets stored under accelerated temperature and humidity conditions to mimic the changes observed, referred to as Intermediate Aged, and freshly manufactured tablets (Control); the respective dissolution profiles are shown in Fig. 1 (methodology provided below). SIL TQ was synthesized to contain the stable isotopes 13C and 15N in positions that are not susceptible to metabolism ([13C415N]-TQ). The SIL TQ (30 mg total) was prepared as a 0.25 mg/mL aqueous solution for oral administration. .

Dissolution profiles of Control and Intermediate Aged TQ tablets close to time of dosing. TQ, tafenoquine
Dissolution Method
A dissolution method was developed and validated for TQ 150 mg tablets using a United States Pharmacopoeia (USP) II apparatus at 75 rpm and a medium containing phosphate buffer at pH 6.8 with 0.20% Polysorbate 20 (surfactant).
Study Objectives
The primary objective of the study was to determine the relative bioavailability of TQ from tablets exhibiting different dissolution profiles in healthy subjects, i.e., Intermediate Aged versus Control. Secondary objectives were to compare the maximum plasma concentration (Cmax) of TQ from tablets exhibiting different dissolution profiles in healthy subjects; determine the correlation between relative exposures of TQ after single oral doses of TQ 300 mg using PK samples from venous blood and peripheral blood collection (obtained via microsampling) to support PK sampling in a pediatric population; and assess the safety and tolerability of TQ after single oral doses of 300 mg from tablets exhibiting different dissolution profiles.
Study Design
This single-center, two-arm, randomized, open-label, parallel-group study was conducted in healthy volunteers. Following screening for study eligibility, subjects were randomized 1:1 to receive one dose of TQ as one of two TQ tablet formulations, which differed in their in vitro dissolution profiles: 300 mg TQ Intermediate Aged or 300 mg TQ Control (Fig. 2); subjects completed their meal within 30 mins after which the formulations were administered in an inpatient setting as 2 × 150 mg tablets orally after food on Day 1. Each subject also received 30 mg of TQ SIL solution (as 120 mL of 0.25 mg/mL solution) with the TQ tablets. A rinse of the SIL dosing container was permitted using approximately 20 mL or less of water. The maximum dose of TQ did not exceed 330 mg in a single dose (300 mg TQ and 30 mg SIL TQ). Subjects returned for outpatient visits on Days 7, 14, 21, 28, and 56 (Fig. 2).

Study design schematic. SIL, stable isotope label; TQ, tafenoquine
The study protocol, any amendments, the informed consent, and other information were reviewed and approved by an institutional review board (Aspire IRB, Santee, California USA), in accordance with the International Conference on Harmonization Good Clinical Practice and the Declaration of Helsinki. All subjects provided written informed consent prior to participation in the study.
Randomization and Blinding
Subjects were randomized to treatment in accordance with the schedule generated by GlaxoSmithKline (GSK) Quantitative Sciences using validated internal software; randomization was stratified by baseline body weight (< 80 kg, ≥ 80 kg). No subject or study center staff blinding was performed.
Study Population
Eligible subjects were healthy males, based on medical assessment, 18–55 years of age (inclusive) with a body weight between ≥ 35 kg and ≤ 100 kg. Subjects were not enrolled in the study if they had a QT interval corrected for heart rate according to Fridericia’s formula (QTcF) > 450 msec, or if they had any documented phenotypic glucose-6-phosphate dehydrogenase (G6PD) deficiency, with < 70% of local median G6PD activity level derived from G6PD normal males.
Subjects had to abstain from taking prescription or non-prescription drugs (including vitamins and dietary or herbal supplements) within 7 days (or 14 days if the drug was a potential enzyme inducer) or 5 half-lives (whichever was longer) prior to the first dose of study medication until completion of the follow-up visit. The full list of exclusion and withdrawal criteria is provided in the online Supplementary material.
Study Endpoints
The primary endpoint of this study was the ratio of the geometric means (90% confidence interval [CI]) for the area under the concentration-time curve (AUC) for Intermediate Aged TQ to that of Control (AUC(0-t) and AUC(0-∞)). Secondary endpoints included the ratio of geometric means for the Cmax for Intermediate Aged TQ to Control and PK parameters comparing venous and peripheral (via microsampling) blood samples, adverse events, vital signs, and laboratory parameters.
