
Abstract
Background
Emulsomes are a type of lipid-based nanoparticle that consists of a solid lipid core surrounded by a phospholipid bilayer and have shown promise as drug delivery systems for a variety of applications. The major advantageous aspect of employing lipid-based carriers is their ability to enhance the solubility and bioavailability of poorly water-soluble drugs, mandatorily in case of intranasal drug delivery. These structures have portrayed significant ability to encapsulate drug with poor water solubility and low oral bioavailability, further leading to a completely enhanced drug delivery systems for achieving stability and controlled release of drug. The selection of lipid components and their physiochemical properties can be tailored to optimize drug solubility, blood brain barrier permeability, and enhanced targeting.
Main body of abstract
Intranasal drug delivery systems offer several advantages over other routes of administration. Intranasal delivery of drugs can provide rapid and efficient absorption into the bloodstream, bypassing first-pass metabolism in the liver and potentially reducing the risk of systemic side effects. Nasal mucosa comprises of dense network of blood vessels, that allow much enhanced rapid drug absorption and direct systemic delivery once the medication is being insufflated through the nasal route. Emulsomes can be used to encapsulate a wide range of drugs, including hydrophobic compounds that are difficult to formulate using traditional delivery methods. By incorporating targeting ligands or other components into the emulsome structure, it is possible to create formulations that are highly selective for specific tissues or cells. The characterization parameters majorly particle size, zeta potential, and encapsulation efficiency play a significant role while demonstrating the effectiveness of emulsome formulation and further its nasal route of administration. Therefore, by assessing and evaluating the parameters, researchers could effectively gain insights into the quality, stability, and enhanced therapeutic effects of emulsome drug carrier, leading to impactful information which would help in future intranasal emulsome preparation preparations, optimization and ensuring the overall effectiveness of the drug delivery systems.
Short conclusion
This review discusses the idea of emulsomes drug delivery systems, reviews the effectiveness of emulsomes for the delivery of small molecules, and pays particular attention to its structural and formulation design including benefits of intranasal emulsome delivery with recent advancements, stability aspects, and various considerations related to drug delivery and comprising of future prospects.
Similar content being viewed by others
Background
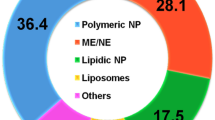
Lipid-based drug delivery systems are nanotechnology-based systems that have gained popularity because of their size-dependent characteristics, high level of biocompatibility, and versatility when compared to other nano-carriers [1]. Lipid-based carriers are a desirable candidate for the formulation of drugs, vaccines, diagnostics, and nutraceuticals due to their shown safety and efficacy [2, 3]. In the last few years, lipid-based carriers have been used to transport a specific class of drugs (biopharmaceutical classification system, BCS Classes II and IV) that are lipophilic in nature and have low water solubility, which acts as a barrier to their solubility in biological fluids and affects their absorption, which in turn affects their bioavailability [3]. These systems are thought to be beneficial for oral administration of BCS Class-II pharmaceuticals, increasing the bioavailability of medications with low water solubility but high permeability [4].
