
Abstract
Background
Non-synchronized pods shattering in the Brassicaceae family bring upon huge yield losses around the world. The shattering process was validated to be controlled by eight genes in Arabidopsis, including SHP1, SHP2, FUL, IND, ALC, NAC, RPL, and PG. We performed genome-wide identification, characterization, and expression analysis of shattering genes in B.napus and B. juncea to gain understanding into this gene family and to explain their expression patterns in fresh and mature siliques.
Results
A comprehensive genome investigation of B.napus and B.juncea revealed 32 shattering genes, which were identified and categorized using protein motif structure, exon-intron organization, and phylogeny. The phylogenetic study revealed that these shattering genes contain little duplications, determined with a distinct chromosome number. Motifs of 32 shattering proteins were observed where motifs1 and 2 were found to be more conserved. A single motif was observed for other genes like Br-nS7, Br-nS9, Br-nS10, Br-jS21, Br-jS23, Br-jS24, Br-jS25, and Br-jS26. Synteny analysis was performed that validated a conserved pattern of blocks among these cultivars. RT-PCR based expressions profiles showed higher expression of shattering genes in B. juncea as compared to B.napus. SHP1, SHP2, and FUL gene were expressed more in mature silique. ALC gene was upregulated in fresh silique of B. napus but downregulation of ALC were observed in fresh silique of B. juncea.
Conclusion
This study authenticates the presence of shattering genes in the local cultivars of Brassica. It has been validated that the expression of shattering genes were more in B. juncea as compared to B.napus. The outcomes of this study contribute to the screening of more candidate genes for further investigation.
Similar content being viewed by others


Background
Brassicaceae is one of the important families, with ~ 360 genera and 3700 species around the globe [1]. Species from this family are very significant from an economic, and agricultural point of view. A few examples of species from this family are Brassica napus, Brassica juncea (oilseed crops); Brassica oleracea (cabbage, cauliflower, kale, broccoli); Brassica rapa (turnip, leaf vegetable); Raphanus sativus (vegetable); and Arabidopsis thaliana (model plant). The most cultivated species of the Brassica genus includes those with three diploid genomes like B. nigra (BB, 2n =16) B. oleracea (CC, 2n = 18), and B. rapa (AA, 2n = 20), together with three amphidiploid species like B. napus (AACC, 2n = 38) B. carinata (BBCC, 2n = 34), and B. juncea (AABB, 2n = 36). Hybridization and cytogenetic studies have determined that amphidiploid species are natural hybrids of diploids and all species are interconnected [2].
Non-synchronous pod shattering remained a major problem of Brassica that results in yield loss. It also causes seed loss due to the dispersion of a silique following complicated physiological and biological mechanisms [3]. However, premature and unsynchronized pods shattering like the dehiscence results in a huge loss in crop yield [4]. Pod’s shattering occurs when the adhesions among walls change into fragile and internal forces apply them to the moveable position [5]. Seed valves are responsible for the attachment and internal force creation that contributes to the necessary protection of the seed [6]. Seeds of B. juncea and B. napus are very important equally 14% of oil around the world is produced by these crops. Moreover, rapeseed is considered the third most important oilseed crop worldwide [7]. Distinct nutrients and biological molecules are reported to be involved in the evolution of shattering in canola [8].
Previous investigation over shattering revealed that shattering occurred because of molecular components excess production and enrichment in the valve margin and cellar portion around the pods in siliques. Lignin and cellulose play a key role in the hardening of pod walls, which lowers water content during the later development stages of rapeseed and Brassica species [9]. The shattering mechanism of B. napus and B. juncea are controlled by eight different genes, like SHATTERPROOF1 (SHP1), SHATTERPROOF2 (SHP2) FRUITFULL (FUL), INDEHISCENT (IND), ALCATRAZ (ALC), NAC, (NST1 and NST2) REPLUMLESS (RPL), and POLYGlACTOURANAZE [10]. For the development of shattering genes, distinct transcription factor binding sites are involved which are important both structurally and functionally [11]. Other genes like SHP1/2, FUL, IND, ALC, NAC, RPL, and PG of canola and Indian mustard were also reported [12]. In one study, a comparative analysis was performed to unveil the genomic maintenance for the evolutionary and functional correlation among shattering genes SHP1/2, FUL, IND, ALC, NAC, RPL, and PG [13] having functional and genetic conservation among them. The pattern of conservation in these shattering gene sequences was also found with comparative synteny approach by Krzywinski et al. [14].
