
Abstract
Advanced prostate cancer has significant long-term morbidity, and there is a growing interest in alternative and complimentary forms of therapy that will improve the outcomes of patients. Azadirachta indica (common name: neem) contains multiple active compounds that have potent anti-inflammatory and anticancer properties. The present study investigates the novel targets of the anticancer activity of ethanol extract of neem leaves (EENL) in vitro and evaluates the in vivo efficacy in the prostate cancer models. Analysis of the components in the EENL by mass spectrometry suggests the presence of 2′,3′-dehydrosalannol, 6-desacetyl nimbinene, and nimolinone. Treatment of C4-2B and PC-3M-luc2 prostate cancer cells with EENL inhibited the cell proliferation. Genome-wide expression profiling, using oligonucleotide microarrays, revealed genes differentially expressed with EENL treatment in prostate cancer cells. Functional analysis unveiled that most of the up-regulated genes were associated with cell death, and drug metabolism, and the down-regulated genes were associated with cell cycle, DNA replication, recombination, and repair functions. Quantitative PCR confirmed significant up-regulation of 40 genes and immunoblotting revealed increase in the protein expression levels of HMOX1, AKR1C2, AKR1C3, and AKR1B10. EENL treatment inhibited the growth of C4-2B and PC-3M-luc2 prostate cancer xenografts in nude mice. The suppression of tumor growth is associated with the formation of hyalinized fibrous tumor tissue and the induction of cell death by apoptosis. These results suggest that EENL-containing natural bioactive compounds could have potent anticancer property and the regulation of multiple cellular pathways could exert pleiotrophic effects in prevention and treatment of prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is one of the most prevalent malignant neoplasms and the third leading cause of cancer-related death of men of the Western countries (1). The mainstay of treatment of advanced prostate cancer is focused on suppression of intraprostatic testosterone and dihydrotestosterone (DHT) actions (2). However, after an initial response, therapy-resistant clones can appear and result in cancer progression and metastasis with high mortality (3). First-line chemotherapy for advanced prostate cancer has not demonstrated significant improvement in overall survival but could provide disease control and palliation (4). Novel treatment modalities are therefore needed to treat hormone-resistant tumors and to prevent the progression of hormone-sensitive prostate cancer to hormone-refractory stage. The search for compounds with few or no adverse effects that will prevent cancer progression and protect against the adverse biological effects of chemotherapeutic agents as compared with the agents currently in use is therefore of greatest relevance.
Herbal plants and plant-derived medicines have been used as the source of potential anticancer agents in traditional cultures all over the world and are becoming increasingly popular in modern society (5). The potential natural product-derived anticancer agents are known to possess various bioactive phytochemicals. Terpenoids constitute one of the largest families of natural products accounting for more than 40,000 individual compounds of both primary and secondary metabolisms (6). Many herbal plants contain terpenoids, and several terpenoids have been shown to be available for pharmaceutical applications, for example, artemisinin and taxol as malaria and cancer medicines, respectively (7). Neem is one such medicinal plant, the extract of which has been used for thousands of years for most acute and chronic diseases in India and Africa. The major biologically active constituents of neem leaves are limonoids, triterpenoids, nonterpenoids, phenolics, flavonoids, and meliacins (8,9), potentially targeting multiple signaling pathways of cancer cells (10–12). Extract of neem leaves have been reported to be non-toxic, non-mutagenic, and found to possess immunomodulatory, anti-inflammatory, and anticarcinogenic properties (13,14). Neem leaf glycoprotein exhibited antitumor activity by activation of cytotoxic T lymphocytes and natural killer cells in patients with head and neck squamous cell carcinoma (15). To date, there is only one report of neem on prostate cancer which showed the in vitro inhibition of PC-3 cell proliferation and Bcl-2 expression after neem treatment (12). No further studies of any neem compounds or extracts were reported on prostate cancer. The above reasoning promoted use to explore the antitumor effects of neem leaves on human prostate cancer cells which could lead to future clinical trials for prostate cancer patients.
Our study is designed to identify and evaluate the molecular targets of anticancer activities of ethanol extract of neem leaves (EENL) in prostate cancer models. We performed liquid chromatography/time-of-flight mass spectrometry (LC/TOF-MS) analyses to identify the components in the EENL. To unravel the molecular effects of EENL in androgen-refractory metastatic prostate cancer cells, we used gene expression microarrays and identified target genes regulated in prostate cancer cells after treatment with EENL. We then confirmed the alterations in mRNA and protein expression levels of the genes. The antitumor activity of EENL was further evaluated in the prostate cancer mouse models using C4-2B and PC-3M-luc2 cells.
Materials and Methods
Ethanol Extraction of Neem Leaves
Neem tree leaves harvested during the summer season were obtained from Neem Tree Farms (Brandon, FL). Neem leaves of the same age were washed with distilled water, air-dried, and 10 g of pulverized leaves were passed through a Soxhlet extractor for 4 h with 250 mL of 100% ethanol. All the alcohol was evaporated at low temperature using Rotavapor R-200 (Buchi, New Castle, DE) under vacuum. The residue was freeze-dried and yielded approximately 1.0 g of the dried powder. This extracted powder was stored at −20°C. An aliquot of 100 mg of this powder was dissolved in 250 μL of dimethyl sulfoxide (DMSO) plus 250 μL of 100% ethanol (stock 200 μg/μL). The suspension was filtered using a 0.22 μm filter and stored at −20°C. The stock solution was further diluted with ethanol for all the experiments. The final concentration of DMSO in the culture medium never exceeded 0.01%. The effect of the extract on cell viability and gene expression levels described below were assessed to standardize the method of extraction. We obtained consistent results with different lots of the extract.
