
Abstract
Background
Hepatitis E virus (HEV) is the cause of a liver disease hepatitis E. The translation product of HEV ORF2 has recently been demonstrated as a protein involved in multiple functions besides performing its major role of a viral capsid. As intrinsically disordered regions (IDRs) are linked to various essential roles in the virus’s life cycle, we analyzed the disorder pattern distribution of the retrieved ORF2 protein sequences by employing different online predictors. Our findings might provide some clues on the disorder-based functions of ORF2 protein that possibly help us in understanding its behavior other than as a HEV capsid protein.
Results
The modeled three dimensional (3D) structures of ORF2 showed the predominance of random coils or unstructured regions in addition to major secondary structure components (alpha helix and beta strand). After initial scrutinization, the predictors VLXT and VSL2 predicted ORF2 as a highly disordered protein while the predictors VL3 and DISOPRED3 predicted ORF2 as a moderately disordered protein, thus categorizing HEV-ORF2 into IDP (intrinsically disordered protein) or IDPR (intrinsically disordered protein region) respectively. Thus, our initial predicted disorderness in ORF2 protein 3D structures was in excellent agreement with their predicted disorder distribution patterns (evaluated through different predictors). The abundance of MoRFs (disorder-based protein binding sites) in ORF2 was observed that signified their interaction with binding partners which might further assist in viral infection. As IDPs/IDPRs are targets of regulation, we carried out the phosphorylation analysis to reveal the presence of post-translationally modified sites. Prevalence of several disordered-based phosphorylation sites further signified the involvement of ORF2 in diverse and significant biological processes. Furthermore, ORF2 structure-associated functions revealed its involvement in several crucial functions and biological processes like binding and catalytic activities.
Conclusions
The results predicted ORF2 as a protein with multiple functions besides its role as a capsid protein. Moreover, the occurrence of IDPR/IDP in ORF2 protein suggests that its disordered region might serve as novel drug targets via functioning as potential interacting domains. Our data collectively might provide significant implication in HEV vaccine search as disorderness in viral proteins is related to mechanisms involved in immune evasion.
Similar content being viewed by others

Background
Hepatitis E virus (HEV) is a major zoonotic pathogen causing acute hepatitis E worldwide. HEV is a single-stranded RNA virus belonging to the family Hepeviridae [1]. According to recent data, about 15–110 million of the individuals worldwide are still experiencing infections and about 939 million of the world populations have already (past HEV) experienced infection from HEV [2]. In India, the reporting of hepatitis infections to the CBHI (Central Bureau of Health Intelligence) is exceptionally low, as mostly of the people share a common belief that the disease is characterized with lack of cure in allopathy, thus, establishing inaccurate burden of HEV infections. However as suggested, India has been reported with 10–40% cases of acute hepatitis and 15–45% cases of acute liver failure in its population [3, 4]. As reported in studies, the HEV is segregated into 8 genotypes (genotype 1–genotype 8). Orthohepevirus A species of the Orthohepevirus genus is segregated into 8 genotypes (genotype 1, genotype 2, GT genotype 3, genotype 4, genotype 5, genotype 6, genotype 7, and genotype 8). The genotypes 1 and 2 infect only humans and are major reasons for waterborne outbreaks in endemic regions of Africa, parts of Asia (South and Southeast) and Mexico while the genotypes 3 and 4 infect diverse mammals, such as humans, rabbits, swine and deer, and cause sporadic of hepatitis E cases in mainly developed countries of East Asia and Europe [5–9]. The genotypes 5 and 6 have been identified in Japan from wild boars [10, 11] while genotype 7 from dromedaries in Middle East countries and genotype 8 from China in Bactrian camels [12, 13]. The meat product (raw or undercooked) consumption from animals in developed nations is the chief cause of sporadic infections [14]. Moreover, transmissions such as blood-mediated [15], person-to-person [16], animal (pet) to human [17, 18] have been reported in patients, thus categorizing HEV as major concern of health issue. The diagnostic tests for HEV largely target mostly anti-HEV IgG and anti-HEV IgM antibodies. In diagnosis, initially HEV RNA and anti-HEV IgM antibodies are detected, which is followed up by anti-HEV IgG antibody detection. Presence or positive detection of anti-HEV IgM antibodies in serum are considered as important marker for acute HEV infection. Anti-HEV IgG antibodies have long-lasting persistence (duration is uncertain), thus are considered as markers for an individual who has experienced past infection [19, 20]. In contrast to this, anti-HEV IgM antibodies persist for a short duration of time (3 to 4 months), and thus are considered as markers in an individual who is undergoing recent infection [19, 20].
The three open reading frames (ORFs) altogether forms the HEV genome in which the first, second, and third reading frames encoded proteins ORF1, ORF2, and ORF3 codes for the HEV’s nonstructural polyprotein, capsid, and regulatory protein respectively [21]. Though earlier it was revealed that ORF2 acts as a capsid protein [22], later its role was described in multiple crucial functions of HEV [14, 23–27].
Intrinsically disordered regions (IDRs) [including intrinsically disordered proteins (IDPs) or intrinsically disordered protein regions (IDPRs)] constitute the fraction of a proteome that are known as “dark proteomes.” This dark proteome does not have noticeable similarity to any PDB structure. The IDRs (IDP or IDPR) lack unique structures as they are not folded into three dimensional structures within the viral proteomes [28]. The IDRs exhibit specific functions due to lack of definite 3D structure [29–31]. Additionally, due to possession of intrinsic disorder phenomenon, the IDRs (IDPs or IDPRs) have been correlated with a number of implications in various human diseases (cancer, etc.) [32]. Interestingly, most of the viral proteins possess MoRFs (molecular recognition features), i.e., short protein regions within IDRs that upon binding to partners (interacting partners) undergo disorder-to-order transition [33]. Therefore, the unique characteristics possessed by IDRs assist proteins in their interaction with diverse biological partners and thus form an important requirement for completion of multiple cellular pathway regulation through protein–protein interaction networks [34]. Moreover, the disordered regions in proteins constitute potential drug targets due to their association with important biological processes [35, 36]. As IDP/IDPR is indispensable for carrying out several important crucial functions in viruses, thus they are analyzed using computational approaches [29–31]. It has been revealed recently that besides performing capsid function, the ORF2 plays crucial role in other processes, such as viral replication, immune response regulation, cellular signalling, host tropism, and pathogenesis of HEV. Moreover, it has been suggested that ORF2 has the potential in forming the development of vaccine against HEV [27].