The most desirable solution to the shattering problem of B. napus and B. juncea is to delay pod shattering by knocking out SHPS genes and stimulating the expression of FUL up to the susceptible crop is ready for harvesting. Therefore, before developing genome modified plants it is essential to study these genes elaborately in local plants B.napus and B. juncea. Therefore, this study was carried out to identify the orthologous of shattering genes in the local cultivars of B. napus and B. juncea and to study their expression pattern in fresh and mature siliques. This study further identified the syntenic and evolutionary relationship of shattering genes in the studied cultivars based on phylogenetic analysis with NJ algorithm.
Methods
Identification of shattering genes
BRAD database (http://brassicadb.org/brad/) was used to retrieve protein, genomic, CDS and cDNA sequences of the 8 shattering genes SHP1/2, FUL, ALC, NAC, IND, RPL, and PG and their orthologs in B.napus and B. juncea [15]. Other databases like NCBI (https://www.ncbi.nlm.nih.gov/), TAIR (https://www.arabidopsis.org/), and Plants Ensembl (http://plants.ensembl.org/) were also consulted. A web tool from EMBL was used to identify different protein domains (http://smart.embl.de/smart/set_mode.cgi). The Basic local alignment search tool (BLAST) (htpp://www.ncbi.nlm.nih.gov/BLAST/) was used to search the homology of the shattering genes in B.napus and B.juncea. ProtParam tool was used to study the primary structure of shattering genes (http://expasy.org/tools/protparam.html). The gene structure display server (GSDS) web tool was used to align the CDS sequences of shattering genes with genomic sequences to identify exons and introns [16].
Phylogenetic analysis of shattering proteins
B. napus and B. juncea shattering protein sequences were obtained from the BRAD database using reference sequences of shattering genes obtained from the TAIR database and the other plants protein sequences were obtained from the NCBI database and then aligned using the Clustal X program [17]. Using the Neighbor Joining (NJ) algorithm, a phylogenetic tree was constructed with MEGA 11 software [18]. The implication of nodes was calculated using a bootstrap study of 1000 replicates. For the surety of different domains that show the topology of NJ tree, pairwise gape deletion mode was used.
Analysis of conserved motifs in shattering proteins
MEME software (Multiple Em for Motif Elicitation, V4.9.0) was used to analyze MADS-box shattering genes protein sequences as described by Bailey et al. [19]. MEME search was run with the following parameters: (1) maximum number of motif identification = 10; (2) optimum motif width > 6 and < 200.
Chromosomal locations analysis
To find out identical genes, all shattering genes of B. napus and B. juncea were BLAST searched (htpp://www.ncbi.nlm.nih.gov/BLAST/) against each other, with a query coverage and similarity percentage of candidate genes of more than 80% [20]. The Brassica database (http:// brassicadb.org/brad/) was used to acquire positional information for all putative shattering genes along the 10 chromosomes of B. napus and B. juncea [15]. All genes were mapped along the 10 chromosomes, and the gene’s location was observed.
Analysis of syntenic relationships
The comparative genomic synteny was performed to find the relationship among distinct shattering genes like SHP1/2, FUL, ALC, NAC, IND, RPL, and PG in B.napus and B. juncea using the circoletto program; genome visualization tool circoletto [14].
Plant collection and sample preparation
Seeds of two Brassica varieties canola (Punjab Sarson) and Indian mustard (Super Raya) were collected from the plant Bioresources Conservation Institute (BCI) and Crop Sciences Institute (CSI) of the National Agricultural Research Centre (NARC), Islamabad Pakistan. The seeds were cultivated at National Institute for Genomics and Advanced Biotechnology (NIGAB), National Agriculture Research Centre (NARC), Islamabad, Pakistan under pods in a glasshouse. Forty days old samples of pre-mature and mature siliques of B. napus (Punjab Sarson) and B. juncea (Super Raya) were collected and stocked at – 80 °C for gene expression analysis. Morphological analysis was performed, and the data of plants were recorded in triplicates.
RNA extraction and cDNA synthesis
Total RNA from the fresh and mature siliques of B. napus and B. juncea was extracted using a Pure LinkTM RNA Mini kit (Invitrogen). The RNAs were quantified by using BioSpec-nano Micro-volume UV-Vis Spectrophotometer (Shimadzu). The quality and integrity of RNA was checked on 1.5% agarose gel. cDNA was synthesized by using RevertAidTM reverse transcriptase enzyme (FermentasTM Cat.No. K1621) following the manufacturer’s guidelines.