LC/TOF-MS Analyses
High-performance liquid chromatography (HPLC)-grade acetonitrile, water, isopropanol, and methanol were purchased from Burdick and Jackson (Muskegon, Michigan). Formic acid was obtained from Fluka (Fluka/Sigma-Aldrich St. Louis, Missouri). 2′,3′-dehydrosalannol, a known component of neem leaves, was obtained from the Asthagiri Herbal Research Foundation (Channai, Tamil Nadu, India) for use as a standard. The analytes were separated using an HPLC system (Agilent series 1100, Agilent Technologies, Palo Alto, CA) equipped with a reversed-phase C18 analytical column (Zorbax Eclipse 300SB-C18 1.0 × 150 mm, 3.5 mm). The column temperature was maintained at 45°C. The makeup of the LC mobile phases was as follows: mobile phase A water:acetonitrile:isopropanol:formic acid (98:1:1:0.1), mobile phase B acetonitrile:water:isopropanol:formic acid (80:10:10:0.1). Separation was achieved by using a linear gradient from 5% B to 100% B over 45 min. The flow rate was 0.05 mL/min, and 5 μL injections were made of the 2 μg/μL standards and neem extract solutions dissolved in mobile phase A. The HPLC system was connected to a time-of-flight mass spectrometer (MSD-TOF, Agilent Technologies) equipped with an electrospray interface. The instrument was operated under the following operating parameters: capillary 4,000 V, nebulizer 15 psig, drying gas 7 L/min, gas temperature 325°C, fragmentor 225 V, skimmer 60 V, Oct dc1 37.5 V, Oct rf V 250 V. The instrument was calibrated using the calibrant mixture provided by the manufacturer over the 50–3200m/z range. The scan range for data acquisition was 300–1,500m/z range.
Cell Line and Cell Culture
C4-2B, originated from LNCaP cell line, is a castration-resistant prostate cancer cell line purchased from ViroMed Laboratories (Minnetonka, MN). PC-3M-luc2, originated from PC3, is a luciferase-expressing metastatic prostate cancer cell line which was stably transfected with firefly luciferase gene (luc2), was purchased from Caliper LifeScience (Hopkinton, MA). C4-2B cells were grown in RPMI 1640 medium and PC-3M-luc2 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) media as described previously (16).
Cell Viability Assay
C4-2B and PC-3M-luc2 cells were seeded into 96-well plates at a density of 3 × 103 and 1.5 × 103 per well respectively, as previously described (17). C4-2B cells were treated with 5.0 to 15.0 μg/mL and PC-3M-luc2 cells were treated with 5.0 to 50.0 μg/mL of the EENL or with the vehicle control (ethanol + DMSO) for 24, 48, and 72 h. Cell viability was then determined by the colorimetric MTS assay using CellTiter 96 AQueous One Solution Proliferation Assay System from Promega (Madison, WI, USA)
RNA Extraction, Microarray Hybridization, and Data Analysis
C4-2B cells were treated with 8.0 μg/mL of EENL or vehicle control for 24 and 48 h. Total RNA from each biological replicate was isolated using Trizol (Invitrogen, San Diego, CA) as per the manufacturer’s instructions. On-column DNase treatment was performed followed by RNA cleanup using RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacture’s protocol as described previously (17). cDNA for each sample was synthesized by using the high capacity cDNA archive kit (Applied Biosystems, Foster City, CA). Complementary RNA was prepared, labeled, and hybridized to Human Genome-U133-Plus2 oligonucleotide arrays (Affymetrix, Santa Clara, CA) representing >47,000 transcripts as described previously (17). The experiments were performed in duplicate and the CEL files were imported into Partek Genomics Suite software (Partek Inc., St. Louis, MO), and data were normalized using the Robust Multichip Averaging algorithm. One-way analysis of variance (ANOVA) with nominal alpha value set to 0.05 was used to determine probe sets significantly different between the EENL and vehicle-treated cells, followed by a Benjamini and Hochberg Multiple testing correction to reduce the false positive rate. These results were then separated by significant up-regulated or down-regulated genes, and used for further validation. Differentially expressed genes were evaluated for biological function using Ingenuity Pathway Analysis (Ingenuity, Mountain View, CA).
Quantitative Real-Time PCR for Microarray Data Validation
To confirm the differential expression of genes from microarray data, we selected 40 up-regulated genes for validation using custom TaqMan® Low Density Arrays in C4-2B and PC-3M-luc2 cells. The arrays were preloaded with gene-specific primers, FAM and MGB probes (Applied Biosystems, Foster City, CA). The cDNA isolated as described above was mixed with the Taqman universal master mix (1:1) and loaded on to the microfluidic cards. The reactions were performed in ABI 7900 HT system and the quantity of cDNA was normalized using the housekeeping gene GAPDH. mRNA levels were calculated as fold change compared to control as described previously (18).
Protein Extraction and Western Blotting
C4-2B and PC-3M-luc2 cells were plated in 10-cm plates and after reaching 60–70% confluency, were treated for 24 and 48 h with EENL. Both the cell lines were treated with two different concentrations, 45% (8.0 μg/mL for C4-2B and 20.0 μg/mL for PC-3M-luc2) and 55% (10 μg/mL for C4-2B and 30 μg/mL for PC-3M-luc2) inhibitory concentrations of EENL as determined by the viability assay. Proteins were extracted from cells in modified RIPA buffer and western blotting was performed using primary antibodies against heme oxygenase-1 (HMOX1), aldo-keto reductases AKR1C2, AKR1C3 and AKR1B10 from Abcam Inc., (Cambridge, MA) and horseradish peroxidase-conjugated secondary antibodies as described previously (19). Immunodetection was performed by LumiGLO chemiluminescence detection system (Cell Signaling, Danvers, MA), in line with the manufacturer’s instructions. GAPDH was used as loading control.