Thus, in this regard, the present study carried out the intrinsic disorder analysis of the HEV ORF2 proteins by employing a set of bioinformatics predictors through evaluating its functional significance. The disorder analysis predicted ORF2 as either moderately disordered or highly disordered protein revealing them as IDPR or IDP variants. The presence of MoRF regions in ORF2 sequences revealed its propensity to bind to interacting partners. Additionally, we also carried out the structure-based analysis of ORF2 protein (using sequences obtained from different host and genotypes) in order to reveal its molecular functions. The identified ion binding, protein binding, and metal binding sites in conjunction with diverse biological processes, such as viral replication, RNA biosynthetic process in the ORF2 protein signified its importance in interaction with the host cell membrane. Our study could shed some novel light on the understanding more ORF2 protein functions beyond its role as a viral capsid protein.
Methods
Sequence retrieval
The HEV ORF2 protein sequences were obtained from GenBank at NCBI (National Center for Biotechnology Information). The present analysis included those sequences that encompassed different GTs and hosts and are listed in Table 1.
Three dimensional (3D) structure
The elements of secondary structure and 3D tertiary structure of the HEV ORF2 proteins were predicted using Phyre2 (Protein Homology/AnalogY Recognition Engine) (PHYRE2 Protein Fold Recognition Server (ic.ac.uk) and I-TASSER (Iterative Threading ASSEmbly Refinement) (itasser - Search (bing.com)) webserver, respectively.
Disorder plot evaluation
The disorder character of the HEV ORF2 proteins was examined using the online tool PONDR (Predictor of Natural Disordered Regions) by considering its default parameters. Different predictors of PONDR like PONDR-VLS2 [37], PONDR-VL3 [38], and PONDR-VL-XT [39], in combination with DISOPRED3 (PSIPRED Workbench (ucl.ac.uk)), were used to identify the disordered regions in the ORF2 proteins.
MoRFs (molecular recognition features) prediction
The disorder-based binding residues within ORF2 protein segments were identified using three different online predictors, DISOPRED3 (PSIPRED Workbench (ucl.ac.uk)), IUPred3 (IUPred3 (elte.hu)), and IUPred2A (IUPred2A (elte.hu)). The cutoff score was set as 0.5 for all the predictors (cutoff = ≥ 0.5).
Phosphorylation pattern evaluation
DEPP (disorder enhanced phosphorylation prediction) (http://www.pondr.com/cgi-bin/depp.cgi), an online predictor, was used to predict the phosphorylated residues (Ser, Thr and Tyr) in the HEV ORF2 sequences.
Structure-based function prediction
COFACTOR algorithm [40, 41] was employed to identify the probable molecular functions and biological processes of the HEV ORF2 proteins by utilizing their 3D structured models.
Results
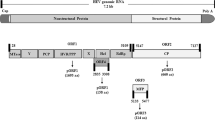
The HEV genome encoded ORF2 starts at the 5147th nucleotide and terminates at the 7127th position of nucleotide. The schematic illustration of the genome of HEV (with reference to Sar55 strain with accession number AF444002) is represented in Fig. 1 [42].

Schematic representation of the genome of HEV. The genomic organization is organized into three open reading frames (ORFs), i.e., ORF1, ORF2, and ORF3. The nucleotide numbers are with reference to the strain Sar55 (GenBank Accession Number: AF444002)
3D structures with predicted disorder
The HEV ORF2 protein 3D structures modeled through I-TASSER (based on homology modeling) consisted of alpha helix, beta strand and coil (Fig. 2A–H). The results showed the dominance of coils in comparison to both helices and strands. The predicted percentage of helices, strands, and disorder (evaluated through Phyre2) are mentioned in Table 1.

Generated 3D models of the HEV ORF2. A JF443720 (GT 1); B M74506 (GT 2); C AB222182 (GT 3); D GU119961 (GT 4); E AB573435 (GT 5); F AB602441 (GT 6); KJ496143 (GT 7); and H KX387865 (GT 8). The prediction was carried out using I-TASSER
Thus, our results clearly revealed that the HEV ORF2 proteins consisted of significant fraction of disordered regions. This further prompted us to analyze its intrinsic disorder content using computational approach.
Intrinsic disorder distribution
The HEV ORF2 proteins intrinsic disorder analysis was evaluated (using different disorder predictors) as mentioned in Table 2. The disorder graphs of the ORF2 proteins are shown in Fig. 2A–H. As suggested, the proteins are termed as structured, moderately disordered, or highly disordered due to their overall predicted intrinsic disorder fraction [43]. Additionally, disorder variants like ORDP (ordered protein), IDPR (intrinsically disordered protein region), and IDP (intrinsically disordered protein) are termed as due to the disordered domain’s length and disordered residue (overall) fraction [44, 45].
ORF2 protein (JF443720)
The disorder distribution analysis of the ORF2 polypeptide sequence (JF443720) categorized it into a highly disordered protein as it contained >30% of disordered residues (43.25% by VLXT and 35.66% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (28.53% by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 59 to 132 consecutive amino acid residues, categorized it into IDP, i.e., protein possessing significant fraction of disordered regions (as predicted by VLXT and VSL2) or IDPR, i.e., structured protein possessing intrinsically disordered segments (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 20.33% (<30%) of disordered residues with long continuous stretch of disordered domain (about 45 amino acid residues).
ORF2 protein (M74506)
The disorder distribution analysis of the ORF2 polypeptide sequence (M74506) categorized it into a highly disordered protein as it contained >30% of disordered residues (43.55% by VLXT and 36.57% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (25.19% by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 60 to 150 consecutive amino acid residues, categorized it into IDP (as predicted by VLXT and VSL2) or IDPR (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 20.48% (<30%) of disordered residues with long continuous stretch of disordered domain (about 48 amino acid residues).
ORF2 protein (AB222182)
The disorder distribution analysis of the ORF2 polypeptide sequence (AB222182) categorized it into a highly disordered protein as it contained >30% of disordered residues (40.30% by VLXT and 37.12% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (28.48% by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 57 to 91 consecutive amino acid residues, categorized it into IDP (as predicted by VLXT and VSL2) or IDPR (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 19.39% (<30%) of disordered residues with long continuous stretch of disordered domain (about 48 amino acid residues).
