
ABSTRACT
Early prediction of human clearance is often challenging, in particular for the growing number of low-clearance compounds. Long-term in vitro models have been developed which enable sophisticated hepatic drug disposition studies and improved clearance predictions. Here, the cell line HepG2, iPSC-derived hepatocytes (iCell®), the hepatic stem cell line HepaRG™, and human hepatocyte co-cultures (HμREL™ and HepatoPac®) were compared to primary hepatocyte suspension cultures with respect to their key metabolic activities. Similar metabolic activities were found for the long-term models HepaRG™, HμREL™, and HepatoPac® and the short-term suspension cultures when averaged across all 11 enzyme markers, although differences were seen in the activities of CYP2D6 and non-CYP enzymes. For iCell® and HepG2, the metabolic activity was more than tenfold lower. The micropatterned HepatoPac® model was further evaluated with respect to clearance prediction. To assess the in vitro parameters, pharmacokinetic modeling was applied. The determination of intrinsic clearance by nonlinear mixed-effects modeling in a long-term model significantly increased the confidence in the parameter estimation and extended the sensitive range towards 3% of liver blood flow, i.e., >10-fold lower as compared to suspension cultures. For in vitro to in vivo extrapolation, the well-stirred model was used. The micropatterned model gave rise to clearance prediction in man within a twofold error for the majority of low-clearance compounds. Further research is needed to understand whether transporter activity and drug metabolism by non-CYP enzymes, such as UGTs, SULTs, AO, and FMO, is comparable to the in vivo situation in these long-term culture models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
In drug discovery programs, the predictions of human pharmacokinetics and recommendation of dose and dosing regimen are essential tasks to achieve safe and efficacious treatments. For physiologically based modeling and simulation efforts, human clearance prediction from in vitro data is an important component. Determinations of in vitro intrinsic clearances are routinely conducted in pharmaceutical research via in vitro liver models, such as microsomes or hepatocytes (1,2). However, the activity of drug metabolizing enzymes in these widely used in vitro models declines within hours (3), and therefore, depletion of compounds with low turnover (<10%) cannot be determined with confidence. As the number of low-clearance compounds in pharmaceutical programs increases due to the application of highly effective metabolic clearance reduction strategies in lead optimization to allow once-daily administration of moderate drug quantities (4), more sophisticated in vitro liver models are required (5). For example, models where the metabolic activity is prolonged for up to 2–4 weeks can extend the sensitive range for clearance determination. Long-term in vitro liver models, such as the hepatic stem cell line HepaRG™ (6), micropatterned HepatoPac® co-cultures of human hepatocytes with mouse embryonic 3T3 fibroblasts cells (7), and HμREL™ hepatocyte/fibroblast co-culture plates (8), as well as alternative approaches for low clearance determination, such as the relay method with suspension hepatocyte cultures (9), are being intensively explored. Chan et al. (10) studied the prediction of in vivo clearances of reference compounds in HepatoPac® co-cultures, which are engineered to have extended cell viability and metabolic activity. Here, they reported the superiority of the HepatoPac® system compared to suspension cultures for predicting low in vivo clearances. Recently, Bonn et al. (11) reported the successful application of plated hepatocytes, HepaRG™ and HμREL™ co-cultures, for low intrinsic clearance determination and found slightly better performance of the HμREL™ system. In this study, we report the metabolic activity across a diverse set of phase I and phase II enzyme markers in different in vitro liver models as this is an important prerequisite for successful clearance determination across structural classes. Additionally, for the first time, we applied pharmacokinetic (PK) modeling using a nonlinear mixed-effects approach for low in vitro clearance estimations with a long-term liver model for reference and in-house Roche compounds. The nonlinear mixed-effects approach is routinely applied for modeling clinical PK data to quantify population mean kinetics, inter-individual variability, and residual variability, including intra-individual variability and measurement error (12). The nonlinear mixed-effects approach is preferred as it estimates residual variability in contrast to the naive pooled data approach, where all data are fitted together, or the two-stage approach, where individual data are fitted separately and then the individual parameter estimates are combined (13). By applying the nonlinear mixed-effects approach to in vitro data, where the statistical unit is the well, the confidence in the clearance estimate is increased as the pharmacokinetic parameters and residual variability can be derived. Furthermore, we compare clearance predictions in man for reference compounds in the long-term in vitro liver model, HepatoPac®, and in suspension cultures using the same hepatocyte donor. Finally, we describe for the first time human clearance predictions for clinical development candidates with different physicochemical properties and clearance pathways than reference compounds, applying the well-stirred model and different protein binding assumptions based on the HepatoPac® system.
MATERIALS AND METHODS
Materials
Midazolam, 1′-hydroxymidazolam, and Roche compounds RO1–RO9 were synthesized at F.Hoffmann-La Roche Ltd. (Basel, Switzerland). Dextromethorphan (D-2531), diclofenac (D-6899), tolbutamide (T0891), bupropion (B-102), daunorubicin (30450), benzydamine (B5524), O 6-benzyl guanine (B2292), sulfamethazine (S6256), quinidine (Q-3625), warfarin (A2250), dextrophan (D127), 4-hydroxydiclofenac (H3661), 8-oxo-O 6-benzylguanine (B2292), 7-hydroxycoumarin glucuronide (UC263), 7-hydroxycoumarin sulfate (UC283), chlorpromazine hydrochloride (C8138), Trypan blue (T8154), William’s E medium (W-1878), hydrocortisone (H-0888), insulin (I-1882), penicillin/streptomycin (P-0781), bovine serum albumin standard 2 mg/mL (BCA ampules from Pierce, no. 2320), BCA Protein Assay Reagent A (Pierce, no. 23223), and Reagent B (Pierce, no. 23224), human serum albumin (A3782), α1-acid glycoprotein (G9885), and flat-bottom Nunc 96-well plates were purchased from Sigma-Aldrich (St. Louis, MO, USA). SN-38 (S589950), SN-38 glucuronide (S589980), hydroxybupropion (H830675), benzydamine N-oxide (B209960), hydroxytacrine (A629900), daunorubicinol (D194510), and N-acetyl-sulfamethazine (A187850) were purchased from Toronto Research Chemicals (Toronto, Canada). Tacrine (no. 70240, Cayman, Ann Arbor, Michigan, USA), 7-hydroxycoumarin (Acros no. 12111, Thermo Fisher Scientific, New Jersey, USA), nifedipine (Alfa Aesar no. J62811, Thermo Fisher Scientific), and gentamycin (no. 4-07F00-H, Amimed, London, UK) were purchased from different sources. Penicillin/streptomycin (no. 15140-122), GlutaMax-I (no. 31966), l-glutamine (no. 25030-024), fetal calf serum (no. 16000), and collagen I-coated 96-well plates (no. A11428-03) were purchased from Gibco/Life Technologies (Grand Island, NY, USA). Matrigel matrix phenol red-free (no. 356237) was obtained from BD Bioscience (Franklin Lakes, NJ, USA). For urea quantification, the urea nitrogen test (0580-250) from StanBio Laboratory (Boerne, TX, USA) was used. For human albumin quantification, the two-site enzyme-linked immunoassay (ELISA) from GenWay Biotech, Inc (San Diego, CA, USA) was employed. For human, commercially available primary pooled cryopreserved hepatocytes from non-transplantable liver tissues were purchased (no. X008001, Lots ECO (ten donors) and URK (five donors); no. X008052, Lot JJR (ten donors)) from BioreclamationIVT (NY, USA). HepaRG™ cells (no. HPR101) and growth medium supplement (no. ADD710) were received from Biopredics International (Rennes, France). HepG2 cells (lot no. 121205) were purchased from Mitosciences (Oregon, USA). Cryopreserved induced pluripotent stem cell (iPSC)-derived hepatocyte-like cells (iCell® hepatocytes) were purchased from Cellular Dynamics International (Wisconsin, USA). Human HepatoPac® cultures were acquired from Ascendance Corporation (Medford, MA) and prepared from a lot (unpooled) of cryoplateable hepatocytes (Lot 3121A, BioreclamationIVT, Lot TLQ). Human HμREL™ cultures were acquired from HμREL™ Corporation (North Brunswick, NJ) and prepared from pooled (five donors) cryoplateable hepatocytes (Lot HU1007, BioreclamationIVT, Lot YMD). The pooled and frozen plasma from human (Lot PLA022C0AK113) was obtained from BioreclamationIVT. Zeiss Axio Vision SE64 Rel. 4.9.1 microscope (Carl Zeiss AG Corporate, Oberkochen, Germany) was used for cell images. Teflon equilibrium dialysis plate (96-well, 150 μL, half-cell capacity) and cellulose membranes (12- to 14-kDa molecular weight cutoff) were purchased from HT-Dialysis (Gales Ferry, CT, USA). Human fresh blood was obtained internally (F.Hoffmann-La Roche Ltd.). DMSO stock solutions for the compounds were used with final DMSO concentrations of 0.1% in the incubation samples.