Xenograft Tumor Growth
All experiments involving mice were conducted with the approval of Executive Subcommittee of the Institutional Animal Care and Use Committee of Mayo Clinic in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care International’s expectations for animal care and use/ethics committees and the investigators strictly followed the National Institutes of Health guidelines for humane treatment of animals. Male athymic nu/nu mice, 4–5 weeks of age, were obtained from Charles River Laboratories (Wilmington, MA), and were housed at the animal care facility as descried previously (16). After acclimatization for 1 week, C4-2B and PC-3M-luc2 cells (1.5 × 106 and 3.0 × 106 single cell suspension respectively, in 0.1 mL/mouse) suspended in 50% Matrigel in RPMI and DMEM medium were injected subcutaneously on the left flank of the animals. The animals challenged with C4-2B and PC-3M-luc2 cells were randomly assigned to three groups of six each and two groups of six each, respectively. Animals having palpable tumors after 2 weeks of challenge with PC-3M-luc2 cells and 4 weeks of challenge with C4-2B cells, were injected intraperitoneally with vehicle or EENL, 6 days a week for 8 to 11 weeks. Animals in both the groups received the same amount of vehicle (phosphate-buffered saline:polyethylene glycol:DMSO + ethanol in 1:2:1 ratio) or leaf extract + vehicle in 100 μL. Group 1 animals with C4-2B and PC-3M-luc2 cells were vehicle controls. Group 2 and 3 animals with C4-2B cells were treated with 100 and 200 mg/kg body weight of EENL respectively. Group 2 animals with PC-3M-luc2 cells were treated with 200 mg/kg body weight of EENL. The tumor volume of mice was measured every week by external caliper measurements in two dimensions and calculated as follows: length/2× width2. PC-3M-luc2 tumor growth was also monitored weekly using IVIS imaging system (Caliper LifeSciences). Luciferin was delivered intraperitoneally at 150 mg/kg in 200 μL and mice were imaged 5 min post injection. Animals were weighed once every week to monitor the effect of EENL toxicity on body weight. At the end of the study, all the mice were killed by CO2 inhalation; xenograft tumor tissue and the heart, lungs, liver, kidneys, and spleen were excised, weighed, and placed in phosphate-buffered formalin for fixation and hematoxylin and eosin (H&E) staining. Slides were stained for DNA fragmentation using ApopTag peroxidase in situ oligo ligation apoptosis detection kit (Millipore, Billerica, MA) per manufacturer's protocol.
Determination of DHT Levels in the Tumor Tissues of Mice
DHT levels were measured in the EENL- and vehicle-treated tumor tissues from mice as described (20). In brief, prostate tumor tissues were thawed, weighed, and homogenized in 1.0 mL of phosphate-buffered saline. All samples were mixed with deuterated stable isotope (d[4]-DHT) as internal standard and then extracted with 5 mL of dichloromethane. Samples were vortexed at a low speed, centrifuged at 3,000 ×g for 10 min; the organic phase was transferred to a new glass tube and then evaporated to dryness under nitrogen. This was followed by conventional LC on a multiplexed liquid chromatography system and analyzed on a tandem mass spectrometer equipped with an electrospray interface. The inter-assay (n = 24) % coefficient of variation was 18% at 53 pg/mL, 12.0% at 487 pg/mL, and 9% at 1,248 pg/mL
Statistical Analysis
Statistical analysis was carried out by using Student's t test, one-way ANOVA, Fisher's exact test and Kruskal–Wallis non-parametric ANOVA based on ranks with a Dunn's multiple comparison tests were used to compare the different experimental groups. P value < 0.05 was considered significant. Fifty percent inhibition concentration (IC50) values were calculated by Probit regression. Partek Genomics suite 6.4 was used to analyze the genomic data.
Results
LC/TOF-MS Analysis of Neem Compounds in the EENL
LC/TOF-MS analysis was performed to identify the potential active components in the EENL. Our analysis resulted in mass spectral peaks that appear to match the calculated masses of know neem leaf components including the 2′,3′-dehydrosalannol standard. The theoretical monoisotopic M + H+ value for 2′,3′-dehydrosalannol is M + H+1 = 555.2958m/z (molecular formula: C32H42O8). The observed 2′,3′-dehydrosalannol standard showed a mass spectrum with a major peak of 555.3055m/z and retention time of 28.78 min. The mass accuracy between the theoretical and the observed monoisotopic peak for the 2′,3′-dehydrosalannol standard is 17 ppm. The total ion chromatogram of the EENL depicts seven significant, based on intensity, peaks. The three most intense peaks are labeled 1, 2, and 3; the associated mass spectra to each peak is dominated by the monoisotopic M+H+1 values of 453.2364m/z, 441.2342m/z and 555.3041m/z, respectively. Peak 3 has a retention time of 28.82 min, this is in agreement with the retention time and observed mass between the 2′,3′-dehydrosalannol standard and suggests this component of EENL is 2′,3′-dehydrosalannol. The calculated monoisotopic mass (M + H+1) for the compound nimolinone (C30H44O3) is 453.3368m/z, and 6-desacetyl nimbinene (C26H32O6) is 441.2277 m/z. The mass accuracy measurements for the dominant monoisotopic masses observed for peaks 1 and 2, assuming that they are suggestive of nimolinone and 6-deacetyl nimbinene, are 221 and 15 ppm, respectively. The EENL and 2′,3′-dehydrosalannol standard were analyzed in duplicate, representative chromatograms and spectra were shown (Fig. 1).