A lipid-based drug delivery device called “emulsome” was primarily devised for parenteral administration of medications with poor water solubility. Nanoscale lipid assemblies with polar cores in emulsomes made up nano-size lipid particles (bio adhesives nano-emulsion) [5, 6]. They are therefore regarded as nano-lipoidal carriers stabilized by high soy lecithin multilayer concentrations [5]. A high concentration of lecithin is added to stabilize the triglycerides and fats that make up the internal core as an o/w emulsion. Emulsomes combine the characteristics of liposomes and emulsions. The ability to load lipophilic drugs in high concentration while simultaneously achieving a controlled release is made possible by the interior oily core that has solidified or partially solidified. The ability to encapsulate water-soluble drugs in aqueous compartments of surrounding phospholipid layers is another benefit of these materials [1, 7]. A lipid in a solid state rather than an oil in a fluid phase makes up the internal core of emulsomes, which is solid at 25 °C and has a transition temperature from a solid to a liquid state that is close to physiological temperature, is a key feature of emulsomes. They can include water-soluble drugs in the aqueous compartments of the outer phospholipid layers and hydrophobic drugs in the interior lipid core. The system's stability was increased by the bilayer structure [8]. Additionally, due to their distinctively small size, emulsomes can offer site-specificity and thus, boost medication concentrations in the targeted tissues. Emulsomes can be used to administer medications via parenteral, ophthalmic, oral, rectal, intranasal, vaginal, or topical routes in addition to others [6]. Emulsomal drug administration is a method of administering medication through liquids. Totally, water-soluble parenteral medication delivery can be employed for a variety of therapeutic goals. Due to their limited water solubility, lipophilic medications require a lot of surfactants and co-solvents, which could be dangerous [7, 8]. Emulsomes are an oil kind used in water emulsions; it is distinct from regular oil. The phospholipid monolayer that covers the lipid core at the interface has a high phospholipid concentration, which aids in emulsion stabilization. Emulsomes are suitable for intravenous delivery due to their 10–250 nm particle size distribution. The drug release profile of the emulsomal formulation is 12–15% after 24 h, which is a good rate [3, 9].
Emulsomes can contain high concentrations of hydrophobic medications in their cores while encapsulating therapeutics that are water soluble that is found in the aqueous compartments of the outer phospholipid layers [8]. Emulsomes improve the bioavailability and solubility of lipophilic medicines, and their structure allows for sustained or regulated drug release. To create microscopic emulsomes, the medication is loaded and then sonicated [9, 10]. Emulsomes are anticipated to prevent lysosomal breakdown and ensure that the medicine is internally absorbed because of their positive charge [10,11,12]. Emulsomes have been shown to transport some protein and peptide medications, including Amphotericin B (AmB), Azidothymidine (AZT), Methotrexate (MTX), Curcumin, and Dithranol, among others, effectively [10, 13, 14].
Main text
Structure of emulsomes
Emulsomes, which are nanoparticles made of lipid, are frequently utilized as drug delivery mechanisms. Their structure can be compared to a cross between liposomes and solid lipid nanoparticles because they are made up of an oil phase enclosed by a phospholipid bilayer. A head-to-tail arrangement of two layers of phospholipids, with their hydrophilic heads facing outward and their hydrophobic tails facing inside, forms the phospholipid bilayer in emulsomes. This bilayer gives the emulsomes a stable structure and works to keep the encapsulated oil phase from deteriorating [12, 15].
Depending on the required qualities of the final formulation, the oil phase in emulsomes can be made up of a range of different oils. Triglycerides, fatty acids, and other lipid-based substances are examples of frequently used oils. The oil phase forms a core that can enclose hydrophobic medications or other active components as it is disseminated throughout the phospholipid bilayer [16]. The phospholipid bilayer and oil phase are not the only components that may be present in emulsomes; additional substances including surfactants, stabilizers, and targeting ligands may also be present [17, 18]. These extra ingredients may aid in enhancing the emulsomes' efficacy and durability as well as their capacity to target particular tissues or cells [19]. Emulsomes overall structure is quite adaptable and can be customized to fit the unique requirements of various drug delivery applications. It is feasible to make emulsomes that are stable, efficient, and well-tolerated by the body by carefully choosing the ingredients and perfecting the formulation [13, 20] (Fig. 1).