Expression analysis of Shattering genes qRT-PCR
The expression pattern of shattering genes (SHP1/2, FUL, ALC, NAC, RPL, PG, and IND) was determined in fresh and mature silique of B. napus and B. juncea using comparative ΔCT method in real-time PCR (Applied Biosystems) with StepOnePlus software. For the execution of a relative expression, the Elongation factor (EF) was used as endogenous control. No template control (NTC) was also used as negative control in the assay. In total, 10 μl reaction volume, 5 μl Maxima SYBER Green (Thermo Fisher) genes specific primers (1 pmol of each), and 1 μl cDNA as a template were used. Real-time PCR conditions set were; denaturation at 94 °C for 10 min, the second stage followed by 40 cycles at 95 °C for 40 s, 58 °C for 32 s 72 °C for 32 seconds. Finally, a melt curve study was carried out at 52 °C to 95 °C. The statistical analysis of results was carried out by mean of relative fold expression of transcript ± standard deviation (SD). All the primers used in the qRT-PCR analysis listed in Table 1 were designed manually through the conserved region from the A and C subgenome of B. napus. The length of the amplified fragment ranged between 100 and 130 bp.
Results
Identification and sequence analysis of shattering genes
A set of 32 individual shattering genes orthologues from B. napus and B. juncea genome was retrieved and their annotations were checked using keyword gene id to search Swissport annotations at the Brassica database (BRAD) (http://brassicadb.org/brad/). These genes were in greater number than those of model plant Arabidopsis thaliana as shown in Tables 2 and 3. The domain of these shattering genes was also identified using EMBL (http://smart.embl.de/smart/set_mode.cgi). The first six shattering genes of B. napus (Br-nS1-Br-nS6) contain the MADS-box domain whereas, 7–10 contain HLH, 11, 12, Pfam, 13, 14 pox/Hox 15–17 contain PbH1 domain. In B. juncea, 18–22 contain MADS-box domain while 23–26 HLH, 27, 28 Pfam, 29, 30 pox/Hox and 31, 32 PbH1 domain. We selected 32 annotated shattering genes of B. napus and B. juncea as Br-nS1 and Br-jS1 followed by Arabic numbers 1–32.In total shattering genes, 11 were mad box genes having mad box domain whereas the other 21 genes did not possess mad box domain. The genes that lacked mad box domain shared a large sequence resemblance with mad box protein of other crop varieties that also lack this domain and are considered to be mad box or shattering genes. Sequence analysis showed that all shattering genes of B. napus and B. juncea have introns except the gene IND. The maximum numbers of introns were identified in MADS-box shattering genes. These appearances are persistent with shattering genes previously determined in Arabidopsis thaliana and Brassica rapa.
Phylogenetic analysis of shattering genes
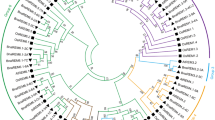
The identified shattering genes protein sequences were used to analyze the phylogenetic relationship of the shattering gene family in B. napus, B. juncea, Arabidopsis, citrus, tomato, wheat, cotton, and rice. The unrooted phylogenetic tree characterizes the length of clades and the level of the evolutionary relationship with well-supported bootstrap values. The sequences of shattering genes SHP1, SHP2, FUL, IND, ALC, NAC, RPL, PG and their orthologous determined into B. juncea, B. napus, Arabidopsis, citrus, tomato, wheat, cotton and rice were aligned to generate the NJ phylogenetic tree (Fig. 1). Every individual shattering gene organized in a distinct clade with various color, characterize their functional and sequential conservation. The light green and dark brown colour in the tree is Clade I which contains a duplication of SHP1 and SHP2 in various plant like B. napus, B. juncea, citrus, tomato, wheat, cotton and rice except SHP2 gene where no duplication was observed in B. napus and B. juncea plants. However, clade II with yellow color consists of FUL genes where duplications were observed. This shows that clades I and II are closely related to each other as compared to other clades. Clade III indicated with blue color, shows duplication and triplication of IND and ALC in B. juncea, B. napus and other plants like citrus, tomato, wheat, cotton and rice that indicates divergence in sequences and clade IV with red colour, duplication of NAC genes was observed. It is clear from the resulting tree that clade III and clade IV are closely related to clade I and II. Similarly, clade V with purple colour and clade IV with dark blue colour contains PG and RPL genes in a duplicated form in B. napus, B. juncea, and all other plants. The clade comprising SHP1 and SHP2 genes contains a greater number of genes as compared to others. Genes from these two clades are present on different chromosomes indicate that every individual gene bear duplication and whole genome triplication events before reaching this level. Environmental, physiological, and chromosomal rearrangement at the development level brought changes in the genome. These results authenticate that every individual gene of B. napus, B. juncea, citrus, tomato, wheat, cotton, and rice under observation are shattering genes having a close resemblance to each other and with a model plant Arabidopsis thaliana as shown in Fig. 1.