Mass spectrometric analysis of the standard 2′,3′-dehydrosalannol and ethanol extract of neem leaves (EENL). a The total ion chromatogram for the 2′,3′-dehydrosalannol shows a retention time of 28.85 min. a1 The mass spectrum of the 2′,3′-dehydrosalannol depict the monoisotopic M + H+1 ion at 555.3055m/z. b The total ion chromatogram of EENL. b1 The mass spectrum of the peak 1, depict a monoisotopic M + H+1 ion at 458.2364m/z, at a retention time of 27.32 min. This is possibly suggestive by mass alone as nimolinone. b2 The mass spectrum of the peak 2, depict a monoisotopic M + H+1 ion at 441.2342m/z, at a retention time of 28.07 min. This is possibly suggestive by mass alone as 6-desacetyl nimbinene. b3 The mass spectrum of the peak 3, depict a monoisotopic M + H+1 ion at 555.3041m/z, at a retention time of 28.75 min. This is suggestive by mass and retention time as 2′,3′-dehydrosalannol. The difference between the measured value in the EENL and the measured value from the 2′,3′-dehydrosalannol standard were <3 ppm with a difference in retention time of only 0.1 min
EENL Inhibits the Growth of Prostate Cancer Cells in Vitro
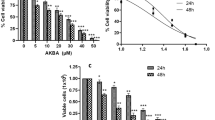
In an initial approach to analyze the potential of whole EENL for anticancer activity, we performed viability assays employing 2 frequently used human androgen-refractory C4-2B and PC-3M-luc2 prostate cancer cell lines. The antiproliferative activity of EENL was measured by MTS assay. Vehicle-treated cells were included as a control. EENL exhibited a dose-dependent inhibition of C4-2B and PC-3M-luc2 cell growth over a broad range of concentrations (Fig. 2), with an IC50 of 9.0 and 25.0 μg/mL respectively, where IC50 is the inhibition concentration at which a 50% inhibition of cell growth is observed at 24 h of treatment.

Inhibition of prostate cancer cell proliferation by treatment with ethanol extract of neem leaves (EENL). The antiproliferative effect of EENL on human prostate cancer cells C4-2B and PC-3M-luc2 was evaluated by using the MTS viability assay. The cells were treated for 24, 48, and 72 h with varying concentrations of EENL or vehicle as control. Experiments were performed in triplicate; data are expressed as the mean ± SD of the triplicate determinations of a representative experiment in % cell viability of untreated cells (100%). *p < 0.05
EENL Alters the Gene Expression Profiles
To identify the molecular targets of the anticancer effects of EENL, we performed high-resolution whole genome profiling using an Affymetrix microarray platform. The gene expression profiling of C4-2B prostate cancer cells treated with EENL for 24 h showed significant up-regulation of 191 genes and down-regulation of 97 genes (greater than twofold), whereas the 48-h EENL-treated cells showed significant up-regulation of 129 genes and down-regulation of 965 genes (greater than twofold). Using the Ingenuity Pathways Knowledge Base, the dataset was used to map independently up- and down-regulated genes for the molecular and cellular functions. The most enriched functions of the up-regulated genes are cell death, cellular development, cellular growth and proliferation, lipid metabolism, and small molecule biochemistry. The majority of the up-regulated genes are associated with cell death function which indicates that EENL could play a vital role in promoting cell death. The most significant down-regulated gene functions are cell cycle, DNA replication, recombination, and repair, cellular assembly and organization, cellular movement, and gene expression. Greater than 70% of the down-regulated genes are associated with cell cycle and DNA replication, recombination, and repair functions which indicate that EENL could play a significant role in inhibition of tumor growth.
To validate the observed changes in gene expression, we chose 40 up-regulated genes for Taqman real-time PCR analysis and analyzed in both C4-2B and PC-3M-luc2 prostate cancer cells. For all genes tested, the direction of gene expression change measured by RT-PCR and microarray analysis agreed, although the magnitude of expression change was not always the same using these 2 different analytical methods. The RNA expression levels of 40 validated genes in C4-2B cells treated with 8.0 μg/mL and PC-3M-luc2 cells treated with 20.0 μg/mL of EENL for 24 and 48 h relatively increased (Table I). These results support the findings obtained from the microarray experiments. Though we only validated up-regulated genes, we believe these results can support the validity of down-regulated genes of our microarray data. The 40 most significantly down-regulated genes were shown from our microarray data of the C4-2B prostate cancer cells after EENL treatment (Supplementary Table S1).
EENL Increases the Protein Expression Levels of HMOX1, AKR1C2, AKR1C3, and AKR1B10
We selected four significantly up-regulated genes to confirm the protein expression by Western blot analysis. Our results revealed significant increase of the HMOX1, AKR1C2, AKR1C3 and AKR1B10 protein levels in C4-2B and PC-3M-luc2 cells after 24 and 48 h of treatment with EENL (Fig. 3). These results were consistent with the increase in the mRNA expression levels of the genes after EENL treatment.

Over-expression of HMOX1, AKR1C2, AKR1C3 and AKR1B10 in C4-2B and PC-3M-luc2 prostate cancer cells after treatment with ethanol extract of neem leaves (EENL) for 24 and 48 h. Protein levels were measured with specific antibodies by Western blot analysis; GAPDH was the loading control. Vehicle-treated cells were used as control. The experiments were repeated thrice and the representative blot was shown
EENL Inhibits the Growth of Prostate Cancer Xenografts in Nude Mice
To evaluate the antitumor efficacy of EENL in vivo, we used xenograft tumor models. Immunodeficient nu/nu mice were subcutaneously injected with C4-2B and PC-3M-luc2 prostate cancer cells and were randomly divided into different groups. The rates of xenograft tumor take for C4-2B and PC-3M-luc2 cells were 100%. After 2 weeks of challenge with PC-3M-luc2 cells and 4 weeks of challenge with C4-2B cells, EENL was administered intraperitoneally. Tumor growth was significantly inhibited in all the EENL-treated groups. Significant inhibition of tumor size was observed in PC-3M-luc2 (greater than fivefold) and C4-2B (greater than tenfold) tumor mice treated with 200 mg/kg body weight of the EENL compared to the vehicle-treated mice (Fig. 4a–c). These findings elucidate that EENL can suppress tumor growth. There was no significant change in body weight in any of the groups after treatment which suggests that current EENL extract causes no major toxicity to mice (Fig. 4d).