Structure of Emulsome
Comparative advantage over other lipidic drug delivery systems
Drugs that are poorly soluble in water are made more soluble and bioavailable by emulsomes. Triglycerides make up these micelles, which are arranged as lipid bilayers with the hydrophilic head group facing the water on both sides and the hydrophobic tails aligned against one another [21]. Phospholipids are best suited to be employed as excipients for medications that are not very water soluble because of these special characteristics [22, 23]. Emulsome-based technology demonstrated outstanding targeting potential. By extending the duration of the drug’s activity at relatively low doses, the formulations could greatly reduce toxicity issues caused by the drug's complementary localization in target cells [24, 25].
Emulsomes offer a sustained and regulated release of the medication. Emulsomes offer a longer drug release than liposomes, up to 24 h, while liposomes exhibit only a release for up to 6 h. Emulsomes are nanoscale in size when compared to various vesicular delivery systems including noisome, pharmacosomes, and ethosomes [25, 26]. They drastically alter the pharmacokinetics of medications. Additionally, they prevent the emergence of multidrug resistance, which is frequently linked to an overexpressed cell membrane glycoprotein that causes drug efflux from the cytoplasm and inadequate drug concentrations inside cellular compartments [27, 28].
If selective absorption is possible, it is projected that drug encapsulation in vesicular structures will prolong the drug's time in systemic circulation and possibly minimize toxicity [29]. Additionally, pre-clinical studies frequently demonstrate that excipients employed in the manufacture of emulsomes change epithelial permeability in animal models [3, 29].
Emulsomes are less expensive than other traditional lipid formulations. Emulsomes are distinct from liposomes because of their inside lipid core, while an aqueous compartment makes up the liposome's interior core. They also possess a high drug loading capacity in comparison with alternative vesicular drug delivery methods. Utilizing specialized emulsification and manufacturing methods, the internal core of the emulsome can be made of stable lipid particles by using particular lipid combinations. The diameter of 50–250 nm has been attained. Apart from that to spread and bind vaccination antigens to target mucosal tissue cells, a bio adhesive polymer has been added in the emulsome preparations in an amount adequate to give the lipoidal particles mucoadhesive characteristics surfaces and making it more effective [14, 30]. The better understanding could about the emulsomes as a stable and efficient drug delivery system for the intranasal drug administration could be impactfully interpreted from Table 1, given below.
Components of emulsomes
Lipid core
The interior lipid core or hydrophobic core made up of lipid is a crucial part of emulsomes. At ambient temperature (25 °C), this core shows solid, lipid, mixed solid/liquid, or lipid crystal phases. Lipids and excipients that resemble lipids are widely available in commerce. In the pharmaceutical industry, all of these are referred to as lipids. We can employ a single lipid or a combination of lipids. They are chemicals that are biologically and functionally connected to fatty acids and their derivatives. 23–25 Since lipids are often soluble in oil but not water, their melting point, fatty acid content, and hydrophilic–lipophilic balance are frequently used to identify them hydrophilic lipophilic balance (HLB). Lipids with a high melting point and a low HLB are appropriate for prolonged release [17]. While the excipients that are semisolid and have a high HLB act as excipients for immediate release and bioavailability augmentation. Triglycerides such as tristearin, tripalmitin, tricaprin and trilaurin that solidify at 25 °C are thought to be good core materials since they reduce the amount of time that o/w emulsion may be stored in an acceptable manner. The triglycerides are utilized to prepare an emulsome with unbranched fatty acid chains that range in length from c-10 to c-18 [29]. Additionally, lipid core makes it simple to alter their surface characteristics, enabling the attachment of ligands or targeting molecules for precise tissue or cell targeting and boosting their therapeutic potential.
Surfactant
One of the phospholipid molecules in the layer that envelops the lipid core. The surface tension is reduced by the stabilizing or surface-active role of the phospholipid layer. It is thought that a monolayer of surface-active phospholipids forms around the lipid core of the particles, with a polar head group at the interface. Using extra phospholipids, lipid cores can be encased by one or more roughly concentric bilayers, with the number of bilayers that surround the core varying. This bilayer envelope contains one or more watery components that could include a medication that is water soluble. A water-soluble medication may be present in this bilayer envelope’s one or more aqueous components. The ability of the particle to carry a high load of medications that are both lipid and water soluble is explained by the usage of several concentric bilayer models in emulsome structure. Drug entrapment in vesicles is influenced by the transition temperature of surfactants as well. The drug is most effectively trapped in starts with the lowest temperature at the phase transition, and vice versa [30].
Negatively charged particle
To increase the composition's zeta potential and stabilize the particles, phospholipids with negative charges such as phosphatidic acid, phosphatidylinositol, and phosphatidylserine, or lipid compounds comprising of negative charge such as oleic acid can be added to emulsomes. Furthermore, the inclusion of these negatively charged lipid molecules in emulsomes causes the creation of phospholipid bilayers with an opposite charge [12]. As a result, the phospholipid bilayers that surround the lipid core increase the loading capacity of the aqueous compartment. The electrical repulsion between the bilayers in the aqueous space between them is what causes this action. Positive charge increases particle dispersion, which lessens coalescence, flocculation, or fusion [19, 30].