Suspension Cultures
Primary pooled cryopreserved hepatocytes from different lots were reconstituted in warm William’s E media containing 10% fetal calf serum, 0.048 mg/mL hydrocortisone, 0.004 mg/mL insulin, 0.05 mg/mL streptomycin, 50 U/mL penicillin, 0.01 mg/mL gentamycin, and 0.4 mM l-glutamine. Viability of hepatocytes after reconstitution was at least 80%. Suspension cultures of 1 million (Mio) cells per milliliter were then prepared using the thawing medium. After the addition of test compounds at different concentrations to the wells (100 μL), the 96-well hepatocyte suspension cultures plates were shaken (900 rpm) in a 5% CO2 atmosphere at 37°C and sampling was done at different time points up to 2 h.
HepaRG™
HepaRG™ cells were cultured at 37°C in a humidified atmosphere with 5% CO2 in complete HepaRG™ growth medium consisting of William’s E medium, growth medium supplement, and 1% (v/v) GlutaMAX-I. To initiate differentiation, 0.9% (v/v) DMSO was added to the growth medium on confluent cells. After 1 week, the medium was replaced by complete differentiation medium (HepaRG™ growth medium supplemented with 1.8% (v/v) DMSO), in which cells were maintained for approximately 4 weeks with differentiation medium renewal every 7 days. Differentiated HepaRG™ cells displayed hepatocyte-like cell islands surrounded by a monolayer of biliary-like cells. For metabolic experiments, differentiated HepaRG™ cells were seeded into collagen I-coated 96-well plates at 60,000 cells per well.
HepatoPac®
HepatoPac® cultures were received from Ascendance and recovered for 2 days after shipment by changing the medium and keeping them at 10% CO2 atmosphere and 37°C. The hepatic functionality of the HepatoPac® cultures was assessed in detail by Khetani et al. (14). For the used donor lot 3121A, the albumin and urea production was provided in the Ascendance specification sheet, e.g., 32 μg/day per Mio cells for albumin and 290 μg/day per Mio cells for urea at day 8 of culture. Similar results for albumin and urea production were found after receiving the HepatoPac® cultures after shipment using an ELISA test for albumin quantification and a standard urea nitrogen kit for urea quantification. The metabolic functionality was evaluated by studying the clearance of midazolam at different days of the long-term cultures, confirming data from the provider that the metabolic activity is conserved up to day 15 in culture. Prior to the addition of test substances, the cultures were washed with 64 μL serum-free Dulbecco’s modified Eagle’s medium specially formulated for HepatoPac® cultures (Ascendance Corporation). Incubations containing only mouse fibroblast cells served as the controls.
HμREL™
Co-culture plates were shipped in maintenance medium at 37°C from HμREL™. After arrival, the cultures were recovered in HμREL™ PlatinumHeps maintenance medium overnight at 5% CO2 atmosphere and 37°C. Prior to the addition of test substances, the cultures were washed with 100 μL HμREL™ PlatinumHeps media specially formulated for HμREL™ cultures.
iPSC-Derived Hepatocyte-Like Cells
Cryopreserved iPSC-derived hepatocyte-like cells (iCell®) were thawed according to the manufacturer’s instruction, resuspended in medium D (provided by the manufacturer) containing 0.25 mg/mL Matrigel, and seeded on collagen-coated 96-well plates at 140,000 cells per well. Cells were cultured at 37°C in a humidified atmosphere with 5% CO2. The culture medium was replaced 24 h post-plating and every 2 days before conducting metabolic activity studies.
HepG2
HepG2 cells were cultured at 37°C in a humidified atmosphere with 5% CO2 in complete HepG2 medium consisting of Dulbecco’s modified Eagle’s medium, GlutaMAX-I containing penicillin/streptomycin, and 10% heat-inactivated fetal calf serum. For experiments, HepG2 cells were trypsinized and seeded into collagen I-coated 96-well plates at 25,000 cells per well.
Metabolic Activity of In Vitro Liver Models
For metabolic activity determination, the following enzyme substrates were added to the in vitro liver models in a 96-well format and incubated at 5% CO2 atmosphere and at 37°C. Midazolam, dextromethorphan, diclofenac, bupropion, benzydamine, daunorubicin, 7-hydroxycoumarin, O 6-benzyl guanine, and tacrine were incubated at 1 μM and sulfamethazine and SN-38 at 20 and 50 μM, respectively. At defined time points, samples were drawn and quenched with acetonitrile (volume ratio, 1:2) containing 0.1 mM chlorpromazine as internal standard. Samples were then cooled, centrifuged, and quantified by using liquid chromatography and mass spectrometry (LC-MS/MS). The parameter settings for LC/MS analyte detection in positive or negative ion multiple reaction monitoring (MRM) mode are summarized in Supplemental 1. The Shimadzu HPLC (high-pressure liquid chromatography) system consisted of 10ADVP pumps connected to a 5000 AB Sciex mass spectrometer equipped with a TurboIonSpray source (IonSpray Voltage 5500 V in positive mode and −4500 V in negative mode) and a HTS CTC PAL autosampler. For 1′-hydroxymidazolam, benzydamine N-oxide, daunorubicinol, N-acetyl-sulfamethazine, SN-38 glucuronide, 8-oxo-O 6-benzylguanine, hydroxybupropion, 4-hydroxydiclofenac, dextrorphan, and hydroxytacrine, a 50 × 2-mm Phenomenex Gemini C18 110A resin analytical column with 5 μm particle size was used. 7-Hydroxycoumarin glucuronide and sulfate were chromatographed using a 50-cm × 2-mm Phenomenex Synergi Hydro-RP 80 Å resin analytical column with 4 μm particle size at 40°C. Mobile phase A was 0.2% formic acid in water and mobile phase B was 0.1% formic acid in 95:5 water/methanol. Aliquots (1 μl) of the centrifuged sample solutions were injected and transferred onto the analytical column at a flow rate of 0.50 mL/min using 95% mobile phase A. To elute the compounds, a high-pressure linear gradient from 5 to 95% B in 98 s was applied. The precision and accuracy of the standard and quality control (QC) samples were between 80 and 120%. Data analysis was performed using weighted (1/x 2) linear regression on analyte/internal standard area ratios. For data analysis, Analyst software (version 1.6.2, SCIEX, Framingham, USA) was used. To derive metabolic rates, the determined concentrations of the metabolites were plotted against time and a linear fit made to the data with emphasis upon the initial linear rate. The linear rate was then used to derive at the metabolite formation rate (in picomoles per minute per Mio cells) after normalization by the protein content of the cells. For the long-term liver co-culture model HepatoPac®, metabolic rates were also determined in mouse embryonic 3T3 fibroblast control plates only.
In Vitro Clearance Assessment
For in vitro clearance assessment, incubations of the test compounds at different concentrations were performed in the 96-well format in vitro liver models (5% CO2 atmosphere and at 37°C). At defined time points, either an aliquot of the cell incubation medium was taken or the whole well (for suspension cultures) was quenched with acetonitrile (volume ratio, 1:2) containing 0.1 mM chlorpromazine as internal standard. The samples were then cooled and centrifuged before analysis by LC-MS/MS. LC-MS/MS was used for the quantification of the reference and Roche compounds. The HPLC system consisted of Shimadzu pumps. An API6500, QTRAP 5500, or a QTRAP4000 AB Sciex mass spectrometer equipped with a TurboIonSpray source (IonSpray Voltage 5500 V in positive mode and −4500 V in negative mode) and a HTS CTC PAL autosampler were used. The analytical column was a 20-cm × 2.1-mm Supelco Ascentis Express C18 with 2.7 μm particle size at different temperatures for the analytes. For RO1, mobile phase A was 10 mM ammonium formate adjusted to pH 3.7 with formic acid in 95:5 water/methanol and mobile phase B was 100% acetonitrile. For RO2, RO4, RO6, RO7, RO9, midazolam, dextromethorphan, tolbutamide, and warfarin, mobile phase A was 0.5% formic acid in 95:5 water/methanol and mobile phase B was methanol. For RO3, mobile phase A was 0.5% formic acid in water and mobile phase B was 0.5% formic acid in acetonitrile. For quinidine, diclofenac, and nifedipine, mobile phase A was 20 mM of ammonium bicarbonate in 95:5 water/methanol and mobile phase B was 100% methanol. Aliquots (1 or 5 μL) of the centrifuged sample solutions were injected and transferred at the analytical column at flow rates between 500 and 600 μL/min. To elute the compounds, high-pressure linear gradients were applied. Detections were achieved in positive or negative ion MRM mode with the parameter settings given in Supplemental 1. Data analysis was performed using quadratic regression with 1/x 2 weighting on peak area ratios. The precision and accuracy of the standard and QC samples were between 80 and 120%. Analyst software (version 1.6.2, SCIEX) was used for data processing. For determination of the intrinsic clearance, either individual curve fitting (medium to high clearance) or pharmacokinetic modeling (low clearance) using a nonlinear mixed-effects approach was applied.