Neighbor joining consensus phylogenetic tree of 181 shattering genes from B. napus (17), B. juncea (15), Arabidopsis (At 8), citrus (Ct 30), cotton (Cn 29), rice (Ri 27), wheat (Wt 35), and tomato (TO 20)
Gene structure organization and conserved motifs analysis of shattering proteins
We compared the coding DNA sequences of exons and introns to their genomic DNA sequences to facilitate phylogenetic reconstruction. As shown in Fig. 2a, the distribution, number, and length of exons and introns were not highly diverse among all genes. Br-nS1 was the longest sequence and Br-nS10 was the shortest among all shattering genes. Br-nS1 contain 8 introns. While Br-nS2, Br-jS18, and Br-jS19 contain six introns. Br-nS4, Br-nS5, and Br-nS6 contain seven introns while in B. juncea divergence is found Br-jS21 contains 4 and Br-jS22 contains 7 introns for the same gene. Br-nS9 contain a single intron, whereas the same gene Br-nS10, Br-jS25 and Br-jS26 didn’t contain any introns and showed a divergence in the gene sequence. Br-nS13 and Br-nS14 contain four introns whereas the same genes in B. juncea, Br-jS29, and Br-jS30 contain three introns showing a clear difference among B. napus and B. juncea. Similarly, Br-nS15, Br-nS16, and Br-nS17 genes contain three introns, while in other species B. juncea Br-jS31 contain three and Br-jS32 contains two introns for the same genes, which also showed some variance in genes sequences.

a Exon–intron structure of shattering genes in B. napus and B. juncea. Exons are shown as yellow boxes, introns are shown as a thin black line, and UTRs are shown as blue boxes. b Motif distribution analysis, 10 motifs are shown as colored boxes
MEME (Multiple Em for Motif Elicitation) motif search tool was used to identify 10 conserved motifs of 32 shattering protein sequences of B. napus and B. juncea (Fig. 2b). Motifs 1 and 2 exhibit the MADS-box domain which was found in 11 genes whereas other shattering genes did not show motif 1 or 2 features. The genes which exhibit the characteristics of motifs 1 or 2 were Br-nS1-Br-nS6 and Br-jS18-Br-jS22. These genes did not contain other representative motifs of Mads-box family such as motifs 4, 5, 6, 7, 8, 9, and 10. Motif 4 and 5 comprised of PbH1 domain found in 5 genes which were Br-nS15, Br-nS16, Br-nS17, Br-jS31, and Br-jS32. Br-nS7, Br-nS9, Br-nS10, Br-jS21, Br-jS23 Br-jS24, Br-jS25, and Br-jS26 genes consists of single motif whereas Br-nS8 gene did not contain any motif. Motif 8 and 10 showed pox/Hox domain which was found in Br-nS13, Br-nS14, Br-jS29, and Br-jS30 gene. Br-nS15, Br-nS16, Br-nS17, Br-jS31, and Br-jS32 comprised PbH1 domain with motif 5 and 6 features. Motif 1 and Motif 2 were conserved among genes which is the characteristic feature of shattering genes. The different motifs are represented by different colors that showed similarities among B. napus and B. juncea as shown in (Fig. 2b). The number of motifs found in both species is similar except for Br-nS7, Br-nS9, Br-nS10, Br-jS21, Br-jS23 Br-jS24, Br-jS25, and Br-jS26 which shows single motif and revealed similarities and differences with other shattering genes among brassica species.
Chromosomal distributions of shattering genes
According to the available Brassica genome database, 32 shattering genes were mapped onto 10 chromosomes of B. napus and B. juncea. The chromosome localization of Brassica gene ID was confirmed by ensemble Plant Browser. There were 17 shattering genes distributed on both the A and C subgenome of B. napus while on the B and A sub-genome of B. juncea, 15 shattering genes were observed. Similarly, on the A subgenome of both plants, a total of 13 genes were identified and on the B sub-genome of B. juncea, 11 genes were observed, while on the C genome of B. napus, 8 genes were observed.