Phosphatidylcholine
Lecithin contains a lot of phosphatidyl choline. Water does not readily dissolve phosphatidylcholine. Depending on temperature and hydration levels, the phospholipids in this solution can form lamellar, micelle, or bilayer sheets. A surfactant that is typically categorized as amphipathic is produced as a result of this. They are readily available from a range of conveniently accessible sources, like egg yolk or soy beans, and they are an important part of biological membranes. They are known as soya lecithin and egg lecithin, respectively, depending on the source from which they are made. Lecithin incorporation increased the percentage of drug entrapment to 96.1% and caused the size of the vesicles to be reduced as a result of an increase in hydrophobicity [20, 26].
Cholesterol
Emulsomes function as vesicles and require cholesterol as a major component. The stability of the vesicles is largely affected by cholesterol insertion. It was also reported that it enhances the buffering action in fluidity of the overall components of the combined formulation [10]. Cholesterol was added to all formulations as a stabilizing agent since it can cause the creation of the liquid crystal phase by changing the fundamental packing structure. Additionally, it has the ability to stabilize the outer phospholipid layers, which increases drug entrapment effectiveness and decreases drug leakage. Cholesterol also plays a crucial role in increasing the entrapment efficiency of the emulsomes. According to certain findings, the effectiveness of trapping rises as cholesterol concentration does. A very high cholesterol content was found to have a negative impact on drug entrapment in the vesicles. This may be because cholesterol levels above a certain point begin to disrupt the normal bilayer structure, which results in a loss of drug entrapment [31, 32].
Absorption mechanism of emulsome after intranasal administration
There are only two pathways connecting the brain to the external world: olfactory and trigeminal [33]. The human olfactory region, which contains the olfactory and trigeminal nerve terminations, is around 60 m thick and covers an area of 2–12.5 cm2, or 1.25–10% of the nasal cavity’s total surface area [34]. Drug targeting can be accomplished by applying formulations to the nasal mucosa [35, 36]. Further the drug entity reaches to the endoneurial micro-vessels within the nerve fascicle and the investing perineurium which make up the blood-nerve barrier (BNB) [37]. These micro-vessels actively participate in the mechanisms that control the permeability of the perineurium and endoneurial capillaries, and they undoubtedly depict a significant role in accomplishing transportation of chemicals from the trigeminal and olfactory pathways into the central nervous system [38].
The increased drug concentration in the brain after emulsome administration may be due to the nano-vesicle size, which enables drug particles to be transported deeper into the olfactory epithelium cellular layers and easily translocate from one cerebral compartment to another [35]. The nasal membrane's capacity to permit molecules to pass through it via olfactory neurons in the olfactory bulb is increased by chitosan's permeation-enhancing qualities and the lipid structure of emulsomes [36]. Emulsomes have site selectivity due to their small size, which results in higher medication levels at target tissues [16]. The intranasal emulsomes delivery mechanism further shows that the olfactory pathway is a primary pathway for drug transport to the brain [34]. Emulsomes are highly effective at delivering pharmaceuticals to the brain directly through the nose, as evidenced by their high drug targeting efficiency (DTE%) and nose-to-brain direct transport (DTP%) rates [39]. Three mechanisms—two extracellular transport-mediated channels that allow medications to enter the brain quickly, within minutes of intranasal administration, and one intracellular transport-mediated route that accounts for slowly absorbing pharmaceuticals—are based on direct nose-to-brain drug delivery [33].
Although the precise mechanism of drug delivery by nanoparticles across the BBB is still unknown, some theories include increased nanoparticle retention in brain capillaries, transcytosis and endocytosis of these particles by brain endothelial cells, and the fluidizing effect of surfactants and phospholipids on endothelial cell membranes, which increases drug permeability [40]. Drug transport to the brain following nasal administration has been investigated using nanosized lipid-based vesicles as a viable platform. Several methods, such as surface cationization, surface tailoring by targeting ligands, and activating the release of the medication by a magnetic field, temperature, ultrasound, or any other external component, have been suggested to support brain targeting of such systems [39, 41].
Emulsomes could be administered through intranasal route to increase drug molecule concentration in the brain and plasma, assists much more in getting the desired therapeutic result as depicted in Fig. 2 [42]. As a result, the availability of drug at the brain level has improved after intranasal delivery methods [43]. For instance, contrary to the oral route, the nasal one allows for direct vinpocetine (VNP) absorption into the systemic circulation while avoiding the pre-systemic metabolism, increasing the quantity of drug that may reach the targeted sites of brain. Additionally, olfactory route enables direct medication absorption into cerebrospinal fluid and brain tissues from the nasal cavity. Another aspect that needs to be taken into account is the high lipophilicity of the lipid-rich emulsomes, which may increase VNP's systemic absorption and blood brain barrier (BBB) uptake [41].