Determination of Intrinsic Clearance Values by Individual Curve Fitting for Medium- to High-Clearance Compounds
A linear fit was applied to the ln(concentration)–time profiles for each dose level using XLfit software (version 5.3.1.3, ID Business Solutions Limited, Guildford, UK). The slope of the fit was then used to calculate the intrinsic clearance value (Eq. 1).
Determination of Intrinsic Clearance Values for Low-Clearance Compounds in Long-Term Liver Models by Pharmacokinetic Modeling Using a Nonlinear Mixed-Effects Approach
The objective of this modeling was to improve the accuracy and precision of the intrinsic clearance determination by performing a simultaneous compartmental model fit to the in vitro time–concentration profiles from multiple dose levels. Data were analyzed by using a nonlinear mixed-effects PK modeling approach in the Monolix® software (version 4.22, Lixoft–Incuballiance, Orsay, France) (15). The statistical unit was the well. Two types of PK models were tested: one with linear (Eq. 2) and one with nonlinear elimination (Eq. 3).
Linear model:
Nonlinear model:
where C′ is the uncorrected concentration, k e is the elimination rate, K m the Michaelis–Menten constant, α a correction factor (volume), V m the maximal velocity, and C 0 the theoretical initial concentration.
In both cases, the simulated concentration is described by the following formula (Eq. 4).
where α is a correction between the theoretical initial concentration and the model projected initial concentration.
In addition, the derived \( {V}_{\mathrm{m}}\left(\frac{\upmu \operatorname{mol}}{ \min}\right) \) parameter was normalized by the protein content of the cells (Eq. 5).
The selection of an appropriate PK and error model was performed considering statistical criteria (improvement of the objective function and Akaike criterion), precision of parameter estimates, and diagnostic plots (observed versus predicted, prediction distribution, visual predictive check, and distribution of residuals).
For the long-term liver co-culture model HepatoPac®, elimination rates were also determined in fibroblast control plates only. The fibroblasts were treated in the same manner as the HepatoPac® plates. Because each well in the HepatoPac plates has 75% surface area as fibroblasts and 25% surface area as hepatocytes (14), the determined elimination rates from the fibroblast cultures were multiplied by 0.75 and then subtracted from the values determined in the HepatoPac plates to correct for compound adsorption phenomena and non-hepatic clearance by fibroblasts in the co-cultures.
The intrinsic clearance value was then calculated with the following equations (Eqs. 6 and 7):
Linear model:
Nonlinear model (Clint is a function of the concentration c x ):
Protein Content of the Different In Vitro Liver Models
For the protein content determination, cultures were washed twice with PBS and centrifuged in the case of suspension cultures. The supernatant was discarded and 1% Triton-X-100 (SURFACT-AMPS X-100) in water was added to the cells. After mixing, the cell lysates were kept for 1 h at 4°C and vortexed from time to time before they were stored overnight at −20°C. The samples were then thawed and centrifuged; the supernatant was used for the protein content determination. The protein content was measured by the Pierce BCA assay kit following the standard protocol. For 1 Mio cells of hepatocyte suspension cultures, the protein content was 1 mg/mL. Protein contents of 0.6 and 1.5 mg/mL were found for the HepaRG™ and HepG2 96-well plate cultures, respectively. The iPSC-derived hepatocyte-like cells (iCell®) had a protein content of 1.4 mg/mL. The micropatterned HepatoPac® co-culture plates contained 3200 (in a 96-well plate) hepatocytes per well, as specified in the HepatoPac® donor specification (3121A) sheet provided by the supplier. A protein content of 0.05 mg/mL (0.032 mg per well) was therefore applied for the 96-well HepatoPac® format. For the HμREL™ long-term hepatocyte co-cultures in 96-well plates, a protein content of 0.3 mg/mL was used as specified by the supplier.
Plasma Protein Binding Determination and Binding to Plasma Proteins
The determination of unbound compound was performed in human plasma and in plasma protein buffer solution (45 g/L human serum albumin and 0.7 g/L α1 acid glycoprotein in phosphate buffer, pH 7.4, respectively) using a 96-well format equilibrium dialysis device with a molecular weight cutoff membrane of 12–14 kDa, as described elsewhere (16,17). The derived samples from each dialysis were then quenched with acetonitrile (ratio, 1:2) containing an internal standard, centrifuged, and quantified by LC-MS/MS.
Blood-To-Plasma Ratios
Freshly drawn blood was centrifuged at low speed (600×g) for 3 min to generate a small erythrocyte-free plasma layer. DMSO stock solutions of compounds were then spiked into the erythrocyte-free plasma layer and immediately mixed and put on a KS130C Mixer™ (IKA® Werke GmbH & Co., Staufen, Germany) at 37°C. After 30 min, a whole blood sample was drawn and the rest of the blood sample was centrifuged (3000×g for 3 min). The derived blood and plasma samples were then quenched with an acetonitrile/water mixture containing an internal standard, centrifuged, and quantified by LC-MS/MS.
LogD and pK a Determination
The logD values were determined as published previously (18). The pK a determinations were done by mean of the Sirius Technology according to the Sirius technical applications notes (19).
In Vitro to In Vivo Extrapolation of In Vitro Clearance Assessments
Scaling of In Vitro Intrinsic Clearance to Whole Liver and In Vivo Total Blood Clearance
The in vitro intrinsic clearance value Clint (in microliters per minute per milligram protein) was scaled to the in vivo equivalent whole liver intrinsic clearance Liver Clint (in liters per hour) using an average liver weight and the hepatocellularity (Eq. 8), whereas 1 mg protein corresponds to 1 Mio cells. The in vitro whole liver intrinsic unbound clearance In vitro Clint (in milliliters per minute per kilogram) normalized by body weight is given in Eq. 9. The predicted in vivo total blood clearance Clblood (in liters per hour) was then derived by applying the well-stirred model, as given in Eq. 10. The physiological data used to scale in vitro intrinsic clearances were 23 mL min−1 kg−1 for liver blood flow, 70 kg for body weight, and 1.8 kg for liver weight for human. In addition, a scaling factor of 120 million hepatocytes per gram of liver was applied.
Well-stirred model:
Different Protein Binding Assumptions
Conventional (20,21) and direct scaling (22,23) approaches and the scaling approach by Poulin et al. (24,25) were explored. For the conventional approach, the fuinc values of the studied compounds were experimentally determined. Direct scaling assumes that the fraction unbound in plasma is equal to the fraction unbound in the in vitro system. The scaling approach by Poulin et al. (24,25) introduces the fraction unbound in liver, which is then used instead of the fraction in plasma. The derivation of the fraction unbound in liver is described elsewhere (24).
Determination of Apparent In Vivo Intrinsic Clearance
Apparent in vivo unbound intrinsic clearance In vivo Clint (in liters per hour) was calculated from the observed in vivo plasma clearance Clplasma (in liters per hour), fraction unbound in plasma, and average hepatic blood flow using the well-stirred model (Eq. 11). The apparent in vivo intrinsic clearance In vivo Clint (in milliliters per minute per kilogram) normalized by body weight is given in Eq. 12. Blood clearance Clblood (in liters per hour) and unbound fraction in blood (fub) were determined using the blood/plasma concentration ratio.
Accuracy and Precision of Predictions
The accuracy of Clint predictions for a set of compounds was assessed using the maximal fold error, average fold error (afe), and absolute average fold error metric using Eqs. 13, 14, and 15. The precision of the prediction for a data set was assessed using the root mean squared error (rmse) applying Eq. 16. The size of the data set is represented in Eqs. 14 and 15 as n. In addition, the concordance correlation coefficient global (CCC) was calculated as described by Lin (26).