ORF2 protein (GU119961)
The disorder distribution analysis of the ORF2 polypeptide sequence (GU119961) categorized it into a highly disordered protein as it contained >30% of disordered residues (40.06% by VLXT and 33.23% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (24.04% by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 60 to 131 consecutive amino acid residues was also observed, thus was categorized it into IDP (as predicted by VLXT and VSL2) or IDPR (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 21.22% (<30%) of disordered residues with long continuous stretch of disordered domain (about 53 amino acid residues).
ORF2 protein (AB573435)
The disorder distribution analysis of the ORF2 polypeptide sequence (AB5734351) categorized it into a highly disordered protein as it contained >30% of disordered residues (39.02% by VLXT and 32.64% by VSL2). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 56 to 130 consecutive amino acid residues, categorized it into IDP (as predicted by VLXT and VSL2). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 21.81% (<30%) of disordered residues with long continuous stretch of disordered domain (about 55 amino acid residues).
ORF2 protein (AB602441)
The disorder distribution analysis of the ORF2 polypeptide sequence (AB602441) categorized it into a highly disordered protein as it contained >30% of disordered residues (41.97% by VLXT and 38.18% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (26.97% by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 57 to 147 consecutive amino acid residues, categorized it into IDP (as predicted by VLXT and VSL2) or IDPR (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 18.93% (<30%) of disordered residues with long continuous stretch of disordered domain (about 43 amino acid residues).
ORF2 protein (KJ496143)
The disorder distribution analysis of the ORF2 polypeptide sequence (KJ496143) categorized it into a highly disordered protein as it contained >30% of disordered residues (41.52% by VLXT and 36.82% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (26.21% by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 90 to 108 consecutive amino acid residues, categorized it into IDP (as predicted by VLXT and VSL2) or IDPR (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 18.78% (<30%) of disordered residues with long continuous stretch of disordered domain (about 46 amino acid residues).
ORF2 protein (KX387865)
The disorder distribution analysis of the ORF2 polypeptide sequence (KX387865) categorized it into a highly disordered protein as it contained >30% of disordered residues (41.06% by VLXT and 34.70% by VSL2), and moderately disordered protein as it contained <30% of disordered residues (27.192 by VL3). Additionally, the inclusion of long disordered domain at N-terminus in the polypeptide sequence, i.e., up to 64 to 148 consecutive amino acid residues, categorized it into IDP (as predicted by VLXT and VSL2) or IDPR (as predicted by VL3). The DISPOPRED3 predicted the ORF2 as a moderately disordered protein or IDPR as it contained 19.39% (<30%) of disordered residues with long continuous stretch of disordered domain (about 47 amino acid residues).
On combining the results, obtained from the aforementioned predictors (VLXT, VL3, VSL2, and DISOPRED3), it was inferred that the HEV exhibited significant intrinsic disorder character in the ORF2 proteins.
Categorizing protein variant on the basis of predicted disorder percentage
Next we combined the results of obtained disorder predictors for the HEV ORF2 protein sequences to categorize them into a specific category of disorder variant, i.e., ORDP, IPD, or IDPR. The mean PPID (predicted percentage of intrinsic disorder) score was obtained by summing up the individual percentage disorder score predicted by different predictors (VLXT, VL3 VSL2, and DISOPRED3) and dividing by 4. The mean PPID scores of the ORF2 proteins are mentioned in Table 3.
As it could be interpreted from Table 3, most of the HEV ORF2 were predicted as highly disordered proteins or IDPs due to the presence of more than 30% of the disordered residues in its polypeptide chain. However, two proteins (GU119961 and AB573435) were categorized as moderately disordered proteins or IDPRs as they consisted of less than 30% of the disordered residues in its polypeptide chain along with disordered domains (as seen in Table 2). But it is important to mention that these sequences also possessed IDP character as the percentages were only almost equivalent to 30. All the ORF2 protein sequences obtained from different genotypes showed that significant proportion of their fraction consisted disordered character. Thus, it can be assumed from these results that ORF2 belonged to the IDP category.
Disorder-based binding site in protein
In addition to intrinsic disorder, we have also computationally estimated the presence of disorder-based binding sites, MoRFs, in each HEV ORF2 protein. The predicted MoRFs for individual ORF2 protein, by different computational tools (DISOPRED3, and IUPRED3 ANCHOR and IUPRED2A ANCHOR), are listed in Table 4.
Analysis of phosphorylation sites
The ORF2 sequences were predicted with several phosphorylation sites (P-sites). The predicted phosphorylated residues, i.e., Ser, Thr, and Tyr in HEV ORF2 sequences with the DEPP score are summarized in Table 5 (Fig. 4).
Our results revealed that Ser was found in higher fractions in comparison to other phosphorylated residues (Thr and Tyr) (Fig. 4). Our analysis revealed that most of the P-sites were present within the disordered ORF2 regions which clearly indicated the correlation between disordered regions and phosphorylation sites (Figs. 3 and 4).

Analysis of intrinsic disorder predisposition of HEV ORF2. A JF443720 (GT 1); B M74506 (GT 2); C AB222182 (GT 3); D GU119961 (GT 4); E AB573435 (GT 5); F AB602441 (GT 6); KJ496143 (GT 7); and H KX387865 (GT 8). Graphs A–H represent the intrinsic disorder profiles of ORF2 sequences of HEV. Disorder probability was calculated using three members of the family PONDR (Prediction of Natural Disordered Regions), i.e., VLXT, VL3, and VSL2. A threshold value of 0.5 was set to distinguish between ordered and disordered region along the genome (line). Regions above the threshold are predicted to be disordered

Prediction of phosphorylation sites showing the scores of phosphorylated residues (Ser, Thr, Tyr) along with the depicted scores within ORF2. A JF443720 (GT 1); B M74506 (GT 2); C AB222182 (GT 3); D GU119961 (GT 4); E AB573435 (GT 5); F AB602441 (GT 6); KJ496143 (GT 7); and H KX387865 (GT 8). Graphs A–H represent the phosphorylation patterns of the ORF2 sequences of HEV. The score was computed using DEPP (disorder enhanced phosphorylation prediction). A threshold value of 0.5 was set to distinguish between ordered and disordered region along the genome (line). The predicted phosphorylated residues above the threshold are represented as Ser (S): Blue, Thr (T): Green, and Tyr (Y): Red
Prediction of consensus GO terms
The putative 3D modeled structure-based molecular functions and biological processes (using COFACTOR algorithm) are summarized in Table 6.