Pharmacokinetic Assessments
Systemic TQ PK samples were collected over a period of 8 weeks. Samples were analyzed using a validated liquid chromatography-tandem mass spectrometry (LC-MS-MS) method. The characteristic precursor [M + H]+ to product ion transitions, for TQ, TQ SIL ([13C415N]-TQ), and the bioanalytical internal standard were used as multiple reaction monitoring (MRM) transitions to ensure high selectivity within the LC-MS-MS assay. As part of the assay, validation selectivity of the method for TQ and TQ SIL ([13C415N]-TQ) was demonstrated by analyzing six replicates of one validation sample spiked at 1.5 ng/mL with TQ on its own, TQ SIL on its own, and the two spiked together. The accuracy was comprised between 85 and 115% and precision values were within 15% indicating that there were no interferences between TQ and TQ SIL. Furthermore, the stability of TQ and TQ SIL was demonstrated in plasma, whole blood, processed samples, and solvent and all supported the maximum storage duration of the study. The AUC and Cmax for TQ and TQ SIL were estimated in each subject as described elsewhere [8]. Further details of PK assessments and bioanalytical assay are provided in the online Supplementary material.
Microsampling Assessments
To enable the application of peripheral microsampling in planned pediatric studies, a comparison was performed for PK exposures (AUC and Cmax for TQ) in peripheral blood samples (obtained via microsampling) versus venous blood samples. Two peripheral microsamples were collected for each plasma sample within 5 min of the venous plasma sample collection.
Safety and Tolerability Assessments
Safety assessments included the monitoring of adverse events (AEs), clinical laboratory tests, vital signs, and physical examinations.
Statistical Analyses
The primary analysis of this study was the treatment comparison of TQ 300 mg Intermediate Aged and Control using TQ PK parameters AUC(0-∞) and AUC(0-t) derived from venous samples. PK parameters were determined from the TQ plasma concentration-time data and derived separately for TQ (venous sample), TQ SIL (venous sample), and TQ (peripheral sample), as data permitted. AUC(0-∞), AUC(0-t), and Cmax were log-transformed prior to statistical analysis. The primary comparison of interest is the treatment difference in the TQ PK parameters. The statistical comparisons of PK parameters were conducted using an analysis of covariance model (ANCOVA), adjusting for the corresponding log-transformed TQ SIL PK parameter. This was done to reduce the between subject variation for the comparison of the TQ PK parameters, given the high correlation between the TQ and TQ SIL. Separating out this variation with the ANCOVA model allows better assessment of the treatment effect on the TQ PK parameters. Treatments were compared using a ratio of geometric least squares (LS) means and 90% CIs.
Simulations were conducted to determine the appropriate sample size for this study. Assuming an underlying between-subject variability of 27% in the AUC and a correlation of 0.95 between enriched and non-enriched PK parameters, a sample size of 6 subjects per arm provided a reduction of the between-subject variability to around 12% or less for statistical comparisons (mean CV 8%; 5th and 95th percentiles 5 and 12%, respectively). With a between-subject variability of 12% and sample size of 6 subjects per group, the confidence limits for a point estimate of geometric mean ratio of 1.0 are 0.88 to 1.13. Further details of the statistical analysis are included in the online supplement. The primary analysis was conducted in the PK population, which included all subjects who received their dose of study medication and had any valid PK parameters derived. The safety population included all subjects who had received at least one dose of study treatment.
RESULTS
Subject Disposition
A total of 63 subjects were screened, of whom 14 subjects were randomized 1:1 to receive the Intermediate Aged and Control TQ 300 mg. All randomized subjects were dosed and completed the study follow-up. Subjects receiving TQ 300 mg Control were older on average than those receiving TQ 300 mg Intermediate Aged. However, with such a low number of subjects, small imbalances in baseline characteristics are not unexpected. Other demographic and baseline characteristics were reasonably well balanced across treatment groups (Table I).
Pharmacokinetic Results
The exposures for TQ Intermediate Aged compared to Control are summarized in Table II, which shows the ratio of geometric LS means and their 90% CIs for AUCs and Cmax, as well as the between-subject variability (percentage of geometric coefficient of variation, CVb).
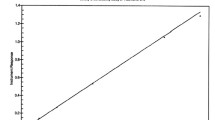
The point estimate and 90% CIs of the ratio of PK parameters fell within the standard bioequivalence limits of 0.80 to 1.25 (Table II, Fig. 3).

Exposure ratios across the two TQ formulations (PK population). Comparison of exposure between Intermediate Aged and Control tablet formulations with the SIL approach. AUC, area under the concentration-time curve; Cmax, maximum plasma concentration; PK, pharmacokinetic; SIL, stable isotope label; TQ, tafenoquine
A high correlation was observed between the TQ and TQ SIL PK parameters (Fig. 4), with correlation coefficients for each treatment group, and each parameter, greater than 0.9, indicating that the SIL approach was appropriate for the intended comparative assessment.