According to our results, Br-nS4 and Br-nS10 genes were observed on similar chromosome A03, whereas Br-nS3 lies on chromosome A05. Similarly, Br-nS1 and Br-jS24 were identified on the same chromosome A07, while Br-nS17 and Br-jS31 were located on chromosome A08. Genes like Br-nS5 and Br-nS16 were located on the A09 chromosome, whereas Br-nS11, Br-nS13, Br-jS27, and Br-jS30 were observed on chromosome A10. Similarly, on chromosome B01, gene Br-jS20 was observed while on the B02 chromosome, gene Br-jS22 was located. Br-jS28 and Br-jS32 genes were identified on the same chromosome B03, whereas on B04 chromosome Br-jS21 gene was located. Hence genes Br-jS18, Br-jS19, and Br-jS23 were observed on similar chromosome B06. Similarly, on the B08 chromosome, genes like Br-jS25, Br-jS26, and Br-jS29 were identified. The genes observed on chromosome C02 were Br-nS6, Br-nS8 and Br-nS14, whereas on other chromosomes like C03, C05, C06, C07, and C08, genes located were Br-nS9, Br-nS12, Br-nS2, Br-nS7, and Br-nS15 respectively. Hence, all the shattering genes were scattered on Brassica chromosomes as shown in Figs. 3 and 4.

Logos of tens motifs discovered in shattering genes

Gene localization of shattering genes on B. napus and B. juncea chromosomes
Syntenic relationship among shattering genes of B. napus and B. juncea
Comparative genomic synteny analysis was performed by circoletto tool (tools.bat.inspire.org/circoletto/) for genome conservation visualization. The orthologs’ relationship and conservation were determined for the shattering gene family in B. napus and B. juncea. Synteny diagram represents a remarkable relationship among these species in the context of duplication, triplication, evolution, function, and expression (Fig. 5) showed a unique relationship among B. juncea and B. napus. It was observed that B. napus Br-nS13 and Br-nS14 gene sequence showed synteny with B. juncea sequence Br-jS29 and Br-jS30, while B. napus gene sequence Br-nS15, 16, and 17 showed synteny with B. juncea gene sequence Br-jS31, 32 and gene sequence Br-nS11 and 12 showed synteny with Br-jS27 and Br-jS28. In Addition, Br-nS7 and Br-nS8 gene sequence showed synteny with Br-jS23 and Br-jS24 gene sequences while Br-nS9 and Br-nS10 showed synteny with Br-jS25 and Br-jS26 gene sequences. Similarly, Br-nS1 and Br-nS2 showed synteny with Br-jS18 and Br-jS19 gene sequences, while Br-nS3 showed synteny with Br-jS20. B. napus gene Br-nS4, 5, 6 sequences showed synteny with Br-jS21 and Br-jS22. In comparative synteny analysis inward tangling ribbons color intensity exhibited the rate of conservation while outward tangling ribbons showed duplication events. Genomic dynamicity and evolutionary improvement along mobile elements in the genome of B. napus and B. juncea were determined in syntenic circles. In chromosomal shuffling, duplication, and triplication mobile elements play an important role. A permanent position was adopted by the blocks at a specific position in genome initiate expression that involve another biological pathway disturbance (Fig. 5).

Representation of synteny of B. napus and B. juncea identifying the level of conservation at the sequence level in 4 colors. The red, green, orange, and blue colors signify the level and intensity of evolutionary conservation among distinct shattering genes, e.g. maximum intensity is from orange to green
qRT-PCR expression of shattering genes in fresh and mature siliques
The expression levels of shattering genes in fresh and mature siliques of B. napus and B. juncea were confirmed by qRT-PCR. Our results inferred that the expression level of shattering genes was higher in B. juncea as compared to B. napus in both fresh and mature siliques. Strong signals of shattering genes were observed in mature siliques in both species, while in fresh silique, the transcripts levels were low (Fig. 6). The correlation is completely noticeable in the evidence that shattering genes play a major role in shattering associated pathways by devoting to developmental pathways of lignification and valve margin associated transcriptional activity. Moreover, ALC gene expression was upregulated in fresh silique of B. napus while down regulation of ALC gene was observed in fresh silique of B. juncea. Similarly, higher expression of ALC genes was observed in mature silique of B. juncea compared to B. napus. PG gene downregulation was observed in fresh silique of B. napus while it expressed more in B. juncea. The expression of SHP1, SHP2, FUL and RPL were observed more in B. juncea in both fresh and mature siliques showed a difference expression patterns.