The molecular functions included structural molecule activity, ion binding, metal ion binding, transition metal ion binding, ion-sulfur cluster binding, and oxidoreductase activity. In this regard, our gene ontology findings clearly revealed that binding interactions in conjunction with catalytic activities were attributed to ORF2. The binding interactions, such as metal cluster binding (GO:0051540), protein binding (GO:004280), transition metal ion binding (GO:0046914), and iron-sulfur cluster binding (GO:0051536) revealed the propensity of ORF2 to bind to a variety of molecules (ion, metal, protein). Furthermore, the involvement of ORF2 proteins in different predicted biological processes, such as, electron transport chain (GO:0022900), oxidoreductase activity (GO:0016638), DNA replication (GO:0006260), and cell differentiation regulation (GO:0045595) revealed the significant mitochondrial functions as well as significant processes attributed to ORF2 (Table 6).
Discussion
The three ORFs (ORF1, ORF2 and ORF3) constitute the genome of HEV [21]. The ORF2 encoded protein polypeptide comprises 660 amino acids [46] and codes for the viral capsid [22, 23]. The intrinsic disorder occurrence in diverse viral proteins has been predicted through different bioinformatics tools [47–49]. The disordered segments (IDRs) in viral proteins perform indispensable functions like accommodation and adaptation of the virus in unsympathetic habitats, and assist in helping proper management of virus and invasion of the host cell pathways [50, 51]. IDPs are often frequently associated with the progression of diseases and they constitute druggable-targets [35, 36, 52, 53]. The ORF2 protein performs various regulatory roles in addition to its role in viral replication and pathogenesis [54]. Also, its application in vaccine development has been documented recently [54]. Thus, targeting the ORF2 protein is ideal for devising treatment against the HEV. Some of our recent investigations have shown varied levels of disorderedness in different HEV ORF encoded proteins [55–60], however, irrespective of the significance of the ORF2 protein role in the virus life cycle, its disorder character has not been explored in different GTs and hosts. In this regard, it is essential to investigate the disorder status of the ORF2 protein (sequences that encompassed different hosts and genotypes) to understand its functions based on its disorderness. The presented study employed different computational tools to shed light on the ORF2 disorderedness in HEV functionality through utilizing GenBank data.
As detailed examination of a protein’s structure provides knowledge on its functional aspects, therefore, we scrutinized the homology modeled ORF2 structures generated through I-TASSER webserver (a portal for protein modeling and analysis). The homology modeled structures comprised major secondary elements (in form of α-helix, β-strand) and coils. As defined by Kabsch and Sander in 1983 [61], in a study [62], that though coils/loops are not necessarily found within disorder protein segments, necessarily disorderness of proteins exists in loops only [62]. On examining the ORF2 constructed homology models, we found that the structures possessed significant disordeness that initially revealed ORF2 proteins with significant percentage of IDRs within loops. Further, detailed examination of the ORF2 proteins was undertaken by employing various disorder predictors. The presented study utilized three PONDR family members VLXT, VL3, and VSL2 [37–39], to examine the ORF2 proteins related to HEV. The predictor VL3 was chosen as it predicts disorderness of long segments with high accuracy [63], whereas VLXT was shown as it is very sensitive [64, 65]. DISOPRED3 was utilized as it predicts disordered segments within protein sequences precisely [66].
The complete life cycle of a virus is achieved by establishing a variety of interactions with the different components of host cell. The various stages of virus life cycle, such as, its attachment, entry, commandeering host machinery, viral component synthesis, and assembly till its exit from hosts as new infectious particle, heavily depend on the intrinsic disorder prevalent in their proteomes [67]. Importantly, studies have shown the relation of intrinsically disorder protein to specific roles [68] in viruses like HCV (hepatitis C virus) [69], MeV (Measles virus) [70], and Hendra virus [71]. The nonstructural HEV ORF1 domains like PPR (Polyproline region) [72] and Y-domain [55] in addition to other proteins [56–60] have also been linked to regulation of HEV due to its characteristic intrinsic disorder property. The HEV ORF2 proteins were initially categorized into structured proteins, moderately disordered proteins, and highly disordered proteins, on the basis of the overall degree of intrinsic disorder [43]. Next, the ORF2 proteins were categorized into ORDPs, IDPRs, and IDPs on the basis of the disordered domain’s length with the overall disordered fraction of residues [44, 45]. These three categories of intrinsic disorder variants are briefly described as follows: ORDPs are variants which consist of less than 30% of disordered residues with the absence of disordered domain at either terminus (C- or N-) or in positions distinct from both terminals. IDPRs are variants which consist of less than 30% of disordered residues with the presence of disordered domain at either terminus (C- or N-) or in positions distinct from the terminals. IDPs are variants which consist of more than 30% of disordered residues. On exploiting these criteria, our intrinsic disorder propensity analysis revealed that ORF2 as a highly disordered protein or moderately disordered protein categorizing them as IDP or IDPR. Our results showed the disorderness in ORF2 protein at N-terminals is due to the occurrence of continuous long disordered domains. According to a study, it has been revealed that the N-terminal region arginine-rich motif (from 1st to 111th amino acid residues) of the ORF2 protein inhibits the phosphorylation of IRF3 (Interferon Regulatory Factor 3) via interacting with a multiprotein complex [73]. But the exact domain binding this complex remains to be determined. A recent study has shown the involvement of arginine-rich motifs in nuclear translocation of ORF2 by serving as nuclear location signals [74]. Thus, it is noteworthy to mention that the prevalent intrinsic disordered regions in ORF2 protein could perform crucial regulatory functions by interacting with the other viral and host components.
Further, studies have shown the importance of MoRFs in several viruses [55, 56, 75–77]. The MoRF is defined as short segment within disordered regions of a protein (exists as IDPR or IDP) that upon binding with its partner undergoes a transition from disorder-to-order state [33]. These are segments that are prone to interactions [33]. Our study predicted MoRFs in ORF2 proteins by set of three predictors (DISOPRED3, IUPred3, and IUPred2A). DISOPRED3 server predicts the protein binding disordered regions within given target sequences [66]. We used DISOPRED3 for identifying IDRs as it provides significantly improved results over DISOPRED2 [66]. Additionally, IUPred members (IUPred3 and IUPred2A) were also utilized to predict the disordered binding regions within ORF2 protein sequences [78, 79]. IUPred3 and IUPred2A are webservers that allow identification of both disordered protein regions (using IUPred2/IUPred3) and disordered binding regions (using ANCHOR2) respectively [78, 79]. The identified multiple MoRFs at N-terminus of ORF2 protein further provided compelling evidence that ORF2 show propensity towards interaction with multiple partners with its N-terminus through its order/disorder tendency. According to some reports, ORF2 also contributes to interferon production and immunity recognition [80]. Thus, altogether these findings substantiate our results that the disordered N-terminal of the ORF2 protein interacts with various partners. Further, the ORF2 has also been linked to host tropism [81, 82]. This suggested the involvement of ORF2 in regulation and pathogenesis of HEV which shows consistency with recent report [54].