RESULTS
Metabolic Activity of In Vitro Liver Models
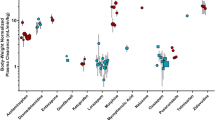
Table I summarizes the metabolite formation rates of a diverse set of enzyme markers in HepG2, iPSC-derived hepatocyte-like cells (iCells®), HepaRG™, HepatoPac® (single donor), HμREL™ (pooled, five donors), and primary pooled cryopreserved hepatocytes in suspension cultures (several lots, five to ten donors). The used primary hepatocyte lots were similar in their cytochrome P450 activities, as specified by the suppliers. The marker reactions were midazolam 1′-hydroxylase (cytochrome P450 3A4, CYP3A4), dextromethorphan O-demethylase (CYP2D6), diclofenac 4′-hydroxylase (CYP2C9), bupropion hydroxylase (CYP2B6), tacrine hydroxylase (CYP1A2), benzydamine N-oxidation (flavin monooxygenase, FMO), daunorubicin reductase (aldo-keto reductase, AKR), O 6-benzylguanine-8-oxidase (aldehyde oxidase, AO), sulfamethazine N-acetylase (N-acetyltransferase 2, NAT2), SN-38 glucuronation (UDP glucuronosyltransferase 1A1, UGT1A1), 7-hydroxycoumarin glucuronidation (UDP glucuronosyltransferase, UGT), and sulfation (sulfotransferases, SULT). The different cell morphologies of the studied in vitro liver models are given in Fig. 1. HepG2 and HepaRG™ cells showed the expected epithelial-like morphology and the typical irregular cell shape, respectively. The typical cubic cell shape of primary hepatocytes often containing two nuclei could be seen in the iPSC-derived hepatocyte-like cells (iCell®) and the co-culture systems (HμREL™ and HepatoPac®). The HepG2 model was included as a negative control as it has very limited metabolic activity, and iPSC-derived hepatocyte-like cells (iCells®) were included due to their future potential as an interesting tool lacking donor-to-donor variability. To compare the in vitro models in terms of metabolic activity, a metabolic activity index was calculated either across the diverse set of phase I and phase II enzyme markers (mean index shown as a line in Fig. 2) or across the five CYP enzymes only (as given in Table II). Figure 2 and Table II show that there is a similar average metabolic activity across all 11 enzyme markers for HepaRG™ (at day 7), HμREL™ (at day 8), and HepatoPac® (at day 8) as compared to the primary pooled cryopreserved hepatocytes (at day 0) in suspension using different lots. For iPSC-derived hepatocyte-like cells (iCell®, at day 4) and HepG2 (at day 4), the metabolic activity was more than tenfold lower on average. In terms of mean CYP activities, the HepatoPac® and primary pooled cryopreserved hepatocytes had a similar CYP index, showing that the studied CYP activities in HepatoPac® at day 8 are equivalent to those of the primary suspension cultures at day 0. HepaRG™ and HμREL™ had a three to fourfold lower CYP activity index compared to suspension cultures, which might be due to donor variability or due to culture conditions. Comparing the single activities for the different enzyme markers in HepaRG™, HμREL™, and HepatoPac® versus primary pooled cryopreserved hepatocytes in suspension, several differences were observed. Importantly, the CYP2D6 activity differed between the three in vitro liver models, e.g., 56-fold lower in HepaRG™, 14-fold lower in HμREL™, and 2.5-fold lower in HepatoPac® as compared to suspension cultures. Furthermore, the AO and FMO activities for HepaRG™, HμREL™, and HepatoPac® were reduced compared to the suspension cultures. The AO activities of HepaRG™ and HμREL™ were five to sixfold lower, whereas the activity for HepatoPac® was threefold lower. For the FMO activity, an eightfold lower activity was found in HμREL™ and a threefold lower activity, on average, in HepaRG™ and HepatoPac®. Furthermore, the AKR activity in HepaRG™ was increased (threefold) and similar in HμREL™ and HepatoPac®. The UGT1A1 activity as determined by SN-38 glucuronide formation was two to threefold lower in HepaRG™, HμREL™, and HepatoPac® as compared to the suspension cultures. In HμREL™ and HepatoPac®, the NAT2 and UGT activities were higher (two and threefold, respectively), whereas in HepaRG™, the NAT2 activity was lower (twofold) and the UGT activity was similar. For the SULT activity using 7-hydroxycoumarin as the enzyme marker, an eightfold higher formation of 7-hydroxycoumarin sulfate was observed in HepatoPac® as compared to the suspension cultures, whereas only a twofold increase or similar activity was seen in HμREL™ and HepaRG™, respectively. As HμREL™ and HepatoPac® are co-culture models containing hepatocytes and non-parenchymal cells, metabolite formation was also studied in mouse embryonic 3T3 fibroblasts, which are co-cultured in the HepatoPac® system. As expected, no CYP-related metabolism in the fibroblast cultures was found, but significant formations of daunorubicinol (AKR enzyme marker, 50% of HepatoPac®), 7-hydroxycoumarin glucuronide (UGT, 50% of HepatoPac®), and SN38 glucuronide (UGT1A1, 20% of HepatoPac®) were observed, which were subtracted from the metabolite formation rates reported in Fig. 2 and Table I for the micropatterned HepatoPac® system as the surface area occupied by fibroblasts and hepatocytes is specified (for details, see “MATERIALS AND METHODS”). In summary, HepaRG™, HμREL™, and HepatoPac® demonstrated metabolic activity similar to primary suspension cultures and could qualify for use in intrinsic clearance determinations of compounds.

Cell morphology of the different studied in vitro liver models. a Human liver cancer cell line HepG2 (at day 4) with its epithelial-like morphology containing one nucleus as compared to often two nuclei of primary hepatocytes. b iPSC-derived hepatocyte-like cells (iCell® at day 4). c Hepatic stem cell line HepaRG™ (at day 7) with its typical irregular cell shape. d Micropatterned co-cultures of human hepatocytes (unpooled) with mouse embryonic 3T3 fibroblast cells (HepatoPac®, at day 8). The micropatterned hepatocyte colony is surrounded by fibroblasts. e Co-cultures of pooled hepatocytes and cells of a non-parenchymal, stromal type (HμREL™, at day 8) showing primary hepatocytes with their typical cubic cell shape and two nuclei. f Primary pooled cryopreserved hepatocytes in suspension cultures (at day 0) showing the rounded cell morphology. Cells were imaged with a ×10 magnification phase contrast by means of a Zeiss Axio Vision SE64 Rel. 4.9.1 microscope (Carl Zeiss AG Corporate)

Metabolic activities of in vitro liver models. The metabolite formation rates (black symbols) of 11 diverse phase I and phase II enzyme markers were determined in 2D cultures of HepG2 (at day 4), iPSC-derived hepatocyte-like cells (iCell®, at day 4), HepaRG™ (at day 7), HepatoPac® (at day 8), HμREL™ (at day 8), and primary pooled cryopreserved hepatocytes in suspension cultures (at day 0). The metabolites were quantified by LC-MS/MS and the formation rates were derived for 1′-hydroxymidazolam (CYP3A4), dextrorphan (CYP2D6), 4-hydroxydiclofenac (CYP2C9), hydroxybupropion (CYP2B6), hydroxytacrine (CYP1A2), benzydamine N-oxide (FMO), daunorubicinol (AKR), 8-oxo-O 6-benzylguanine (AO), N-acetyl-sulfamethazine (NAT2), SN-38 glucuronide (UGT1A1), 7-hydroxycoumarin glucuronide (UGT), and sulfate (SULT). Gray symbols indicate the lower limit of quantification for the studied metabolites (for details, see Table I). The line represents the metabolic activity index calculated by averaging the determined metabolite formation rates of the 11 enzyme markers (see Table II). For the HepatoPac® system, cryopreserved primary hepatocytes from a single donor were used (Lot 3121A, BioreclamationIVT, Lot TLQ). Primary pooled cryopreserved hepatocytes were employed for the HμREL™ model (Lot HU1007, BioreclamationIVT, Lot YMD (five donors)) and for the suspension culture studies (BioreclamationIVT, Lots ECO (ten donors), URK (five donors), and JJR (ten donors)). The different lots of primary hepatocytes showed similar metabolic activities for the CYP and UGT/SULT enzyme markers in the BioreclamationIVT lot characterization sheets (listed in Supplemental 6)
In Vitro Clearance Assessment of Model Compounds: Comparison between Long-Term In Vitro Liver Model and Suspension Cultures from the Same Human Donor
After metabolic activity profiling of the different in vitro long-term liver models, we further evaluated one representative, HepatoPac®, for in vitro clearance assessments using reference compounds. For these reference compounds, midazolam (CYP3A4), quinidine (CYP3A4), nifedipine (CYP3A4), diclofenac (CYP2C9), warfarin (CYP2C9), tolbutamide (CYP2C9), and dextromethorphan (CYP2D6), the intrinsic clearances were determined in the HepatoPac® system and compared to the suspension cultures from the same donor (for details, see “MATERIALS AND METHODS”, Eq. 1). In Fig. 3, the time–concentration profiles for quinidine in the HepatoPac® system are exemplified. In HepatoPac®, quinidine concentrations decreased to 5% after 4 days in comparison to 71% after 2 h in the suspension cultures. The intrinsic clearance of quinidine in HepatoPac® across six studies (different shipments, same donor) was, on average, 12.4 μL min−1 mg−1 protein, with a variability of 29%. This study-to-study variability of ∼30% was deemed to be acceptable. The in vitro clearances were then scaled by applying the well-stirred model using the conventional approach as described in “MATERIALS AND METHODS” (Eqs. 8–12). Fraction unbound values in the incubation media and human plasma were experimentally determined and are listed in Table III together with the blood-to-plasma ratios. The observed plasma clearances for these model compounds were obtained from literature sources and are listed in Supplemental 2 and 3. Figure 4 shows the comparison of the predicted scaled in vitro unbound intrinsic clearance values of these model compounds in HepatoPac® and in the suspension cultures from the same human donor compared to the observed in vivo intrinsic clearance values. Generally, the predictions for medium- to high-clearance compounds, nifedipine (CYP3A4), midazolam (CYP3A4), dextromethorphan (CYP2D6), and diclofenac (CYP2C9), were similar in both in vitro systems, with a tendency to higher predicted intrinsic clearance values of 1.8-fold, on average, in the HepatoPac® incubations. For the low-clearance compounds (quinidine (CYP3A4), warfarin, and tolbutamide (CYP2C9)), hepatic clearances could be predicted with confidence from the HepatoPac®, whereas no clearance values could be determined in the suspension cultures for the very low-clearance compounds warfarin and tolbutamide. Supplemental 2 and 3 list the observed in vivo (intrinsic) clearances and the predicted scaled in vitro intrinsic clearance values for the HepatoPac® (underprediction average of 1.6) and primary pooled cryopreserved hepatocyte suspension cultures (underprediction average of 2.9) from the same human donor, respectively.