Chemical structure of Tristearin
Intranasal emulsome drug delivery process is a upcoming approach for administering medication to the brain through the nasal route [44]. Emulsomes are lipid-based nanocarriers that can encapsulate both hydrophobic and hydrophilic drugs, providing stability and protection from degradation in the body. When delivered intranasally, emulsomes can bypass the blood–brain barrier, allowing for direct transportation of drug into the brain [45]. This method of administration has shown potential in the treatment of neurodegenerative disorders such as Alzheimer's and Parkinson's disease, as well as in the management of pain and inflammation [46]. Intranasal emulsome administration also offers several advantages over traditional drug delivery methods, including improved bioavailability, reduced systemic toxicity, and increased patient compliance due to its non-invasive nature. Further research is needed to optimize the formulation and dosing of emulsomes for intranasal delivery, but the potential benefits make it a promising avenue for drug development (Figs. 3, 4, 5, 6, 7, 8 and 9).

Chemical structure of Span 80 (Sorbitan monooleate)

Chemical structure of Phosphatidic acid

Chemical structure of phosphatidylcholine

Chemical structure of Cholesterol

Emulsomes intranasal absorption mechanism for targeted therapy

Lipid film hydration method

Reverse phase evaporation technique
Preparation techniques of emulsomes
Emulsomes are combination of solid lipid core, cholesterol, and phosphatidylcholine in a ratio of 1:0.5:1, thus leading to a formulation aspect of a highly optimized and stable preparation [37]. Emulsomes could be efficiently prepared from a wide range of nanoparticle formulation techniques. Obtaining particle size within a range of 10–1000 nm and maintaining formulation’s stability is the fundamental objectives of the emulsome preparation [47]. The transition temperature employed in the preparation is in range of (25–45) °C. Organic solvent such as n-hexane, dicloro-methane, toluene and diethyl ether is primarily constituently dissolved along with the basic formulation in vacuum conditions for lipid film incorporation. Thus, it also plays a significant role in poorly water-soluble drug successful encapsulation in the drug delivery system along with other excipients of formulation [48, 49].
Lipid film formation (hand shaking method)
This process involves utilizing a flask rotary evaporator with reduced pressure to cast layers of film made of surfactants and lipids from their organic solution. The cast films are subsequently distributed in aqueous medium. The lipids expand and peel off from the wall of a round bottom flask when surfactants are used for a predetermined period of time (the time of hydration) at a temperature just slightly over their phase transition temperature. Hand shaking or 15 min of exposure to a steam of water saturated nitrogen before allowing the films to swell in an aqueous solution without shaking both supply the mechanical energy required for the swelling of lipids and the dispersion of cast lipid films [19]. It is also inferred while in hand shaking method that the suspension like appearance occurs and further after hydration large unilamellar vesicles (LUV) are formed that could further be made to go under probe sonication and centrifugation as per the formulation design and requirements. The non-shaking method resulted in large unilamellar vesicles, whereas the hand shaking method led to multi lamellar vesicles (MLV) [50].
Reverse phase evaporation
In this process, there are two steps: making an o/w emulsion and adding buffers to an excess organic phase. It is possible to emulsify lipid and aqueous phases mechanically or by sonication. With the second step, an organic solvent was evaporated under vacuum, causing water droplets that were covered in phospholipids to condense and create a matrix that resembled gel. Under lower pressure, additional organic solvent removal enables the gel-like matrix to transform into a smooth paste formulation. This technique can successfully encapsulate small and big compounds, with an encapsulation effectiveness of 60–65% [15]. The main drawback of this approach is that the components and medication are subjected to mechanical agitation and organic solvents. Bioactive compounds like enzymes, protein medicines, and organic solvent exposure and mechanical agitation can cause conformational changes in RNA-type molecules, protein denaturation, or DNA strand breakage [51].
High pressure extrusion technique
When MLV are repeatedly passed through very small pore polycarbonate membranes (0.8–1.0 pm) under high pressure, the average diameter of the vesicles shrinks over time, reaching a minimum of 60–80 nm after 5–10 passes, according to research done by large number of scientists. The vesicles tend to become unilamellar when the average size decreases. Other researchers who used MLV with a microfluidizer reported getting similar outcomes. A microfluidizer is a device that applies intense pressure to the feed material and forces it through a small opening [52, 53]. As MLV are forced through the narrow orifice, layers of bilayers appear to be peeled off of the vesicular structure, much like the layers of an onion skin. It was further asserted that the layer separation process only functions in vesicles with positively charged phospholipids and vesicles larger than 70 pm [17, 54]. The detailed flowchart representation is further illustrated in Fig. 10, where we could properly observe the complete process of homogenization using the microfluidizer.