Further, studies have documented the role of post-translational modifications (PTMs) in various processes, such as folding of proteins, transduction of signals, regulation of transcription, progression of cell cycle, survival, and apoptosis [83]. Also, phosphorylation is essential for the establishment productive infection cycle for majority of intracellular pathogens [84, 85]. In RNA viruses, such as Alphaviruses [86, 87] and Flaviviruses [88–90], literatures have shown the essentiality of phosphorylation in critical protein functions. In this regard, we evaluated the phosphorylation scores using the DEPP online tool by analyzing the ORF2 sequences. Our phosphorylation patterns of ORF2 protein revealed that all the sequences consisted of P-sites. The observations revealed that P-sites were prevalent within disordered segments of the ORF2 polypeptide chains. This inferred strong correlation between protein phosphorylation and intrinsically disordered ORF2 regions. Thus, our findings are in agreement with previous report that has shown the interconnection between phosphorylated residues and overall disorderness in the proteins [91, 92]. It has been suggested that the disordered segment of protein regions displays sites for PTM is perhaps due to the conformational flexibility of display sites provided by the disordered regions in the proteins [93, 94]. Report has revealed that serines’s hydroxyl group act as target for phosphorylation by protein kinases, within the disordered protein segments [95]. Therefore, the higher predicted serine (phosphorylated) residue in ORF2 proteins revealed its interacting ability and flexible nature, ultimately relating its important role to protein regulation and its linked various biological processes.
Furthermore, the predicted molecular functions and biological processes (based on 3D ORF2 structured models) using COFACTOR algorithm [40, 41], such as, ion binding, metal ion binding, transition metal ion binding, ion-sulfur cluster binding, and protein binding, were predicted which clearly revealed the propensity of ORF2 to bind to a variety of molecules, such as ions, metals, and proteins. Such kind of interactive functions have been reported in regulation of various processes, like cellular signal transduction, phosphorylation, transcription, and translation [96]. Electron transport chain occurs inside mitochondria’s matrix and it involves redox reactions which are catalyzed by oxidoreductase enzymes. The predicted ETC and oxidoreductase activity suggested the ORF2 involvement in HEV regulation as mitochondrion serves as signaling hub for immune response [97, 98]. Also, a literature has evidenced the role of complex III (of electron transport chain) in HEV infection [99], which performs assorted biological functions [100, 101]. These predicted biological processes besides structural molecule activity clearly inferred the involvement of ORF2 protein in multiple crucial roles beyond its role as capsid protein [54]. These findings further substantiate our present hypothesis. On summing up our observations, it can be hypothesized that ORF2 is a protein with multiple functions and is involved in cell regulation and pathogenesis in addition to its role as a HEV capsid protein.
Conclusions
The present study provides novel investigation on the biology of HEV ORF2 protein in terms of its disorderedness. This unique study employed disorder predictors to reveal peculiar intrinsic disorder patterns of HEV ORF2 that will help in understanding its behavior. The extent of intrinsic disorder distribution was calculated using different bioinformatics predictors by obtaining the ORF2 protein sequences from publicly available database. The initial comprehensive analysis of the ORF2 structured models showed significant percentages of coil. On analyzing the occurrence of intrinsic disorder extent, the ORF2 protein was revealed as IDP (highly disordered protein), thus suggesting its various significant roles in the life cycle of viruses. Further, the predicted MoRFs in the HEV ORF2 proteins suggested its propensity towards numerous disorder-based binding functions. These identified disordered regions in addition to disorder-based protein binding residues could perform diverse important roles such as viral replication, pathogenicity, and its particle assembly. Furthermore, the presence of several phosphorylation sites signified the involvement of ORF2 protein in various important mechanisms like cellular and signalling pathways. Moreover, structure-based prediction of crucial molecular functions and biological processes indicated multiple functions associated with it beyond its capsid function. Our study is further envisaged to provide critical information in studying the HEV ORF2 behavior.
Availability of data and materials
Not applicable.
Abbreviations
- HEV:
-
Hepatitis E virus
- GT:
-
Genotype
- ORF:
-
Open reading frame
- ORF2:
-
Open reading frame 2
- PONDR:
-
Predictor of natural disordered regions
- MoRFs:
-
Molecular recognition features
- IDRs:
-
Intrinsically disordered regions
- ORDP:
-
Ordered protein
- IDPR:
-
Intrinsically disordered protein regions
- IDP:
-
Intrinsically disordered proteins
References
Khuroo MS (2011) Discovery of hepatitis E: the epidemic non-A, non-B hepatitis 30 years down the memory lane. Virus Res 161(1):3–14
Li P, Liu J, Li Y, Su J, Ma Z, Bramer WM, Cao W, de Man RA, Peppelenbosch MP, Pan Q (2020) The global epidemiology of hepatitis E virus infection: a systematic review and meta-analysis. Liver Int 40(7):1516–1528
MoHFW-WHO-ILBS (2016) Third GoI-WHO-ILBS National Technical Consultation on Viral Hepatitis Towards a National Action Plan for Viral Hepatitis (NAP-VH), New Delhi
Suresh K (2020) Viral Hepatitis in India. Arch Hepat Res 6(1):003–006
Meng XJ (2011) From barnyard to food table: the omnipresence of hepatitis E virus and risk for zoonotic infection and food safety. Virus Res 161(1):23–30