Quinidine concentrations as a function of time in the cell medium of long-term human hepatocyte co-cultures (HepatoPac®) from six independent studies. The line represents the prediction based on nonlinear mixed-effects modeling. The intrinsic clearance of quinidine was, on average, 12.4 μL min−1 mg−1 protein with a study-to-study variability of 30%. For comparison using individual curve fitting (two-stage approach), an intrinsic clearance of 11.6 μL min−1 mg−1 protein was determined with a study-to-study variability of 36%. Thus, the nonlinear mixed-effects and two-stage approaches give rise to similar results for compounds with linear clearance and decent turnover (in general, intrinsic clearance values >3 μL min−1 mg−1 protein)

Comparison of in vitro intrinsic unbound clearances derived from long-term human hepatocyte co-cultures (HepatoPac®, white bars) and cryopreserved primary human hepatocyte suspension cultures (black bars, same human donor) to the observed in vivo intrinsic clearance range (lines) for seven reference compounds in man. na not applicable. Conventional scaling for the clearances was applied as described in “MATERIALS AND METHODS.” The applied fraction unbound in cell medium and human plasma and the blood plasma partitioning values of the model compounds, which were used for the conventional scaling approach, are listed in Table III. The shown in vitro and observed in vivo intrinsic clearance values with their predicted versus observed fold errors for the reference compounds are given in Supplemental 2 and 3
Nonlinear Mixed-Effects Approach to In Vitro Low Clearance Assessments in Long-Term Liver Models
After the evaluation study with reference compounds, we assessed the in vitro clearances for low turnover Roche compounds in the long-term model. Figure 5 exemplifies the observed concentration–time profiles for four of these compounds (RO1, RO2, RO5, and RO8). For the determination of low in vitro intrinsic clearance values, a nonlinear mixed-effects approach (Eqs. 2–7) instead of traditional individual curve fitting (Eq. 1) was applied to further boost confidence in the clearance assessment. For this approach, the well of a 96-well plate was the statistical unit from which a single concentration-time point was derived. To describe the in vitro PK data, i.e., concentration–time profiles, a one-compartment PK model was used with volume and either linear (Eqs. 2 and 6) or nonlinear (Eqs. 3–5 and 7) clearance. The lines in Fig. 5 represent the predictions for the observed concentration–time profiles (symbols), which describe the observed data well. Table IV summarizes the derived elimination rates using the nonlinear mixed-effects approach for three compounds with linear clearance. As shown in Table IV, robust parameters could be derived for the elimination rates in the co-cultures. Compound adsorption phenomena and non-hepatic clearance by fibroblasts in the co-cultures were addressed by determining in addition the elimination rates in fibroblast cultures only (Table IV, controls). To derive the in vitro intrinsic clearance values in hepatocytes, the elimination rates in the co-cultures were corrected by the control rates and normalized by the protein content, as described in “MATERIALS AND METHODS.” For the fourth compound RO2, the clearance was nonlinear and K m and V m values of 2.19 μM (relative standard error (rse) = 0.1%) and 19.6 pmol min−1 mg−1 protein (rse = 2%) were estimated by applying a nonlinear PK model. Table V summarizes the derived intrinsic clearance values for all the compounds in the long-term cultures (Supplemental 4 and 5 list all the derived parameters in co-cultures and control plates). For the clearance parameters, the variability for the whole data set was, on average, 10%. This demonstrates a significant increase in confidence for in vitro intrinsic clearance determinations as compared to the suspension cultures, where the clearance variability was, on average, >80% for low-clearance compounds (Clint < 3 μL min−1 mg−1 protein).

Time–concentration profiles of Roche clinical drug candidates in the long-term in vitro human liver model, HepatoPac®. a RO1 at concentrations of 1 and 3 μM. b RO8 at concentrations of 0.3 and 1 μM. c, d RO5 (c) and RO2 (d) at concentrations of 0.3, 1, 3, and 10 μM. Lines represent the predictions based on pharmacokinetic modeling using a nonlinear mixed-effects approach
IVIVE of In Vitro Clearance Assessments
We then explored the in vitro to in vivo extrapolation (IVIVE) for clearance by applying the well-stirred model assessing different protein binding assumptions, e.g., conventional (20,21), direct (22,23), and the method reported by Poulin et al. (24,25) (see Eqs. 8–16). For that, the fractions unbound in the incubation media and human plasma were experimentally determined and are listed in Table III together with the blood-to-plasma ratios. For the IVIVE of the Roche clinical drug candidates tested in the HepatoPac® system, the direct scaling approach gave rise to reasonable clearance predictions in man with no under- or overprediction (average fold error of 1.0) and a root mean square error (rmse) of 90. In comparison, the conventional scaling approach showed a large underprediction of 6.4 (rmse = 52), whereas the Poulin methodology reduced the underprediction to 1.6, on average, with a very low rmse of 15. The statistical parameters for the human hepatic clearance predictions for the Roche compounds are listed in Table V. Considering all of the statistical parameters, the direct and the Poulin methods performed very similarly, whereas the conventional scaling approach was less favorable. For six out of eight compounds, the predicted versus observed fold errors were, on average, 1.1 and 0.7 for the direct scaling (63% within twofold and 75% within threefold) and the Poulin methodologies (50% within twofold and 75% within threefold), respectively. For RO6 and RO7, a >6-fold underprediction or overprediction was found for all protein binding assumptions, respectively. As the Poulin methodology is reported to perform best for compounds with high binding to plasma proteins, the IVIVE of a subset with fu < 0.15 was explored. For this small data set, however, no further gain in performance could be observed using this scaling approach. Figure 6 exemplifies for the direct scaling approach the correlation between the predicted scaled in vitro and observed in vivo intrinsic clearances of the Roche compounds. In addition, Table VI summarizes the in vitro intrinsic clearance values and the scaled predicted and observed clearances.

Comparison of the in vitro and in vivo intrinsic unbound clearances for Roche clinical drug candidates in man. The in vitro intrinsic clearance values were determined in long-term hepatocyte co-cultures from human (HepatoPac®). The well-stirred model and direct scaling were used as described in “MATERIALS AND METHODS.” The applied fraction unbound in human plasma and the blood plasma partitioning values are listed in Table III. In addition, the line of unity and the twofold (dashed) and fourfold (dotted) error margins are given. Most of the studied compounds are within a twofold error margin
DISCUSSION
In drug discovery programs, the determination of in vitro intrinsic clearances is routinely conducted using microsomes or hepatocytes, which are still the industry standard in vitro liver models.