High pressure extrusion technique
Sonication method
With a few minor tweaks, this technique is similar to the lipid film hydration technique. In a round-bottomed flask, solid lipids such as cholesterol and phosphatidylcholine were added in various molar ratios, and they are then dissolved in a minimum amount of chloroform that contained three or four drops of methanol. An amount of drug that was precisely weighed was dissolved in this solution [55]. A thin lipid layer is formed on the walls of the round bottom flask by rotary evaporating the organic solvent until it was completely dry under reduced pressure. When phosphate buffered saline with a pH of 7.4 is used to hydrate dry film, the phospholipid expands to create a milky emulsion that contains enormous multilamellar vesicles. To create equally distributed nanosized emulsomes vesicles, this emulsion is further sonicated using a sonicator (Probe/Bath) for a set amount of time [56]. The complete process finally results in formation of aggregates of lipids which is also being shown in Fig. 11.

Sonication technique
Cast film method
Using this technique, phospholipids and triglycerides with a solid to liquid phase transition temperature of greater than 25 °C can be mixed to form emulsomes at a weight ratio of 0.5:1.0. In order to create more emulsomes, the suspension is either reduced, or the mixture is suspended in an aqueous solution at a temperature below the point at which solids turn into liquids. The liquid particles that make up these emulsomes have mean particle diameters that range from 10 to 250 nm, are frequently in the range of 20–180 nm, and are frequently in the range of 50–150 nm. When establishing the size range, a weight percentage basis is favored over a particle number basis [1, 57]. Traditionally, the lipid component could be a volatile organic solvent that is chemically unreactive and volatile, like dichloromethane or diethyl ether. In a rotating evaporator or beneath a stream of inert gas, the solvent is normally evaporated under reduced pressure. By wrapping in an aqueous solution and shaking it, the resultant lipid layer is hydrated and dispersed. Aqueous hydration solution may be supplemented with medication components if they were absent from the organic solution. The lipid solution or dispersion is then frequently size-reduced using a high-shear homogenizer working at a pressure of up to 800 bars [58] (Figs. 12, 13 and 14].