The expression levels of shattering genes in B. napus and B. juncea: Graph bar defining the difference and the correlation in the expression level among two tissues of B. napus and B. juncea. A1 A lower level of shattering genes expression in B. napus than B. juncea. A2 A higher level of shattering genes expression in B. juncea than B. napus in the given tissues. ALC gene expressed more in B. napus fresh silique. Lower expression of ALC gene was observed in fresh silique of B. juncea but highly expression was observed in mature silique
Discussion
Brassicaceae is a large plant family consists of ~ 338 genera and 3700 species, important both economically and agriculturally [19]. In addition to this, plants of this family are grown like a weed in different parts of the world including North America, South America, and Austria [21]. Arabidopsis thaliana, a model plant from the family Brassicaceae was the first plant to be entirely sequenced [21]. Plants and vegetables from this family offer essential food nutrients to human and other animals. Due to their great importance, all the Brassica plants have common and commercial value with a positive influence on earth and manhood. Brassica species have inconstant traits and morphological differentiation revealing that the genome of this family is very vibrant and endured a lot of rearrangement and evolutionary measures [22].
In this study, SHP1, SHP2, FUL, IND, ALC, NAC, RPL, and PG when compared at the genomic level showed close similarity. Protein and nucleotide shows an important correlation at the sequence level. It has been showed that these genes are responsible in shattering and seed development of plants [23]. The phylogenetic analysis here showed that SHP1 as compared to other genes have fever dynamicity which is balanced in the connection of genomics but bear duplication. The duplicated genes determined with a distinct chromosome number in B. napus and B. juncea which recommended genomic flexibility as previously reported in Arabidopsis and B. rapa [24, 25] shows similar results with our investigations. SHP2 shattering gene study uses a novel approach to phylogenetic analysis bears no duplication and triplication as previously reported in other Brassica species [26, 27]. FUL is known for fruit development in different Brassica species. The Phylogenomics of FUL affords unusually different results than SHP1 and SHP2. The behavior observed more dynamics among the various species of Brassica family. FUL genes showing duplication and differential location in the genome of B. juncea and B. napus also previously described in B. rapa further strengthen our results [8].
In current research, we have study 32 shattering gene of B. juncea and B. napus which are more in number than the shattering genes reported for A. thaliana [28]. The syntenic analysis performed among B. napus and B. juncea shows the similar sequence feature and whole genome of both species go through triplication events since its divergence from Arabidopsis. The evolutionary and syntenic relationships among Arabidopsis and B. rapa is also supporting our results [29]. On the other hand, we observed the expression of shattering genes SHP1, SHP2, FUL, IND, ALC, NAC, RPL, and PG in B. napus and B. juncea like previously reported in Arabidopsis [30]. Our result also suggest that these genes are the reputed orthologous of Arabidopsis genes AGL1, AGL5, AGL8, AT5G67110, EDA33, At5g22380, BLH9, and At1g45015 might play the similar role, and they are expressed in both plants in fresh and mature siliques.
In previous studies, divergence in expression pattern was observed in shattering genes in B. napus. Wu et al. [31] determined the expression patterns and evolution of MADS-box TF family in B. napus. Becker and Theissen [32] reported that Shatterproof1/2 and genes which are members of MADS box family are engaged in controlling this pod shattering issue. SHP1 and SHP2 genes are involved in opening of silique in B. napus plants when the expression level is low [23, 33, 34].
The expression of these genes started from developed flower to mature silique with lower expression in the late stage of development of seed [31]. SHP1, SHP2, and FUL showed a relationship with IND, ALC that initiate acting to abrogate activity of DZ to forbid dehiscence at the time of seed formation follow indehiscence in the existence of multiple regulatory genes. The present analysis of all shattering genes showed different expression pattern in different tissues such as fresh and mature siliques of both plants as previously reported in Arabidopsis and B. rapa. These genes were expressed in both plant tissues, although in B. juncea they were slightly higher than in B. napus. These different expressions of shattering genes shows that they are important for cellular valve and margin evolution [24, 35].
A similar study was conducted by Yasin et al. [36], Ahmad et al. [37], & Khan et al. [38] whose results agree with our results. They demonstrated higher expression of FUL gene in mature aerial part silique plant as compared to leaves and flowers of B. napus plants. Similarly, SHP1 and SHP2 transcripts were expressed in flower silique whereas; no expression was detected in the leaves. Our findings showed basic gene expression information about shattering cascade genes which can be useful for developing genome edited brassica plants which are resistant to shattering.