Sensitive Range for In Vitro Clearance Assessments and In Vivo Relevance
For intrinsic clearance determinations in microsomes, the sensitive range is generally defined across the pharmaceutical industry as between 10 and 1000 μL min−1 mg−1 protein (27). In this range, the clearance values can be determined with confidence, e.g., with a coefficient of variation (CV) of ≤30% for multiple determinations. As the maximal incubation time of the hepatocyte suspension cultures is 2–4 h as compared to microsomes (0.5–1 h), the sensitive range for in vitro clearance determination in hepatocyte suspension cultures is lower, e.g., between 3 and 200 μL min−1 mg−1 protein (CV ≤ 30%) (4). For the majority of drugs, the volume of distribution is between the blood volume of 0.1 and 10 L/kg (28). Clearance values are between 0.2 and 60 L/h to enable potential twice- or once-daily drug dosing regimens. Thus, in vitro clearance values could be well below 1 μL min−1 mg−1 protein depending on the physicochemical properties of the compounds (BPP and fu values) (4,29). With the current development of engineered in vitro liver models offering increased hepatocyte life span, one can now extend the confidence in clearance predictions towards the low range, which is important for once-daily dosing regimens.
As a prerequisite for clearance prediction in man, the enzyme activities need to be conserved during in vitro long-term culturing of the hepatocytes. We therefore studied the metabolic activities with a diverse set of phase I and phase II enzyme markers across different models.
Metabolic Activity Assessment of In Vitro Liver Models and Comparison
The metabolic profiling study showed similar metabolic activities, on average, for HepaRG™ (at day 7), HμREL™ (at day 8), and HepatoPac® (at day 8) as compared to primary pooled cryopreserved hepatocytes in suspension (at day 0). However, pronounced differences in metabolic activities were observed for CYP2D6. The underprediction for CYP2D6 metabolized compounds in HepaRG™ has already been reported due to a CYP2D6-deficient donor (11,30,31). However, the fold of underprediction seems to vary between the applied enzyme markers due to potential metabolism by multiple CYP enzymes (11,30–32). For example, the intrinsic clearances in HepaRG™ as compared to suspension cultures were decreased more for dextromethorphan and metoprolol than for propranolol (11,30). Several factors, such as differences in the metabolism of enzyme markers, study design, and donor-to-donor variability, complicate a meta-analysis comparing the metabolic activities of new in vitro liver models to those of primary hepatocytes in suspension cultures. Furthermore, reduced activities of FMO and AO were found in all long-term in vitro liver models as compared to suspension cultures in our metabolic profiling study. Interestingly, both enzymes are using flavin adenine dinucleotide as prosthetic group. The effect of time on AO activity, i.e., substantial loss of activity within 24 h after isolation of hepatocytes from human liver tissue, was recently reported by Hutzler et al. (33). Furthermore, substantial donor variability of the AO activity was observed by these authors (33) in primary hepatocyte suspension cultures. For FMO, loss of enzyme function due to either liver tissue preparation (34,35) or with time in plated human primary hepatocytes was found previously (36). Thus, either biological variability or in vitro conditions could explain the reduced AO and FMO activities in the long-term in vitro systems after 7–8 days in culture. It will need further studies to understand whether loss of enzyme function occurs, especially during the long-term culture of 8–15 days, and to which extent it affects the IVIVE of compounds metabolized by these enzymes. In addition, the findings of significant AKR and UGT metabolism in the fibroblasts of the co-culture models, such as HepatoPac® and HμREL™, need to be addressed as they complicate metabolite identification and clearance prediction for compounds with low or non-CYP-related metabolism.
IVIVE of In Vitro Clearance Assessments
To explore the IVIVE for mainly CYP-metabolized model compounds, the intrinsic clearances were determined in one in vitro model, HepatoPac®. The derived intrinsic clearance values were then compared to those in suspension cultures from the same donor after applying the well-stirred model using the conventional scaling approach. The seven model compounds were midazolam (CYP3A4), quinidine (CYP3A4), nifedipine (CYP3A4), diclofenac (CYP2C9), warfarin (CYP2C9), tolbutamide (CYP2C9), and dextromethorphan (CYP2D6). The prediction of hepatic clearances derived from HepatoPac® showed an underprediction of 1.6, on average, compared to those of the suspension cultures with an underprediction of 2.9. The twofold lower underprediction average for predicted clearances in HepatoPac® was in line with the twofold higher metabolic activity in HepatoPac® as compared to the suspension cultures from the same donor. Our results agree well with the finding of Chan et al. (10), who also reported a twofold higher metabolic capacity in HepatoPac® compared to suspended hepatocytes from a different donor and using different model compounds. For low-clearance compounds, warfarin and tolbutamide, the long-term system was clearly superior for the prediction of hepatic clearances as no clearance values could be determined in the suspension cultures. The better performance of HepatoPac® for low-clearance compounds was also shown by Chan et al. (10), who reported the prediction of low-clearance reference compounds within twofold for six out of ten in HepatoPac® cultures. Recently, Bonn et al. (11) explored HepaRG™, plated primary hepatocytes, and HμREL™ for low clearance determination. The authors concluded that plated primary hepatocytes and HμREL™ delivered similar intrinsic clearance values and could predict the in vivo clearance within threefold for 70% of the reference compounds with an average fold error of 2, with slightly superior performance of the HμREL™ system. In their hands, HepaRG™ data were less predictive, giving rise to a prediction within threefold for only 50% of compounds (average fold error, 2.9). The inclusion of CYP2D6 metabolized compounds in their evaluation of HepaRG™ and the selection of different incubation times, 24 h for HepaRG™, 12 h for plated hepatocytes, and 72 h for HμREL™, could, however, have influenced the overall system performance, as Zanelli et al. (32) reported earlier that in vivo clearances of 26 different reference compounds could be predicted within twofold when applying the HepaRG™ system.
To evaluate the performance of HepatoPac® for clearance prediction of compounds occupying a different chemical space compared with mainly CYP-metabolized reference compounds, we assessed the intrinsic clearances and the IVIVE for nine Roche compounds. For the determination of the intrinsic clearances in the long-term model, we employed pharmacokinetic modeling using a nonlinear mixed-effects approach instead of the traditional individual curve fitting. With this approach, significant improvement in precision for low clearance estimations could be gained, giving rise to a variability of ∼10%, on average, for the intrinsic clearance parameter compared to >80% in the suspension cultures. In addition, the nonlinear mixed-effects approach gave information on the validity of the chosen pharmacokinetic model as well as the residual variability in contrast to the traditional curve fitting (two-stage approach). Tsamandouras et al. (37) just recently studied inter-individual variability in drug metabolism in vitro using a 3D liver system and nonlinear mixed-effects modeling. These authors characterized six different donors in their 3D liver system and determined donor variability for the in vitro clearances of reference compounds. This work of Tsamandouras et al. (37) highlighted the influence of donor variability and the need for the development of multiple donor pools to minimize the risk of prediction bias due to donor selection, especially for drugs which are metabolized by non-CYP enzymes. It is therefore an important development that long-term multiple donors systems are now becoming available.