Ethanol extract of neem leaves (EENL) inhibits the growth of human C4-2B and PC-3M-luc2 prostate cancer xenografts in nude mice. Male nu/nu mice were challenged with subcutaneous injection of C4-2B and PC-3M-luc2 cells. The animals challenged with C4-2B and PC-3M-luc2 cells were randomly assigned to three groups of six each and two groups of six each, respectively. After 2 weeks of challenge with PC-3M-luc2 cells and 4 weeks of challenge with C4-2B cells the animals were injected intraperitoneally with vehicle control or 100 or 200 mg/kg body weight of EENL, 6 days a week. Results depict mean tumor volume ± SEM from 6 mice of each group with a C4-2B xenografts and b PC-3M-luc2 xenografts. c Representative IVIS image of control and treated mice with PC-3M-luc2 tumors after 8 weeks. Luciferin was delivered intraperitoneally and mice were imaged 5 min post injection. d Body weight changes of tumor bearing mice with (A) C4-2B xenografts and (B) PC-3M-luc2 xenografts (n = 6 per group). *p < 0.05
EENL Promotes Hyalinization and Apoptosis of the Tumor Tissue
PC-3M-luc2 and C4-2B xenograft tumor mice were killed at the end of 8 and 11 weeks of EENL treatment, respectively. The tumor tissue and the other major organs were collected and fixed in phosphate-buffered formalin, sectioned and stained with H&E to identify the histological changes. Tumors were assessed histologically for fibrosis, coagulative tumor necrosis, and apoptosis. Mice treated with EENL showed greater degree of fibrosis and increased apoptotic activity, whereas vehicle-treated group exhibited greater amounts of coagulative tumor necrosis (Fig. 5). There was no significant change in the histology of the heart, lungs, liver, kidneys and spleen after 8 or 11 weeks of EENL treatment compared to vehicle-treated group which indicates that EENL has no adverse effects on these vital organs. Further, we demonstrated the presence of apoptotic cells in the tumor tissue by ApopTag peroxidase staining. Xenograft tumors in the control group of PC-3M-luc2 and C4-2B mice had 0.58% and 0.62% of ApopTag cellular staining, respectively, which increased to 3.8% (greater than sixfold) and 4.77% (greater than sevenfold) in mice treated with the EENL (Fig. 6). These results indicate that the possible mechanism for regression is by inducing apoptosis of the tumor cells.

Histological changes of C4-2B tumor tissues of mice treated with ethanol extract of neem leaves (EENL; 100 μg/kg body weight). At the end of 11 weeks, xenograft tumor tissue was collected from the mice and stained with hematoxylin and eosin. Two sections of tumor tissue from each mouse and six mice in a group were examined for histological changes. a Control tumor tissue from vehicle-treated mice shows dense tumor cells and the arrow points to the area of coagulative tumor necrosis at ×100. b–f Depicts tumor tissues from mice treated with EENL. b Tumor tissue shows nests of tumor cells separated by hyalinized connective tissue indicated by arrow at 100×, indication of treatment effect. c Tumor hyalinization showing apoptosis, the thin and thick arrows indicate apoptotic bodies and pyknotic nucleus undergoing cell death at ×400. d Tumor hyalinization showing apoptotic nucleus at ×600. e Tumor tissue with hyalinized fibrosis, arrow indicates residual tumor at ×100. f Hyalinized fibrous tissue showing residual tumor cells at ×200 magnification

Detection of apoptotic cells in the tumor tissue. ApopTag peroxidase in situ oligo ligation apoptosis detection kit was used for staining apoptotic cells in the tumor tissue at the end of treatment. a Control C4-2B tumor tissue of vehicle-treated mice shows very few apoptotic cells at ×200. b C4-2B tumor tissue from mice treated with 100 μg/kg body weight of ethanol extract of neem leaves (EENL) shows numerous brown apoptotic nuclei at ×200. c Arrow indicates the fragmented apoptotic nuclei at ×600 magnifications in the C4-2B tumor tissue from mice treated with 100 μg/kg body weight of EENL. d Columns and error bars represent average percentage of apoptotic cells in the maximally staining ×200 field per lesion and standard errors of the mean for control and treated mice with C4-2B and PC-3M-luc2 tumors. The data represents the mean ± SD of two sections from each mouse and four mice from each group of treatment. *p < 0.05
EENL Suppresses the DHT Levels in the C4-2B Tumor Tissues
PC-3M-luc2 and C4-2B xenograft tumor mice were sacrificed at the end of 8 and 11 weeks of EENL treatment, respectively. The tumor tissue and the other major organs were snap-frozen in liquid nitrogen and stored at −80°C. The DHT levels in the tumor tissues were analyzed using LC-MS system as described (20). The DHT concentrations in C4-2B tumor tissues of vehicle-treated mice were 1339 ± 9.89 pg/100 mg (n = 4). No DHT was detected in the PC-3M-luc2 tumor tissues of vehicle-treated mice and in the C4-2B and PC-3M-luc2 tumor tissues of EENL-treated mice (n = 4).
Discussion
In advanced castration recurrent prostate cancer, multiple aberrant pathways can be potentially targeted for the therapeutic effect that would yield better outcomes than monotherapies (21). Neem contains multiple active compounds that work simultaneously via different mechanisms (10–12). The scientific evaluation of neem as an anticancer agent is largely unknown. We used EENL extract to evaluate the anticancer activity. Analysis of the components in the EENL revealed a total of seven peaks by mass spectrometry (Fig. 1). The neem compounds 2′,3′-dehydrosalannol and 6-desacetyl nimbinene were reported before in the neem leaves, whereas nimolinone was reported in the neem flowers (22). Our study suggests that 2′,3′-dehydrosalannol, 6-desacetyl nimbinene, and nimolinone were present as major neem compounds in our EENL. In this study, we evaluated the anticancer effects of EENL in castration-resistant C4-2B and highly metastatic PC-3M-luc2 prostate cancer cells with an intent to use the neem extract for locally advanced and metastatic prostate cancer that is associated with considerable morbidity and mortality.