Conclusion
Conclusively, different orthologous of shattering genes are exists in the local cultivars of Brassica. After comparative phylogenetic study, molecular gene characteristics, motifs/domain identifications, and comparative expression study, it is validated that the sequences were conserved across B. napus, B. juncea as well as in Arabidopsis plant. The redundant expression was observed in fresh and mature siliques of both cultivars. The different expression patterns of shattering genes are also helpful to study the nature of both plants and their pathways related to transcription and regulation. Further analysis of shattering genes is required to uncover their functions involved in the regulation of different pathways.
Availability of data and materials
Not applicable.
Change history
26 September 2022
A Correction to this paper has been published: https://doi.org/10.1186/s43141-022-00419-z
Abbreviations
- μl:
-
Microliter
- μM:
-
Miro molar
- A01:
-
Chromosome number for Brassica A genome
- At:
-
Arabidopsis thaliana
- BJ:
-
Brassica juncea
- BLAST:
-
Basic Local Alignment Search Tool
- Bn:
-
Brassica napus
- Bp:
-
Base pairs
- C01:
-
Chromosome number of Brassica C genome
- cDNA:
-
Complementary deoxyribonucleic acid
- cm:
-
Centimetre
- Ct:
-
Cycle threshold
- NARC:
-
National Agricultural Research Centre
- NCBI:
-
National Centre for Biotechnology Information
- MEGA:
-
Molecular Evolutionary Genetic Analysis
- GL:
-
Glucosinolates
References
Warwick SI, Francis A, Al-Shehbaz IA (2006) Brassicaceae: species checklist and database on CD-ROM. Plant Syst Evol 259:249–258
U, N (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J Bot 7:389–452
Dong Y, Wang YZ (2015) Seed shattering: from models to crops. Front Plant Sci 6:476
Raman R, Raman H, Kadkol GP, Coombes N, Taylor B, Luckett D (2011) Genome wide association analyses of loci for shatter resistance in Brassicas. In: Wagga Wagga NSW (ed) Proceedings of the 11th Australian research assembly on brassicas (ARAB) conference, pp 36–41
Raman H, Raman R, Kilian A, Detering F, Carling J, Coombes N, Diffey S, Kadkol G, Edwards D, McCully M (2014) Genome-wide delineation of natural variation for pod shatter resistance in Brassica napus. PLoS One 9(7):e101673
Balanzà V, Roig-Villanova I, Di Marzo M, Masiero S, Colombo L (2016) Seed abscission and fruit dehiscence required for seed dispersal rely on similar genetic networks. Development 143(18):3372–3381
Basalma D (2008) The correlation and Path analysis of yield and yield components of different winter rapeseed (Brassica napus ssp. oleifera L.) cultivars. Res J Agric Biol Sci 4:120–125
Hu Z, Yang H, Zhang L, Wang X, Liu G, Wang H, Hua W (2015) A large replum-valve joint area is associated with increased resistance to pod shattering in rapeseed. Int J Plant Res 128(5):813–819
Schiessl S, Huettel B, Kuehn D, Reinhardt R, Snowdon R (2017) Post-polyploidisation morphotype diversification associates with gene copy number variation. Sci Rep 7:41845
Zumajo-Cardona C, Ambrose BA, Pabón-Mora N (2017) Evolution of the SPATULA/ALCATRAZ gene lineage and expression analyses in the basal eudicot, Bocconia frutescens L. (Papaveraceae). EvoDevo 8(1):5
Cosio C, Dunand C (2010) Transcriptome analysis of various flower and silique development stages indicates a set of class III peroxidase genes potentially involved in pod shattering in Arabidopsis thaliana. BMC Genomics 11(1):528
Yu K, Wang X, Chen F, Chen S, Peng Q, Li H, Zhang W, Hu M, Chu P, Zhang J (2016) Genome-wide transcriptomic analysis uncovers the molecular basis underlying early flowering and apetalous characteristic in Brassica napus L. Sci Rep 6(1):1-13
Hradilová I, Trněný O, Válková M, Cechova M, Janská A, Prokešová L, Aamir K, Krezdorn N, Rotter B, Winter P (2017) A Combined Comparative Transcriptomic, Metabolomic, and Anatomical Analyses of Two Key Domestication Traits: Pod Dehiscence and Seed Dormancy in Pea (Pisum sp.). Front Plant Sci 8:552
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19(9):1639–1645
Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B et al (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11(1):136
Guo AY, Zhu QH, Chen X, Luo JC, Yi Chuan (2007) GSDS: a gene structure display server. Yi Chuan= Hered 29(8):1023–6
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Bailey TL, Williams N, Misleh C, Li WW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34(suppl 2):369–373