Yugo DM, Cossaboom CM, Heffron CL, Huang YW, Kenney SP, Woolums AR, Hurley DJ, Opriessnig T, Li L, Delwart E, Kanevsky I (2019) Evidence for an unknown agent antigenically related to the hepatitis E virus in dairy cows in the United States. J Med Virol 91(4):677–686
Sanford BJ, Emerson SU, Purcell RH, Engle RE, Dryman BA, Cecere TE, Buechner-Maxwell V, Sponenberg DP, Meng XJ (2013) Serological evidence for a hepatitis E virus (HEV)-related agent in goats in the United States. Transbound Emerg Dis 60(6):538–545
Kamar N, Selves J, Mansuy JM, Ouezzani L, Péron JM, Guitard J, Cointault O, Esposito L, Abravanel F, Danjoux M, Durand D (2008) Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N Engl J Med 358(8):811–817
Wang Y, Chen G, Pan Q, Zhao (2018) Chronic hepatitis E in a renal transplant recipient: the first report of genotype 4 hepatitis e virus caused chronic infection in organ recipient. Gastroenteroloy 154(4):1199–1201
Takahashi K, Terada S, Kokuryu H, Arai M, Mishiro S (2010) A wild boar-derived hepatitis E virus isolate presumably representing so far unidentified “genotype 5”. Kanzo 51(9):536–538
Takahashi M, Nishizawa T, Sato H, Sato Y, Nagashima S, Okamoto H (2011) Analysis of the full-length genome of a hepatitis E virus isolate obtained from a wild boar in Japan that is classifiable into a novel genotype. J Gen Virol 92(4):902–908
Rasche A, Saqib M, Liljander AM, Bornstein S, Zohaib A, Renneker S, Steinhagen K, Wernery R, Younan M, Gluecks I, Hilali M (2016) Hepatitis E virus infection in dromedaries, North and East Africa, United Arab Emirates, and Pakistan, 1983–2015. Emerg Infect Dis 22(7):1249
Woo PC, Lau SK, Teng JL, Tsang AK, Joseph M, Wong EY, Tang Y, Sivakumar S, Xie J, Bai R, Wernery R (2014) New hepatitis E virus genotype in camels, the Middle East. Emerg Infect Dis 20(6):1044
Meng XJ (2016) Expanding host range and cross-species infection of hepatitis E virus. PLoS Pathog 12(8):e1005695
Westhölter D, Hiller J, Denzer U, Polywka S, Ayuk F, Rybczynski M, Horvatits T, Gundlach S, Blöcker J, ZurWiesch JS, Fischer N (2018) HEV-positive blood donations represent a relevant infection risk for immunosuppressed recipients. J Hepatol 69(1):36–42
Teshale EH, Grytdal SP, Howard C, Barry V, Kamili S, Drobeniuc J, Hill VR, Okware S, Hu DJ, Holmberg SD (2010) Evidence of person-to-person transmission of hepatitis E virus during a large outbreak in Northern Uganda. Clin Infect Dis 50(7):1006–1010
Zeng MY, Gao H, Yan XX, Qu WJ, Sun YK, Fu GW, Yan YL (2017) High hepatitis E virus antibody positive rates in dogs and humans exposed to dogs in the south-west of China. Zoonoses Public Health 64(8):684–688
Liang H, Chen J, Xie J, Sun L, Ji F, He S, Zheng Y, Liang C, Zhang G, Su S, Li S (2014) Hepatitis E virus serosurvey among pet dogs and cats in several developed cities in China. PLoS One 9(6):e98068
Aggarwal R (2013) Diagnosis of hepatitis E. Nat Rev Gastroenterol Hepatol 10(1):24–33
Takahashi M, Kusakai S, Mizuo H et al (2005) Simultaneous detection of immunoglobulin a (IgA) and IgM antibodies against hepatitis E virus (HEV) is highly specific for diagnosis of acute HEV infection. J Clin Microbiol 43(1):49–56
Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, Fry KE, Reyes GR (1991) Hepatitis E virus (HEV): molecular cloning and sequencing of the full-length viral genome. Virology 185(1):120–131
Robinson RA, Burgess WH, Emerson SU, Leibowitz RS, Sosnovtseva SA, Tsarev S et al (1998) Structural characterization of recombinant hepatitis E virus ORF2 proteins in baculovirus-infected insect cells. Protein Expr Purif 12:75–84
Montpellier C, Wychowski C, Sayed IM, Meunier JC, Saliou JM, Ankavay M et al (2018) Hepatitis E virus lifecycle and identification of 3 forms of the ORF2 capsid protein. Gastroenterology 154:211–223.e8
Ankavay M, Montpellier C, Sayed IM, Saliou JM, Wychowski C, Saas L et al (2019) New insights into the ORF2 capsid protein, a key player of the hepatitis E virus lifecycle. Sci Rep 9:6243
He S, Miao J, Zheng Z, Wu T, Xie M, Tang M et al (2008) Putative receptorbinding sites of hepatitis E virus. J Gen Virol 89:245–224
Guu TS, Liu Z, Ye Q, Mata DA, Li K, Yin C et al (2009) Structure of the hepatitis E virus-like particle suggests mechanisms for virus assembly and receptor binding. Proc Natl Acad Sci U S A 106:12992–12997
Chen Y, Chen T, Luo Y, Fan J, Zhang M, Zhao Q et al (2020) Synthetic peptides containing three neutralizing epitopes of genotype 4 swine hepatitis E virus ORF2 induced protection against swine HEV infection in rabbit. Vaccines (Basel) 8:178
Van Der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM (2014) Classifcation of intrinsically disordered regions and proteins. Chem Rev 114:6589–6631
Giri R, Kumar D, Sharma N, Uversky VN (2016) Intrinsically disordered side of the Zika virus proteome. Front Cell Infect Microbiol 6:144
Gadhave K, Gehi BR, Kumar P, Xue B, Uversky VN, Giri R (2020) The dark side of Alzheimer’s disease: unstructured biology of proteins from the amyloid cascade signaling pathway. Cell Mol Life Sci 77(20):4163–4208
Gadhave K, Kumar P, Kapuganti SK, Uversky VN, Giri R (2020) Unstructured biology of proteins from ubiquitin-proteasome system: roles in cancer and neurodegenerative diseases. Biomolecules 10(5):796
Garg N, Kumar P, Gadhave K, Giri R (2019) The dark proteome of cancer: intrinsic disorderedness and functionality of HIF-1α along with its interacting proteins. Prog Mol Biol Transl Sci 166:371–403
Vacic V, Oldfield CJ, Mohan A, Radivojac P, Cortese MS, Uversky VN, Dunker AK (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J Proteome Res 6(6):2351–2366
Gianni S, Dogan J, Jemth P (2014) Deciphering the mechanisms of binding induced folding at nearly atomic resolution: the Φ value analysis applied to IDPs. Intrinsic Disord Proteins 2(1):e970900
Ruan H, Sun Q, Zhang W, Liu Y, Lai L (2019) Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov Today 24(1):217–227