The extent of cell growth inhibition was measured by MTS assay which was used to determine the number of viable cells in proliferation. When various concentrations of EENL were used for treatment, EENL showed different sensitization potential; PC-3M-luc2 cells required higher concentrations of EENL compared to C4-2B to achieve the IC50 effect. EENL treatment significantly inhibited the growth of both C4-2B and PC-3M-luc2 cells (Fig. 2). To unravel the molecular targets involved in mediating the effect of EENL on prostate cancer cells, we used genome-wide microarray analysis. C4-2B prostate cancer cells treated with EENL showed a significant deregulation of 288 genes that increased to 1094 genes after 48 h. We further validated the expression of 40 significantly up-regulated genes by quantitative PCR in C4-2B and PC-3M-luc2 prostate cancer cells. PCR results were consistent with the microarray data. There was a significant up-regulation of the genes associated with the cell-to-cell signaling, cell death functions, drug metabolism and oxidative stress response (Table I), suggesting that the EENL is promoting cell death of the prostate cancer cells. Most of these up-regulated genes have been previously shown to be down-regulated in human prostate cancer tissues using microarray analysis (18,23–26). The RNA expression profiles of the 40 most significantly down-regulated genes were shown from our microarray analysis (Supplementary Table S1). All these 40 down-regulated genes were found to be up-regulated in various cancer tissues as shown in the Oncomine microarray data base (27). Most of the down-regulated genes ANLN, ASPM, ATAD2, ATRX, BMPR2, CDC2, CENPF, COL12A1, DLGAP5, DSG2, DTL, GUCY1A3, HELLS, HIST1H4C, HMMR, HNRNPA2B1, HSP90B1, KIF11, KIF14, NRIP1, NUF2, PHLDA1, SFPQ, SMC2, SMC3, SMC4, STAG2, TFPI, TOP2A, TPR, ZAK, and ZNF638 were involved in cell cycle, cellular assembly and organization, DNA replication, recombination, and repair functions (26,27), which implicates the role of EENL in the control of tumor cell proliferation.
We further focused on 4 genes HMOX1, AKR1C2, AKR1C3, and AKR1B10 for validation of protein expression levels. HMOX1, the inducible isoform is a rate-limiting enzyme in heme degradation (28,29). HMOX1 is an important homeostatic factor with pleiotropic effects against metabolic immune/inflammatory and angiogenesis (30–33). Over-expression of HMOX1 decreased the invasive potential of prostate cancer cells by down-regulating MMP9 expression (34). Our results revealed a highly significant increase in the RNA and protein expression levels of HMOX1 following EENL treatment of both C4-2B and PC-3M-luc2 cells (Table I and Fig. 3). Induction of HMOX1 expression through EENL could be a promising strategy to treat prostate cancer.
Numerous studies have focused on the androgen ablation, by decreased testosterone synthesis and blockade of androgen receptor, as the major treatment for hormone-sensitive prostate cancer (35,36). Despite the androgen deprivation therapy in prostate cancer patients, prostatic DHT levels were found to be 25% of the pretreatment levels (37). Steady-state levels of intracellular DHT are maintained through a balance between local synthetic and catabolic rates. However, little emphasis has been placed on the importance of DHT catabolism in the prostate. AKRs are phase I drug-metabolizing enzymes for a variety of carbonyl-containing drugs (38). Compared to the paired benign tissues, prostate cancer tissues showed a reduced metabolism of DHT which corresponded with a loss of AKR1C2 expression (39). Transient expression of AKR1C2 reduced DHT-stimulated proliferation of LAPC-4 prostate cancer cells (40). AKR1C2 and AKR1C3 reduced 5α-DHT to yield either 3α-andostanediol (an inactive androgen) or 3β-androstanediol (a proapoptotic ligand for ERβ; 41). Cellular proliferation experiments showed that increased AKR1C2 expression can reduce DHT-stimulated cell growth, and increased metabolism of DHT can block the activation of AR (13). Thus, androgen catabolism can indirectly regulate the activity of AR and thereby provides new therapeutic targets for the treatment of prostate cancer. The over-expression of AKR1B10 was reported in early stages of well and moderately differentiated tumors and down-regulation in advanced tumor-stages with low grade of differentiation, implicating that AKR1B10 may be a helpful marker for differentiation (42). Our results revealed highly significant up-regulation in the RNA and protein expression levels of AKR1C2, AKR1C3, and AKR1B10 with EENL treatment (Table I and Fig. 3). The increase in AKRs could contribute to the suppression of DHT levels observed in the C4-2B tumor tissues of EENL-treated mice. No DHT was detected in the PC-3M-luc2 tumors which supports previous finding that PC-3 cells do not express 5-reductase type II for conversion of testosterone to DHT (43). We speculate that up-regulation of AKRs expression with EENL treatment could inhibit cellular proliferation by inducing apoptosis and reduce the tumor growth. Further studies are required to evaluate the role of EENL induced AKRs on the DHT catabolism in prostate cancer cells.
We further evaluated the antitumor effect of the EENL using the xenograft prostate cancer models.
In mice injected with PC-3M-luc2 metastatic prostate cancer cells (3.0 × 106 cells) maximum tolerable xenograft tumor growth was attained by 8 weeks of administration, whereas in mice injected with C4-2B cells (1.5 × 106 cells) the xenograft tumor growth with was relatively slow and attained maximum tolerable tumor growth by 11 weeks. Our studies revealed that administration of EENL significantly suppressed the tumor growth (Fig. 4). The most significant histological difference between EENL- and vehicle-treated mice were the presence of hyalinized fibrosis (Fig. 5). We believe this hyalinization is a feature of tumor regression. Also, the control mice exhibited a greater amount of coagulative tumor necrosis, a feature typical of rapidly growing malignancies. Additional studies are necessary in large numbers of cases to further characterize these histological changes. It has been reported that fibrous tissue is related to decreased tumor invasiveness and is an indicator of improved survival after resection (44,45). These results are important to be explored further in the human clinical trials. We have demonstrated that reduction of tumor growth in mice is associated with apoptosis of tumor cells (Fig. 6). There was no significant change in either body weight or histology of any major organs in the EENL-treated group compared to control group, which confirms that EENL at 100 and 200 mg/kg body weight has no adverse effects (46,47). The dose administered in the present study (200 mg/kg body weight) was based on previous reports (48,49). This dose is also far less than the oral median lethal dose LD50 for EENL, which was found to be 4.57 g/kg body weight in acute toxicity studies (50). We speculate that neem, with its low risk of toxicity, could be safely used in the prostate cancer prevention and treatment trials.