Kong X, Lv W, Jiang S, Zhang D, Cai G, Pan J et al (2013) Genome-wide identification and expression analysis of calcium-dependent protein kinase in maize. BMC Genomics 14(1):433
Couvreur TLP, Franzke A, Al-Shehbaz IA, Bakker FT, Koch MA, Mummenhoff K (2010) Molecular phylogenetics, temporal diversification, and princliples of evolution in the mustard family (Brassicaceae). Mol Biol Evol 27:55–71
Li X, Zhang S, Bai J, He Y (2016) Tuning growth cycles of Brassica crops via natural antisense transcripts of BrFLC. Plant Biotechnol J 14(3):905–914
Liljegren SJ, Ditta GS, Eshed Y, Savidge B, Bowman JL, Yanofsky MF (2000) Shatterproof MADS box genes control seed dispersal in Arabidopsis. Nat 404:766–770
Yang J, Liu D, Wang X, Ji C, Cheng F, Liu B, Hu Z, Chen S, Pental D, Ju Y (2016) The genome sequence of allopolyploid Brassica juncea and analysis of differential homeolog gene expression influencing selection. Nat Genet 48(10):1225–1232
Xu Y, Xu H, Wu X, Fang X, Wang J (2012) Genetic changes following hybridization and genome doubling in synthetic Brassica napus. Biochem Genet 50(7-8):616–624
Smaczniak C, Immink RG, Angenent GC, Kaufmann K (2012) Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139(17):3081–3098
Cheng F, Wu J, Liang J, Wang X (2015) Genome triplication drove the diversification of Brassica plants. In: The Brassica rapa Genome. Springer, pp 115–120
Parenicova L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J et al (2003) Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis. Plant Cell Online 15(7):1538–1551
Song XM, Huang ZN, Duan WK, Ren J, Liu TK, Li Y et al (2014) Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol Gen Genomics 289(1):77–91
Lee J, Lee I (2010) Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot 61(9):2247–2254
Wu Y, Ke Y, Wen J, Guo P, Ran F, Wang M, Liu M, Li P, Li J, Du H (2018) Evolution and expression analyses of the MADS-box gene family in Brassica napus. PLoS One 13(7):e0200762. https://doi.org/10.1371/journal.pone.0200762
Becker A, Theissen G (2003) The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29:464–489
Ferrandiz C, Liljegren SJ, Yanofsky MF (2000) Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Sci 289:436–438
Wu H, Mori A, Jiang X, Wang YM, Yang M (2006) The Indehiscent protein regulates unequal cell divisions in Arabidopsis fruit
Ahmada K, Mahideen A, Nasir AK, Azeem S (2021) Quality deterioration of postharvest fruits and vegetables in developing country Pakistan: a mini overview. Asian J Agri Food Sci 9:2 (ISSN: 2321–1571)
Yasin M, Shahzadi R, Riaz M, Afridi M, Ajmal W, Rehman O, Rehman N, Ali GM, Khan MR (2019) Expression pattern analysis of core regulatory module Shps-Ful transcripts in rapeseed pod shattering. Sarhad J Agri 35(3):696–707
Ahmad K, Khan S, Yasin MT, Hussain S, Ahmad R, Ahmad N, Bokhari SAI (2021) Enhanced starch hydrolysis by α-amylase using copper oxide nanowires. Appl Nanosci 11(7):2059–2071.
Khan MR, Ihsan H, Ali GM (2016) WSA206, a paralog of duplicated MPF2-like MADS box family is recruited in fertility function in Withania. Plant Sci 253:215–228
Acknowledgements
This research work was supported by National Institute of Genomics and Advanced Biotechnology, National Agriculture Research Center, Islamabad Pakistan and National Center for Bioinformatics Quaid-I-Azam University, Islamabad, Pakistan.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MA, KA, and SSM designed and performed active experimentations and drafted the manuscript. MRK and NR analyzed the data. AH, SMK, MRK, and MY provided lab facilities and helped in smooth facilitation of research. All the authors critically reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All the authors of this study declare that they have no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: the authors identified an error in the affiliation of Muhammad Yasin which is affiliated with affiliation 4 not 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Afridi, M., Ahmad, K., Malik, S.S. et al. Genome-wide identification, phylogeny, and expression profiling analysis of shattering genes in rapeseed and mustard plants. J Genet Eng Biotechnol 20, 124 (2022). https://doi.org/10.1186/s43141-022-00408-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00408-2