Santofimia-Castaño P, Rizzuti B, Xia Y, Abian O, Peng L, Velázquez-Campoy A, Neira JL, Iovanna J (2020) Targeting intrinsically disordered proteins involved in cancer. Cell Mol Life Sci 77(9):1695–1707
Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z (2006) Lengthdependent prediction of protein intrinsic disorder. BMC Bioinf 7(1):1–17
Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK, Obradovic Z (2005) Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol 3(01):35–60
Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK (2001) Sequence complexity of disordered protein. Proteins Struct Funct Genet 42(1):38–48
Roy A, Xu D, Poisson J, Zhang Y (2011) A protocol for computer-based protein structure and function prediction. JoVE (Journal of Visualized Experiments) 3(57):e3259
Roy A, Zhang Y (2011) Recognizing protein-ligand binding sites by global structural alignment and local geometry refinement. Structure 20(6):987–997
Yang YL, Nan YC (2021) Open reading frame 3 protein of hepatitis E virus: Multi-function protein with endless potential. World J Gastroenterol 27(20):2458
Rajagopalan K, Mooney SM, Parekh N, Getzenberg RH, Kulkarni P (2011) A majority of the cancer/testis antigens are intrinsically disordered proteins. Journal of cellular biochemistry 112(11):3256-67
Deiana A, Forcelloni S, Porrello A, Giansanti A (2019) Intrinsically disordered proteins and structured proteins with intrinsically disordered regions have different functional roles in the cell. PLoS One 14(8):e0217889
Van der Lee R, Lang B, Kruse K, Gsponer J, de Groot NS, Huynen MA, Matouschek A, Fuxreiter M, Babu MM (2014b) Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep 8:18
Xu M, Behloul N, Wen J, Zhang J, Meng J (2016) Role of asparagine at position 562 in dimerization and immunogenicity of the hepatitis E virus capsid protein. Infect Genet Evol 37:99–107
Bhardwaj T, Saumya KU, Kumar P, Sharma N, Gadhave K, Uversky VN, Giri R (2020) Japanese encephalitis virus–exploring the dark proteome and disorder–function paradigm. FEBS J 287(17):3751–3776
Kumar D, Singh A, Kumar P, Uversky VN, Rao CD, Giri R (2020) Understanding the penetrance of intrinsic protein disorder in rotavirus proteome. Int J Biol Macromol 144:892–908
Majerciak V, Pripuzova N, Chan C, Temkin N, Specht SI, Zheng ZM (2015) Stability of structured Kaposi's sarcoma-associated herpesvirus ORF57 protein is regulated by protein phosphorylation and homodimerization. J Virol 89(6):3256–3274
Xue B, Williams RW, Oldfield CJ, Kian-Meng Goh G, Keith Dunker A, Uversky VN (2010) Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept Lett 17(8):932–951
Xue B, Williams RW, Oldfield CJ, Dunker AK, Uversky VN (2010) Archaic chaos: Intrinsically disordered proteins in Archaea. BMC Syst Biol 4(1):1–214
Nonell-Canals A, Sanchez-Martinez (2017) Intrinsically disordered proteins as drug targets. MOJ Proteomics Bioinform 5(2):00157
Neira JL, Bintz J, Arruebo M, Rizzuti B, Bonacci T, Vega S, Lanas A, Velázquez-Campoy A, Iovanna JL, Abián O (2017) Identification of a drug targeting an intrinsically disordered protein involved in pancreatic adenocarcinoma. Sci Rep 7(1):1–5
Zhou Z, Xie Y, Wu C, Nan Y (2021) The hepatitis E virus open reading frame 2 protein: beyond viral capsid. Frontiers in Microbiology 12:739124
Shafat Z, Ahmed A, Parvez MK, Parveen S (2021) Role of “dual-personality” fragments in HEV adaptation—analysis of Y-domain region. J Genet Eng Biotechnol 19(1):1–21
Shafat Z, Ahmed A, Parvez MK, Parveen S (2021) Role of ORF4 in Hepatitis E virus regulation: analysis of intrinsically disordered regions. J Proteins Proteomics 12(4):289–306
Shafat Z, Ahmed A, Parvez MK, Parveen S (2021d) Shedding light on the dark proteome of Hepatitis E Virus. Netw Biol 11(4):295–314
Shafat Z, Ahmed A, Parvez MK, Islam A, Parveen S (2022) The dark proteome of rodent hepatitis E virus: analysis of intrinsically disordered regions. Arch Hepat Res 8(1):005–011
Shafat Z, Ahmed A, Parvez MK, Islam A, Parveen S (2022) Intrinsically disordered regions in the rodent hepevirus proteome. Bioinformation 18(2):111–118
Shafat Z, Ahmed A, Parvez MK, Parveen S (2021) Sequence to structure analysis of the ORF4 protein from Hepatitis E Virus. Bioinformation 17(9):818–828
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers: Orig Res Biomol 22(12):2577–2637
Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB (2003) Protein disorder prediction: implications for structural proteomics. Structure 11(11):1453–1459
Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK (2003) Predicting intrinsic disorder from amino acid sequence. Proteins: Struct, Funct Bioinf 53(S6):566–572
Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK (2005) Comparing and combining predictors of mostly disordered proteins. Biochemistry 44:1989–2000
Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK (2007) Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 46:13468–13477
Jones DT, Cozzetto D (2015) DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 31(6):857–863
Xue B, Blocquel D, Habchi J, Uversky AV, Kurgan L, Uversky VN, Longhi S (2014) Structural disorder in viral proteins. Chem Rev 114(13):6880–91119
Mishra PM, Verma NC, Rao C, Uversky VN, Nandi CK (2020) Intrinsically disordered proteins of viruses: involvement in the mechanism of cell regulation and pathogenesis. Prog Mol Biol Transl Sci 174:1
Foster TL, Belyaeva T, Stonehouse NJ, Pearson AR, Harris M (2010) All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J Virol 84(18):9267–9277
Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, Yanagi Y (2009) The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J Virol 83(20):10374–10383
Habchi J, Longhi S (2012) Structural disorder within paramyxovirus nucleoproteins and phosphoproteins. Mol Biosyst 8(1):69–81
Purdy MA, Lara J, Khudyakov YE (2012) The hepatitis E virus polyproline region is involved in viral adaptation. PLoS One 7(4):e35974