Correlation between TQ and TQ SIL PK parameters: AUC(0-t), AUC(0-∞), and Cmax (PK population). AUC, area under the concentration-time curve; Cmax, maximum plasma concentration; PK, pharmacokinetic; R, Pearson correlation coefficients; SIL, stable isotope label; TQ, tafenoquine
TQ Microsampling Results
There was a good correlation between plasma TQ concentrations obtained from peripheral versus venous samples; however, the correlation weakened at higher plasma drug concentrations (Fig. 5). There was also a good correlation between relative exposures of TQ, as measured by AUC, following venous and peripheral blood sampling, for both the Intermediate Aged and Control formulations (correlation coefficient > 0.7). There was poor correlation for Cmax in the Intermediate Aged group (correlation coefficient ~0.4).

Scatter plot of TQ plasma concentrations obtained by venous and peripheral samples. TQ, tafenoquine
Safety Results
One AE (skin abrasion) was reported in the study and it occurred in the Control group on Day 2. The AE was mild, resolved on the seventh day without any treatment, and was not considered related to the study drug. No other AEs, clinically relevant laboratory findings, or physical examination findings were reported during the study. No deaths were reported.
DISCUSSION
Stable isotope studies enable statistically significant conclusions to be drawn from bioavailability comparisons using fewer subjects [2, 3]. A stable isotope acts as a reference for each subject, reducing the variability of statistical tests and decreasing the number of subjects required to achieve the desired statistical power. The reduced number of subjects offers additional advantages in the design of efficient BA/BE studies [2]. Thus, the SIL approach provides a useful tool for implementing the quality by design paradigm [2]. Similarly, this method can be helpful when conducting smaller PK studies in early-phase drug development and can provide valuable information for internal decision making around formulation development. Though currently under-utilized in drug development, the SIL approach has considerable potential for addressing formulation-related issues in both early- and late-phase drug development, as well as during commercial supply. For the application presented here, a significant advantage of the SIL method was the ability to provide “biorelevant” justification to the proposed commercial dissolution specification for TQ 150 mg tablets.
There are certain considerations when implementing the SIL approach, including the time required for the synthesis of SIL compounds, ability to create an appropriate SIL formulation as a suspension to solution, and application for the comparison of similar formulations only [2, 3]. There may also be a need for improving the blood plasma assay sensitivity for the SIL component (currently, the maximum amount of enriched substance, which determines the analytical sensitivity of the assay, is 10% or less of the target drug dose) [2]. While the reduced number of subjects is an advantage in most cases, especially in populations where recruitment is difficult [2], loss to follow-up of even one subject more than anticipated may significantly impact the results in small studies. The limited awareness about the background preparation work, resources, and lead time needed to plan an SIL-based study as described here may often discourage clinical teams the from applying such a valuable approach.
The SIL study presented here characterized the relative bioavailability of TQ 300 mg Intermediate Aged or Control tablets. In vitro differences in dissolution profiles between the two formulations did not affect the systemic exposure of TQ. The SIL approach dramatically reduced the required sample size from 32 to 6 evaluable subjects per treatment arm by reducing variability in the statistical test and allowing good precision of treatment difference using with a small number of subjects (N = 7 per group). The SIL approach, and resultant decrease in study numbers, also allowed for a parallel study design, an important consideration with TQ, where, due to the drug’s long half-life, a crossover study would require a long duration. The additional 10% TQ dose (SIL component) in addition to the 300 mg dose is not expected to impact the TQ PK profiles as TQ exhibits dose proportional exposures with up to 1200 mg dose level [9].
The geometric mean ratios of the key PK parameters and their 90% CIs for the two TQ formulations fell well within the standard BE limits, indicating that the observed differences in dissolution between the Control and Intermediate Aged tablets have no clinically relevant effect on the systemic exposure of TQ. The high correlation between the PK parameters of the enriched and non-enriched compound significantly reduced the variability in the statistical test to compare the two tablets (to approximately 5%). The correlation observed was in line with the estimate of 0.95 used in the sample size calculation. The between-subject variability was lower than was assumed at study design. No subjects withdrew from the study. These factors led to an even greater precision in estimating treatment differences.
The results obtained from the adjusted analysis, i.e., ANCOVA were also compared to those obtained from the unadjusted analysis and are presented in Table III. There is concordance of results with enriched and non-enriched ANOVA. The results reiterate that the SIL approach-based ANCOVA analysis causes significant improvement in the precision around the point estimate with tighter confidence intervals.
The PK results provided critical data to support clinically relevant dissolution specifications for the TQ formulation underpinning commercial batch release. The dissolutions specifications evaluated in the study can be further utilized to predict the impact of any future changes in TQ formulation, manufacturing process, and control on in vivo drug exposure.