For the IVIVE of clearance, we applied the well-stirred model exploring different protein binding assumptions, i.e., conventional (20,21), direct (22,23), and the method reported by Poulin et al. (24,25), where protein-facilitated uptake is taken into account by replacing the fraction unbound in plasma by the fraction unbound in liver (38–41). The conventional approach showed a large underprediction of 6.4 for the Roche compounds, whereas the direct and Poulin scaling approaches gave rise to reasonable clearance predictions in man with average fold errors of 1.0 and 1.6, respectively. For six out of eight Roche compounds, the predicted versus observed fold errors were, on average, 1.1 and 0.7 for the direct scaling (63% within twofold and 75% within threefold) and the Poulin methodology (50% within twofold and 75% within threefold), respectively. For RO6 and RO7, however, a >6-fold underprediction or overprediction, respectively, was found for all protein binding assumptions. The underprediction for RO6 could be due to reduced FMO metabolism and similar clearance rates in co-cultures and control fibroblasts giving rise to large uncertainty in the human clearance prediction. For RO7, the overprediction from in vitro in man is currently not yet understood. Interestingly, the conventional scaling showed better performance for the mainly CYP-metabolized reference compounds in the long-term co-culture system HepatoPac as compared to the direct scaling approach. The better performance of direct scaling for the in vivo clearance prediction of Roche compounds might be due to the involvement of non-CYP enzymes and transporters, which gives rise to larger underprediction compared to CYP-metabolized reference compounds (42,43). These results demonstrated that the performance of the scaling approach was dependent on the applied in vitro liver model, the chemical space of the selected compound set, i.e., their physicochemical properties, such as plasma protein binding, and the involved clearance routes. This dependency of the scaling approaches and their performance are widely discussed (27,44–46). Recently, Lin et al. (47) reported that HepatoPac® showed a better overall performance in predicting 73% of the drug clearances within twofold when applying conventional scaling for low- to very low-clearance compounds (≤1 mL min−1 kg−1) and direct scaling for medium- to high-clearance compounds (>1 mL min−1 kg−1). For drugs highly bound in plasma and drugs bound to albumin, Poulin et al. (38) could demonstrate that their scaling approach performed better in terms of human clearance predictions from suspension cultures, giving rise to an average fold error (afe) of 1.1, followed by the regression method, direct scaling, and conventional approach with afe values of 1.3, 2.2, and 0.52, respectively, for a data set of 38 reference compounds. The hypothesis of protein-facilitated uptake is further supported by the fact that the presence of serum in the in vitro model influences the performance of the scaling approach. Blanchard et al. (48) showed very early on that prediction of human clearance was improved in the presence of serum. This was confirmed by Parrott et al. (49), who reported human clearance prediction within twofold from intrinsic clearance values in suspension cultures containing 10% serum and physiologically based pharmacokinetic (PBPK) modeling. Furthermore, Chao et al. (50) showed better performance of plated hepatocytes in respect to clearance prediction in the presence of 4% albumin, which is equivalent to the albumin concentration in the human plasma. Thus, the addition of plasma proteins may also benefit clearance prediction from long-term in vitro liver models, as demonstrated for suspension and plated hepatocyte cultures.
Finally, new static and dynamic IVIVE methods are explored, such as the integration of non-hepatic clearance routes (51,52), the extended clearance concept (53), and mechanistic PBPK modeling (54,55), to address the underprediction of clearance for some compound classes. For example, the extended clearance concept takes into account metabolic and transporter clearance for compounds, which are actively taken up into the tissue. For these mechanistic approaches, key input parameters, such as metabolic and transporter clearance, need to be determined from in vitro models with confidence. For low-clearance compounds, which are actively taken up by the liver, the determination of the metabolic intrinsic clearance is however a challenge. On the one hand, as discussed, its determination is beyond the sensitive range of the short-term in vitro liver models, i.e., microsomes or suspension cultures. On the other hand, using long-term in vitro liver models gives rise to apparent intrinsic clearance values as these models show active uptake and biliary efflux of compounds under steady-state conditions. Additional work and detailed characterization of the expression and functional activity for phase II enzymes and transporters are required in order to understand whether these systems can be used to determine the rates of the key clearance processes. Furthermore, research is needed to see whether the derived in vitro rates reflect the in vivo situation and can be applied in mechanistic modeling efforts to further improve clearance predictions in man.
CONCLUSION
In essence, the human liver cancer cell line HepG2, induced pluripotent stem cell-derived hepatocyte-like cells (iCell®), the hepatic stem cell line HepaRG™, and hepatocyte co-cultures (HμREL™ and HepatoPac®) were compared to primary hepatocyte suspension cultures with respect to their key metabolic activities. The long-term in vitro liver models, HepaRG™, HμREL™, and HepatoPac®, showed similar mean metabolic activities across 11 metabolic enzyme markers as compared to primary hepatocyte suspension cultures using different hepatocyte donors and qualify for use in intrinsic clearance determinations of compounds. For iCell® and HepG2, the metabolic activity was more than tenfold lower. As one representative model, the micropatterned HepatoPac® system was further assessed in terms of clearance predictions. The HepatoPac® system performed similarly for medium- to high-clearance drugs (CYP-mediated) as compared to the suspension cultures. For the majority of high metabolically stable compounds, however, it was superior, giving rise to clearance prediction in man within a twofold error using the evaluated donor lot. Thus, clearance predictions for low-clearance clinical drug candidates, which differ in their physicochemical properties and clearance routes from literature compounds, could be reliably improved by applying in vitro long-term models in combination with pharmacokinetic modeling using a nonlinear mixed-effects approach. The increased cost for such models is balanced by the benefits of improved clearance predictions for low-clearance compounds and the enabling of sophisticated in vitro disposition studies under steady-state conditions. Further research is needed to understand whether transporter activity and drug metabolism by non-CYP enzymes, such as UGTs, SULTs, AO, and FMO, are comparable to the in vivo situation in these long-term culture models.
References
Obach RS. Predicting clearance in humans from in vitro data. Curr Top Med Chem. 2011;11:334–9.
Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47:1469–79.
Smith CM, Nolan CK, Edwards MA, Hatfield JB, Stewart TW, Ferguson SS, et al. A comprehensive evaluation of metabolic activity and intrinsic clearance in suspensions and monolayer cultures of cryopreserved primary human hepatocytes. J Pharm Sci. 2012;101:3989–4002.
Di L, Obach RS. Addressing the challenges of low clearance in drug research. AAPS J. 2015;17:352–7.
Hutzler JM, Ring BJ, Anderson SR. Low-turnover drug molecules: a current challenge for drug metabolism scientists. Drug Metab Dispos. 2015;43:1917–28.
HepaRG®. 2016. http://www.heparg.com/. Accessed 16 Aug 2016.
HepatoPac® (Ascendance Corporation). 2016. http://www.hepregen.com/products/human-hepatopac. Accessed 16 Aug 2016.
Hμrel® Corporation. 2016. http://hurelcorp.com/technology/hurel-hepatic-co-cultures/. Accessed 16 Aug 2016.
Di L, Atkinson K, Orozco CC, Funk C, Zhang H, McDonald TS, et al. In vitro-in vivo correlation for low-clearance compounds using hepatocyte relay method. Drug Metab Dispos. 2013;41:2018–23.
Chan TS, Yu H, Moore A, Khetani SR, Tweedie D. Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome P450 using a novel hepatocyte model, HepatoPac. Drug Metab Dispos. 2013;41:2024–32.
Bonn B, Svanberg P, Janefeldt A, Hultman I, Grime K. Determination of human hepatocyte intrinsic clearance for slowly metabolized compounds: comparison of a primary hepatocyte/stromal cell co-culture with plated primary hepatocytes and HepaRG. Drug Metab Dispos. 2016;44:527–33.
Lavielle M. Mixed effects models for the population approach: models, tasks, methods and tools. In: Chow S-C, Peace KE, Liu J-P, Jones B, Turnbull BW, editors. Chapman & Hall/CRC biostatistics series. London: CRC; 2014.
Sheiner LB. The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab Rev. 1984;15:153–71.
Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008;26:120–6.
Kuhn E, Lavielle M. Maximum likelihood estimation in nonlinear mixed effects models. Computational Statistics and Data Analysis. 2005;49:1020–38.
Zamek-Gliszczynski MJ, Ruterbories KJ, Ajamie RT, Wickremsinhe ER, Pothuri L, Rao MVS, et al. Validation of 96-well equilibrium dialysis with non-radiolabeled drug for definitive measurement of protein binding and application to clinical development of highly-bound drugs. J Pharm Sci. 2011;100:2498–507.
Banker MJ, Clark TH, Williams JA. Development and validation of 96-well equilibrium dialysis apparatus for measuring plasma protein binding. J Pharm Sci. 2003;92:967–74.
Wagner B, Fischer H, Kansy M, Seelig A, Assmus F. Carrier mediated distribution system (CAMDIS): a new approach for the measurement of octanol/water distribution coefficients. E J Pharm Sci. 2015;68:68–77.
Sirius technical applications notes. Application note 02/11. 2016. www.sirius-analytical.com.
Halifax D, Foster JA, Houston BJ. Prediction of human metabolic clearance from in vitro systems: retrospective analysis and prospective view. Pharm Res. 2010;27:2150–61.
Halifax D, Houston BJ. Evaluation of hepatic clearance prediction using in vitro data: emphasis of fraction unbound in plasma and drug ionisation using a dataset of 107 drugs. J Pharm Sci. 2012;101:2645–52.
Wan H, Bold P, Larsson LO, Ulander J, Peters S, Lofberg B, et al. Impact of input parameters on the prediction of hepatic plasma clearance using the well-stirred model. Curr Drug Metab. 2010;11:583–94.
Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–9.
Poulin P, Kenny JR, Hop CECA, Haddad S. In vitro–in vivo extrapolation of clearance: modeling hepatic metabolic clearance of highly bound drugs and comparative assessment with existing methods. J Pharm Sci. 2012;101:838–51.
Poulin P, Hop CECA, Ho Q, Halladay JS, Haddad S, Kenny JR. Comparative assessment of in vitro–in vivo extrapolation methods used for predicting hepatic metabolic clearance of drugs. J Pharm Sci. 2012;101:4308–26.
Lin LI-K. A concordance correlation coefficient to evaluate reproducibility. Biometrics. 1989;45:255–68.
Beaumont K, Gardner I, Chapman K, Hall M, Rowland M. Toward an integrated human clearance prediction strategy that minimizes animal use. J Pharm Sci. 2011;100:4518–35.
Obach RS, Lombardo F, Waters NJ. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab Dispos. 2008;36:1385–405.
Grime KH, Barton P, McGinnity DF. Application of in silico, in vitro and preclinical pharmacokinetic data for the effective and efficient prediction of human pharmacokinetics. Mol Pharmaceutics. 2013;10:1191–206.
Kanebratt KP, Andersson TB. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos. 2008;36:1444–52.
Lübberstedt M, Müller-Vieira U, Mayer M, Biemel KM, Knöspel F, Knobeloch D, et al. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J Pharmacol Toxicol Methods. 2011;63:59–68.
Zanelli U, Caradonna NP, Hallifax D, Turlizzi E, Houston JB. Comparison of cryopreserved HepaRG cells with cryopreserved human hepatocytes for prediction of clearance for 26 drugs. Drug Metab Dispos. 2012;40:104–10.
Hutzler JM, Yang Y-S, Brown C, Heyward S, Moeller T. Aldehyde oxidase activity in donor-matched fresh and cryopreserved human hepatocytes and assessment of variability in 75 donors. Drug Metab Dispos. 2014;42:1090–7.
Cashman JR, Zhang J. Interindividual differences of human flavin-containing monooxygenase 3: genetic polymorphisms and functional variation. Drug Metab Dispos. 2002;30:1043–52.
Fisher MB, Yoon K, Vaughn ML, Strelevitz TJ, Foti RS. Flavin-containing monooxygenase activity in hepatocytes and microsomes: in vitro characterization and in vivo scaling of benzydamine clearance. Drug Metab Dispos. 2002;30:1087–93.
Ubeaud G, Schiller CD, Hurbin F, Jaeck D, Coassolo P. Estimation of flavin-containing monooxygenase activity in intact hepatocyte monolayers of rat, hamster, rabbit, dog and human by using N-oxidation of benzydamine. Eur J Pharm Sci. 1999;8:255–60.
Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ, Cirit M. Quantitative assessment of population variability in hepatic drug metabolism using a perfused 3D human liver microphysiological system. JPET. 2016. doi:10.1124/jpet.116.237495.
Poulin P. Prediction of total hepatic clearance by combining metabolism, transport, and permeability data in the in vitro–in vivo extrapolation methods: emphasis on an apparent fraction unbound in liver for drugs. J Pharm Sci. 2013;102:2085–95.
Poulin P, Haddad S. Toward a new paradigm for the efficient in vitro-in vivo extrapolation of metabolic clearance in humans from hepatocyte data. J Pharm Sci. 2013;102:3239–51.
Poulin P, Haddad S. Albumin and uptake of drugs in cells: additional validation exercises of a recently published equation that quantifies the albumin-facilitated uptake mechanism(s) in physiologically based pharmacokinetic and pharmacodynamic modeling research. J Pharm Sci. 2015;104:4448–58.
Poulin P, Burczynski FJ, Haddad S. The role of extracellular binding proteins in the cellular uptake of drugs: impact on quantitative in vitro-to-in vivo extrapolations of toxicity and efficacy in physiologically based pharmacokinetic-pharmacodynamic research. J Pharm Sci. 2016;105:497–508.
Di L. The role of drug metabolizing enzymes in clearance. Expert Opin Drug Metab Toxicol. 2014;10:379–93.
Argikar UA, Potter PM, Hutzler JM, Marathe PH. Challenges and opportunities with non-CYP enzymes aldehyde oxidase, carboxylesterase, and UDP-glucuronosyltransferase: focus on reaction phenotyping and prediction of human clearance. AAPS J. 2016;18:1391–405. doi:10.1208/s12248-016-9962-6.
Wong H, Lewin-Koh S-C, Theil F-P, Hop CECA. Influence of the compound selection process on the performance of human clearance prediction methods. J Pharm Sci. 2012;101:509–15.
Hallifax D, Houston JB. Evaluation of hepatic clearance prediction using in vitro data: emphasis on fraction unbound in plasma and drug ionisation using a database of 107 drugs. J Pharm Sci. 2012;101:2645–52.
Page KM. Validation of early human dose prediction: a key metric for compound progression in drug discovery. Mol Pharmaceutics. 2016;13:609–20.
Lin C, Shi J, Moore A, Khetani SR. Prediction of drug clearance and drug-drug interactions in microscale cultures of human hepatocytes. Drug Metab Dispos. 2016;44:127–36.
Blanchard N, Hewitt NJ, Silber P, Jones H, Coassolo P, Lave T. Prediction of hepatic clearance using cryopreserved human hepatocytes: a comparison of serum and serum free incubations. J Pharm Pharmacol. 2006;58:633–41.
Parrott N, Jones H, Paquereau N, Lave T. Application of full physiological models for pharmaceutical drug candidate selection and extrapolation of pharmacokinetics to man. Basic Clin Pharmacol Toxicol. 2005;96:193–9.
Chao P, Barminko J, Novik E, Han Y, Maguire T, Cheng KC. Prediction of human hepatic clearance using an in vitro plated hepatocyte clearance model. Drug Metab Lett. 2009;3:296–307.
Cubitt HE, Houston JB, Galetin A. Prediction of human drug clearance by multiple metabolic pathways: integration of hepatic and intestinal microsomal and cytosolic data. Drug Metab Dispos. 2011;39:864–73.
Fan PW, Song Y, Berezhkovskiy LM, Cheong J, Plise EG, Khojasteh SC. Practical permeability-based hepatic clearance classification system (HepCCS) in drug discovery. Future Med Chem. 2014;6:1995–2012.
Riede J, Poller B, Umehara K, Huwyler J, Camenischa G. New IVIVE method for the prediction of total human clearance and relative elimination pathway contributions from in vitro hepatocyte and microsome data. Eur J Pharm Sci. 2016;86:96–102.
Galetin A. Rationalizing underprediction of drug clearance from enzyme and transporter kinetic data: from in vitro tools to mechanistic modeling. In: Nagar D, Argikar U, Tweedie D, editors. Methods in molecular biology, vol. 1113. Heidelberg: Springer; 2014. p. 255–88.
Cubitt HE, Yeo KR, Howgate EM, Rostami-Hodjegan AR-H, Barter ZE. Sources of interindividual variability in IVIVE of clearance: an investigation into the prediction of benzodiazepine clearance using a mechanistic population-based pharmacokinetic model. Xenobiotica. 2011;41:623–38.
Sohlenius-Sternbeck AK, Jones C, Ferguson D, Middleton BJ, Projean D, Floby E, et al. Practical use of the regression offset approach for the prediction of in vivo intrinsic clearance from hepatocytes. Xenobiotica. 2012;42:841–53.
Enzyme phenotyping data available on file
Acknowledgements
Sincere thanks to Nicolas Frey (Head of Clinical Pharmacometrics, Roche Innovation Center Basel, Switzerland) for critical discussions. The authors would also like to thank Matthew Shipton from HμREL™ Corporation (North Brunswick, NJ, USA) and Tanja Minz, Sabine Sewing, Dan Turley, and Bjoern Wagner (Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, Switzerland) for providing the cell images of the HμREL™ and iCell® in vitro systems and for conducting the logD and pK a determinations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Christophe Meille was a Roche employee during the conduct of this research. He is now employed at Novartis Pharma, Basel, Switzerland.
ELECTRONIC SUPPLEMENTARY MATERIAL
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 3366 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kratochwil, N.A., Meille, C., Fowler, S. et al. Metabolic Profiling of Human Long-Term Liver Models and Hepatic Clearance Predictions from In Vitro Data Using Nonlinear Mixed-Effects Modeling. AAPS J 19, 534–550 (2017). https://doi.org/10.1208/s12248-016-0019-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-016-0019-7