Lin S, Yang Y, Nan Y, Ma Z, Yang L, Zhang YJ (2019) The Capsid Protein of Hepatitis E Virus Inhibits Interferon Induction via Its N-terminal Arginine-Rich Motif. Viruses 11:1050
Hervouet K, Ferrié M, Ankavay M, Montpellier C, Camuzet C, Alexandre V, Dembélé A, Lecoeur C, Foe AT, Bouquet P, Hot D (2022) An Arginine-Rich Motif in the ORF2 capsid protein regulates the hepatitis E virus lifecycle and interactions with the host cell. PLoS Pathogens 18(8):e1010798
Meng F, Badierah RA, Almehdar HA, Redwan EM, Kurgan L, Uversky VN (2015) Unstructural biology of the dengue virus proteins. FEBS J 282(17):3368–3394
Sharma NR, Gadhave K, Kumar P, Saif M, Khan MM, Sarkar DP, Uversky VN, Giri R (2021) Analysis of the dark proteome of Chandipura virus reveals maximum propensity for intrinsic disorder in phosphoprotein. Sci Rep 11(1):1–7
Mishra PM, Uversky VN, Giri R (2018) Molecular recognition features in Zika virus proteome. J Mol Biol Mol Boil 430(16):2372–2388
Mészáros B, Erdős G, Dosztányi Z (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46(W1):W329–W337
Erdős G, Pajkos M, Dosztányi Z (2021) IUPred3: prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res 49(W1):W297–W303
Nan Y, Wu C, Zhang YJ (2017a) Interplay between janus kinase/signal transducer and activator of transcription signaling activated by type i interferons and viral antagonism. Front Immunol 8:1758
Cordoba L, Feagins AR, Opriessnig T, Cossaboom CM, Dryman BA, Huang YW, Meng XJ (2012) Rescue of a genotype 4 human hepatitis E virus from cloned cDNA and characterization of intergenotypic chimeric viruses in cultured human liver cells and in pigs. J Gen Virol 93:2183
Nguyen HT, Shukla P, Torian U, Faulk K, Emerson SU (2014) Hepatitis E virus genotype 1 infection of swine kidney cells in vitro is inhibited at multiple levels. J Virol 88(2):868–877
Keck F, Ataey P, Amaya M, Bailey C, Narayanan A (2015) Phosphorylation of single stranded RNA virus proteins and potential for novel therapeutic strategies. Viruses 7(10):5257–5273
Zor T, Mayr BM, Dyson HJ, Montminy MR, Wright PE (2002) Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J Biol Chem 277(44):42241–42248
Marks F (1996) Protein phosphorylation. VCH Weinheim, New York, Basel, Cambridge, Tokyo
Foy NJ, Akhrymuk M, Akhrymuk I, Atasheva S, Bopda-Waffo A, Frolov I, Frolova EI (2013) Hypervariable domains of nsP3 proteins of New World and Old World alphaviruses mediate formation of distinct, virus-specific protein complexes. J Virol 87(4):1997–2010
Vihinen H, Ahola T, Tuittila M, Merits A, Kääriäinen L (2001) Elimination of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J Biol Chem 276(8):5745–5752
Lin RJ, Chang BL, Yu HP, Liao CL, Lin YL (2006) Blocking of interferoninduced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J Virol 80(12):5908–5918
Bhattacharya D, Mayuri BSM, Perera R, Kuhn RJ, Striker R (2009) Protein kinase G phosphorylates mosquito-borne flavivirus NS5. J Virol 83(18):9195–9205
Forwood JK, Brooks A, Briggs LJ, Xiao CY, Jans DA, Vasudevan SG (1999) The 37-amino-acid interdomain of dengue virus NS5 protein contains a functional NLS and inhibitory CK2 site. Biochem Biophys Res Commun 257(3):731–737
Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 32(3):1037–1049
Collins MO, Yu L, Campuzano I, Grant SG, Choudhary JS (2008) Phosphoproteomic analysis of the mouse brain cytosol reveals a predominance of protein phosphorylation in regions of intrinsic sequence disorder. Mol Cell Proteomics 7(7):1331–1348
Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, Travé G, Gibson TJ (2008) Understanding eukaryotic linear motifs and their role in cell signaling and regulation. J Front Biosci 13(6580):603
Galea CA, Wang Y, Sivakolundu SG, Kriwacki RW (2008) Regulation of cell division by intrinsically unstructured proteins: intrinsic flexibility, modularity, and signaling conduits. Biochemistry 47(29):7598–7609
Rajagopal, K. A., Indira, & Tan, T. (2021) Structure & function - amino acids. from https://bio.libretexts.org/@go/page/7809
Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6(3):197–208
Wang T, Weinman SA (2013) Interactions between hepatitis C virus and mitochondria: impact on pathogenesis and innate immunity. Curr Pathobiol Rep 1(3):179–187
Mills EL, Kelly B, O'Neill LA (2017) Mitochondria are the powerhouses of immunity. Nat Immunol 18(5):488–498
Qu C, Zhang S, Wang W, Li M, Wang Y, van der Heijde-Mulder M, Shokrollahi E, Hakim MS, Raat NJ, Peppelenbosch MP, Pan Q (2019) Mitochondrial electron transport chain complex III sustains hepatitis E virus replication and represents an antiviral target. FASEB J 33(1):1008–1019
Ma X, Jin M, Cai Y, Xia H, Long K, Liu J, Yu Q, Yuan J (2011) Mitochondrial electron transport chain complex III is required for antimycin A to inhibit autophagy. Chem Biol 18(11):1474–1481
Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG (2010) Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci U S A 107(29):12828–12833
Acknowledgements
The authors would like to acknowledge Maulana Azad National Fellowship (MANF), University Grant Commission (UGC), Council of Scientific and Industrial Research (CSIR) (37(1697)17/EMR-II), and Central Council for Research in Unani Medicine (CCRUM), Ministry of Ayurveda, Yoga and Neuropathy, Unani, Siddha, and Homeopathy (AYUSH) (F.No.3-63/2019- CCRUM/Tech) supported by the Government of India.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SP conceptualized the research. SP and ZS designed the manuscript. ZS was a major contributor in writing the manuscript and performed the biocomputational analysis of the protein. KP and AA proofread the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shafat, Z., Ahmed, A., Parvez, M.K. et al. Intrinsic disorder in the open reading frame 2 of hepatitis E virus: a protein with multiple functions beyond viral capsid. J Genet Eng Biotechnol 21, 33 (2023). https://doi.org/10.1186/s43141-023-00477-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00477-x