The microsampling exposure assessments within the study were exploratory in nature, i.e., intended to support pediatric studies with TQ and/or other compounds, and showed good correlation at lower drug concentrations but with greater differences at higher concentrations. The differences in drug concentrations between the two techniques (peripheral vs. venous) could be attributed to a number of reasons, including the personnel’s limited experience with microsampling, skin thickness (which posed difficulties in collecting microsamples in adults but may not be relevant to the pediatric population in future pilot studies), and hemolysis during sample collection and processing. The microsampling results did not impact the overall study conclusions, but they can provide valuable information for future studies, specifically in the pediatric population.
Consistent with previous studies [5, 6], the safety profile of TQ was found to be acceptable, with only one minor AE (grazed skin) being reported.
CONCLUSIONS
This SIL study investigating the bioavailability of TQ tablets under different storage conditions showed similar exposures with the TQ 300 mg Intermediate Aged and Control formulations. Differences in the in vitro dissolution profiles of the two formulations had no clinically relevant impact on systemic TQ exposure. The TQ exposure data and PK results underpin the dissolution specification for the proposed TQ commercial tablet drug product. The SIL approach has the potential to significantly conserve resources in the BA and some specific BE settings, and help implement the quality by design paradigm in clinical drug development.
References
FDA/CDER, September 1997.Guidance for industry, extended release oral dosage forms: development, evaluation, and application of in vitro/in vivo correlations. Available from https://www.fda.gov/downloads/drugs/guidances/ucm070239.pdf. Accessed February 2018.
Parr A, Gupta M, Montague TH, Hoke F. Re-introduction of a novel approach to the use of stable isotopes in pharmacokinetic studies. AAPS J. 2012;14(3):639–45.
Baillie TA. The use of stable isotopes in pharmacological research. Pharmacol Rev. 1981;33(2):81–132.
Pieniaszek HJ Jr, Mayersohn M, Adams MP, Reinhart RJ, Barrett JS. Moricizine bioavailability via simultaneous, dual, stable isotope administration: bioequivalence implications. J Clin Pharmacol. 1999;39(8):817–25.
Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, Krudsood S, Gupta SK, Kochar SK, et al. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet. 2014;383(9922):1049–58.
Fukuda MM, Krudsood S, Mohamed K, Green JA, Warrasak S, Noedl H, et al. A randomized, double-blind, active-control trial to evaluate the efficacy and safety of a three day course of tafenoquine monotherapy for the treatment of plasmodium vivax malaria. PLoS One. 2017;12(11):e0187376.
Ebstie YA, Abay SM, Tadesse WT, Ejigu DA. Tafenoquine and its potential in the treatment and relapse prevention of Plasmodium vivax malaria: the evidence to date. Drug Des Devel Ther. 2016;10:2387–99.
Green JA, Mohamed K, Goyal N, Bouhired S, Hussaini A, Jones SW, et al. Pharmacokinetic interactions between tafenoquine and dihydroartemisinin-piperaquine or artemether-lumefantrine in healthy adult subjects. Antimicrob Agents Chemother. 2016;60(12):7321–32.
Green JA, Patel AK, Patel BR, Hussaini A, Harrell EJ, McDonal MJ, et al. Tafenoquine at therapeutic concentrations does not prolong QTcF interval in healthy subjects. J Clin Pharmacol. 2014;54(9):995–1005.
Acknowledgments
The authors wish to acknowledge Hanu Ramachandruni from Medicines for Malaria Venture for his technical input
Funding
This study (201780) was funded by GlaxoSmithKline (GSK) and Medicines for Malaria Venture. Medical writing support (in the form of writing assistance, including development of the initial draft based on author direction, assembling tables and figures, collating author comments, and grammatical editing and referencing) was provided by Ileana Stoica, PhD, of Fishawack Indicia Ltd., UK, and was funded by GSK.
Author information
Authors and Affiliations
Contributions
NG, KM, KR, TE, LC, GCKWK, SD, and SS contributed to the study concept and design, and data analysis and interpretation. MT contributed to the study concept, data acquisition, and design, and analysis and interpretation. All authors were involved in preparation and review of the manuscript and approved the final version to be submitted.
Corresponding author
Ethics declarations
Conflicts of Interest
NG, LC, GCKWK, MT, KM, KR, SS, and TE are employees of GSK and hold GSK stocks/shares. SD is an employee of Medicines for Malaria Venture.
Electronic Supplementary Material
ESM 1
(DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Goyal, N., Mohamed, K., Rolfe, K. et al. Application of the Stable Isotope Label Approach in Clinical Development—Supporting Dissolution Specifications for a Commercial Tablet Product with Tafenoquine, a Long Half-life Compound. AAPS J 20, 74 (2018). https://doi.org/10.1208/s12248-018-0234-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-018-0234-5