In summary, we demonstrated that EENL-containing natural bioactive compounds 2′,3′-dehydrosalannol, 6-desacetyl nimbinene, and nimolinone inhibited in vitro cell proliferation and in vivo tumor growth. For the first time, we used genome-wide profiling approach to identify the genes associated with multiple biological pathways as potential targets of the antitumor activity of EENL and demonstrated the up-regulation of the HMOX1 and AKR protein changes. This is the first study to evaluate the antitumor effect of EENL in the preclinical models of prostate cancer. Our data suggests that the deregulated genes identified in our expression profiling could play an important role in the inhibition of tumor growth with the formation of hyalinized fibrous tissue. Further studies are required to unravel the role of these individual compounds in the EENL on tumor growth. The molecular targets identified in our study may be exploited for devising mechanism-based chemopreventive or therapeutic strategies for prostate cancer.
Abbreviations
- AKRs:
-
Aldo-keto reductases
- EENL:
-
Ethanol extract of neem leaves
- HMOX1:
-
Heme oxygenase-1
- DHT:
-
dihydrotestosterone
- AR:
-
androgen receptor
References
Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300.
Fang LC, Merrick GS, Wallner KE. Androgen deprivation therapy: a survival benefit or detriment in men with high-risk prostate cancer? Oncology (Williston Park). 2010;24:790–6. 8.
Damber JE, Aus G. Prostate cancer. Lancet. 2008;371:1710–21.
Hotte SJ, Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol. 2010;17 Suppl 2:S72–9.
Desai AG, Qazi GN, Ganju RK, El-Tamer M, Singh J, Saxena AK, et al. Medicinal plants and cancer chemoprevention. Curr Drug Metab. 2008;9:581–91.
Goto T, Takahashi N, Hirai S, Kawada T. Various terpenoids derived from herbal and dietary plants function as PPAR modulators and regulate carbohydrate and lipid metabolism. PPAR Res. 2010;2010:483958.
Gajria D, Seidman A, Dang C. Adjuvant taxanes: more to the story. Clin Breast Cancer. 2010;10 Suppl 2:S41–9.
Bose A, Chakraborty K, Sarkar K, Goswami S, Haque E, Chakraborty T, et al. Neem leaf glycoprotein directs T-bet-associated type 1 immune commitment. Hum Immunol. 2009;70:6–15.
Sarkar K, Bose A, Haque E, Chakraborty K, Chakraborty T, Goswami S, et al. Induction of type 1 cytokines during neem leaf glycoprotein assisted carcinoembryonic antigen vaccination is associated with nitric oxide production. Int Immunopharmacol. 2009;9:753–60.
Manikandan P, Anandan R, Nagini S. Evaluation of Azadirachta indica leaf fractions for in vitro antioxidant potential and protective effects against H2O2-induced oxidative damage to pBR322 DNA and red blood cells. J Agric Food Chem. 2009;57:6990–6.
Anyaehie UB. Medicinal properties of fractionated acetone/water neem [Azadirachta indica] leaf extract from Nigeria: a review. Niger J Physiol Sci. 2009;24:157–9.
Priyadarsini RV, Manikandan P, Kumar GH, Nagini S. The neem limonoids azadirachtin and nimbolide inhibit hamster cheek pouch carcinogenesis by modulating xenobiotic-metabolizing enzymes, DNA damage, antioxidants, invasion and angiogenesis. Free Radic Res. 2009;43:492–504.
Chakraborty K, Bose A, Pal S, Sarkar K, Goswami S, Ghosh D, et al. Neem leaf glycoprotein restores the impaired chemotactic activity of peripheral blood mononuclear cells from head and neck squamous cell carcinoma patients by maintaining CXCR3/CXCL10 balance. Int Immunopharmacol. 2008;8:330–40.
Haque E, Mandal I, Pal S, Baral R. Prophylactic dose of neem (Azadirachta indica) leaf preparation restricting murine tumor growth is nontoxic, hematostimulatory and immunostimulatory. Immunopharmacol Immunotoxicol. 2006;28:33–50.
Bose A, Chakraborty K, Sarkar K, Goswami S, Chakraborty T, Pal S, et al. Neem leaf glycoprotein induces perforin-mediated tumor cell killing by T and NK cells through differential regulation of IFNgamma signaling. J Immunother. 2009;32:42–53.
Vanaja DK, Grossmann ME, Cheville JC, Gazi MH, Gong A, Zhang JS, et al. PDLIM4, an actin binding protein, suppresses prostate cancer cell growth. Cancer Invest. 2009;27:264–72.
Vanaja DK, Ballman KV, Morlan BW, Cheville JC, Neumann RM, Lieber MM, et al. PDLIM4 repression by hypermethylation as a potential biomarker for prostate cancer. Clin Cancer Res. 2006;12:1128–36.
Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–82.
Gong A, He M, Krishna Vanaja D, Yin P, Karnes RJ, Young CY. Phenethyl isothiocyanate inhibits STAT3 activation in prostate cancer cells. Mol Nutr Food Res. 2009;53:878–86.
Kulle AE, Riepe FG, Melchior D, Hiort O, Holterhus PM. A novel ultrapressure liquid chromatography tandem mass spectrometry method for the simultaneous determination of androstenedione, testosterone, and dihydrotestosterone in pediatric blood samples: age- and sex-specific reference data. J Clin Endocrinol Metab. 2010;95:2399–409.
Wegiel B, Evans S, Hellsten R, Otterbein LE, Bjartell A, Persson JL. Molecular pathways in the progression of hormone-independent and metastatic prostate cancer. Curr Cancer Drug Targets. 2010;10:392–401.
Mitchell MJ, Smith SL, Johnson S, Morgan ED. Effects of the neem tree compounds azadirachtin, salannin, nimbin, and 6-desacetylnimbin on ecdysone 20-monooxygenase activity. Arch Insect Biochem Physiol. 1997;35:199–209.
Su AI, Welsh JB, Sapinoso LM, Kern SG, Dimitrov P, Lapp H, et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61:7388–93.
Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–9.
Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54.
Tomlins SA, Mehra R, Rhodes DR, Cao X, Wang L, Dhanasekaran SM, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet. 2007;39:41–51.
Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6.
Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–55.
Bilban M, Haschemi A, Wegiel B, Chin BY, Wagner O, Otterbein LE. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J Mol Med. 2008;86:267–79.
Bussolati B, Mason JC. Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid Redox Signal. 2006;8:1153–63.
Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation. 2008;117:231–41.
Prawan A, Kundu JK, Surh YJ. Molecular basis of heme oxygenase-1 induction: implications for chemoprevention and chemoprotection. Antioxid Redox Signal. 2005;7:1688–703.
Was H, Cichon T, Smolarczyk R, Rudnicka D, Stopa M, Chevalier C, et al. Overexpression of heme oxygenase-1 in murine melanoma: increased proliferation and viability of tumor cells, decreased survival of mice. Am J Pathol. 2006;169:2181–98.
Gueron G, De Siervi A, Ferrando M, Salierno M, De Luca P, Elguero B, et al. Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol Cancer Res. 2009;7:1745–55.
Buchan NC, Goldenberg SL. Intermittent androgen suppression for prostate cancer. Nat Rev Urol. 2010;7:552–60.
Quon H, Loblaw DA. Androgen deprivation therapy for prostate cancer—review of indications in 2010. Curr Oncol. 2010;17 Suppl 2:S38–44.
Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10:7121–6.
Jin Y, Penning TM. Aldo-keto reductases and bioactivation/detoxication. Annu Rev Pharmacol Toxicol. 2007;47:263–92.
Ji Q, Chang L, VanDenBerg D, Stanczyk FZ, Stolz A. Selective reduction of AKR1C2 in prostate cancer and its role in DHT metabolism. Prostate. 2003;54:275–89.
Ji Q, Chang L, Stanczyk FZ, Ookhtens M, Sherrod A, Stolz A. Impaired dihydrotestosterone catabolism in human prostate cancer: critical role of AKR1C2 as a pre-receptor regulator of androgen receptor signaling. Cancer Res. 2007;67:1361–9.
Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, et al. The androgen derivative 5alpha-androstane-3beta,17beta-diol inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype. Cancer Res. 2005;65:5445–53.
Heringlake S, Hofdmann M, Fiebeler A, Manns MP, Schmiegel W, Tannapfel A. Identification and expression analysis of the aldo-ketoreductase1-B10 gene in primary malignant liver tumours. J Hepatol. 2009;52:220–7.
Negri-Cesi P, Colciago A, Poletti A, Motta M. 5alpha-reductase isozymes and aromatase are differentially expressed and active in the androgen-independent human prostate cancer cell lines DU145 and PC3. Prostate. 1999;41:224–32.
Okano K, Yamamoto J, Kosuge T, Yamamoto S, Sakamoto M, Nakanishi Y, et al. Fibrous pseudocapsule of metastatic liver tumors from colorectal carcinoma. Clinicopathologic study of 152 first resection cases. Cancer. 2000;89:267–75.
Yamamoto J, Shimada K, Kosuge T, Yamasaki S, Sakamoto M, Fukuda H. Factors influencing survival of patients undergoing hepatectomy for colorectal metastases. Br J Surg. 1999;86:332–7.
Kumar S, Suresh PK, Vijayababu MR, Arunkumar A, Arunakaran J. Anticancer effects of ethanolic neem leaf extract on prostate cancer cell line (PC-3). J Ethnopharmacol. 2006;105:246–50.
Subapriya R, Kumaraguruparan R, Nagini S. Expression of PCNA, cytokeratin, Bcl-2 and p53 during chemoprevention of hamster buccal pouch carcinogenesis by ethanolic neem (Azadirachta indica) leaf extract. Clin Biochem. 2006;39:1080–7.
Subapriya R, Bhuvaneswari V, Ramesh V, Nagini S. Ethanolic leaf extract of neem (Azadirachta indica) inhibits buccal pouch carcinogenesis in hamsters. Cell Biochem Funct. 2005;23:229–38.
Subapriya R, Velmurugan B, Nagini S. Modulation of xenobiotic-metabolizing enzymes by ethanolic neem leaf extract during hamster buccal pouch carcinogenesis. J Exp Clin Cancer Res. 2005;24:223–30.
Chattopadhyay RR. Possible biochemical mode of anti-inflammatory action of Azadirachta indica A. Juss. in rats. Indian J Exp Biol. 1998;36:418–20.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is supported by the grants American Cancer Society RSG-09-175-01-CCE and U.S. Department of Defense W81XWH-09-1-0216.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Table S1
C4-2B cells prostate cancer cells treated with 8.0 μg/mL of the EENL for 24 and 48 h. Gene expression profiles were analyzed using Human Genome-U133-Plus2 oligonucleotide arrays. The experiments were performed in duplicate; mean ± SD of the fold decrease in the mRNA expression levels of 40 genes compared with the respective controls were shown. (DOC 72 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mahapatra, S., Karnes, R.J., Holmes, M.W. et al. Novel Molecular Targets of Azadirachta indica Associated with Inhibition of Tumor Growth in Prostate Cancer. AAPS J 13, 365–377 (2011). https://doi.org/10.1208/s12248-011-9279-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-011-9279-4