Cast film technique

Ethanol injection technique

Detergent removal technique
Ethanol injection method
Injection of ethanol was originally suggested in 1973. The benefit of this approach is that it eliminates the requirement for extrusion or ultrasound and allows for the one-step injection of an ethanol lipid solution into water to produce small unilamellar vesicles with a limited particle size distribution. This method's drawbacks include a heterogeneous population, dilute emulsions, the difficulty of completely eliminating all ethanol, and a high likelihood of inactivating bioactive macromolecules. In this method, the lipid or lipid mixture first dissolved in the appropriate alcoholic solvent before an aliquot of 200, 500 or 600 mL then swiftly injected into the dispersant solution (water or saline solution) of 9.8 mL (dilution 1:50), 9.5 mL (dilution 1:20), or 9.8 mL (dilution 1:17), respectively, at room temperature using a one-way, 1-mL syringe. The mixture is then strongly shaken for 20–30 s. Without extrusion or sonication, narrowly distributed vesicles in the nano-size range are produced [59]. Another significant benefit of this technique is its appropriateness for the entrapment of numerous different therapeutic compounds, such as membrane attachment of vaccination antigens or large hydrophilic proteins via passive encapsulation or one-step distant loading which would also be very beneficial in preparation of intranasal vaccine comprising of drug loaded emulsomes [60].
Detergent removal technique
In order to remove detergents, phospholipids are dissolved in an aqueous solution that has detergents present at a particular critical micelle concentration (CMC). The reaction medium releases individual phospholipid molecules once the detergent is taken out, and these molecules then spontaneously self-assemble into bilayer structures. A dialysis bag, polystyrene-based absorber beads, or Sephadex columns are the most common methods for removing detergent (gel permeation chromatography). When the resulting combination is diluted with an adequate aqueous medium, the produced micelles undergo reorganization and develop into emulsomes [61]. In order to ensure the stability, biocompatibility, and appropriateness of emulsomes for drug delivery applications, the elimination of residual detergent is a crucial step in the manufacturing process. To accomplish this, a number of detergent removal approaches have been used. In one often used technique, emulsomes are exposed to ultracentrifugation, whereby high-speed centrifugation causes the heavier emulsomes to sediment and the detergent to stay in the supernatant. The remaining traces of detergent are subsequently removed by gathering and washing the detergent-free emulsomes. Alternately, emulsomes may be used in a semipermeable membrane bag during dialysis, which keeps the emulsomes inside while allowing the detergent to seep into the surrounding buffer. To guarantee effective detergent elimination, this procedure is performed numerous times. These two methods are essential for creating detergent-free emulsomes with the best drug loading properties and increased stability.
Characterization techniques of emulsomes
Characterizing the prepared emulsomes is crucial from the perspective of application. Monitoring physical and chemical variables are necessary to ensure that emulsome preparation is repeatable and serves the intended purpose. Emulsomes' primary properties include average size, size distribution, shape, and polydispersity index, along with surface charge and encapsulation efficiency. Emulsomes are confirmed by transmission electron microscopy (TEM) micrographs to be spherical in shape, making them identical in size and morphology to empty emulsomes [62]. Emulsomes are biocompatible vesicular structures made up of a solid fat core encased in many layers of phospholipids. Emulsomes, which have a solid core, can entrap more lipophilic medicinal molecules with a longer half-life than emulsion formulations that have a liquid core. Emulsomes, which are made of fat and lipids, are biocompatible. Emulsomes are attractive possibilities for poorly water-soluble medicinal substances like curcumin, silybin due to these distinctive features. As was recently shown, a dehydration-rehydration technique followed by temperature-controlled extrusion can be used to assemble phospholipids and triglycerides to create stable dispersed emulsomes [63].
Composition and size assessment of emulsomes
Emulsomes are divided structurally into two parts: the innermost core and the outside shell. The innermost core is made up of lipid such that, at normal temperature, either has mixed solid and liquid crystal phases or displays a solid or liquid crystal physical condition. The interior core is supported by a multilamellar shell that can be made of pure, synthetic, hydrogenated, or partially hydrogenated phospholipids, natural or (partially) hydrogenated lecithin, or both [64]. Because repulsive interactions between the emulsomes essentially hinder aggregation, charged PLs can also be utilized to increase the formulation's stability. The phospholipid shell controls the in vivo behavior of the nano-formulation, including circulation half-life, toxicity, and cellular targeting. The outermost layer controls the emulsome’s interface with the environment [65]. Light scattering and electrophoretic techniques were employed to assess the average vesicle size (z-average) and zeta potential, respectively, using Nano-ZSP (Malvern Panalytical, Malvern, UK) at 25 1 °C. Zeta potential mean and distribution were determined along with electrophoretic mobility using mixed mode measurement with phase analysis light scattering (M3-PALS). Emulsomes were typically suitably diluted Prior to measurement, 100 times with phosphate buffer pH 7.4 [44]. It is possible to measure the size of individual emulsomes using electron microscopy techniques with a micron range. The structure of the liposome could be obtained in detail using a technique called freeze-fracture transmission electron microscopy [46]. The particle size and stability of emulsomes are both critically dependent on the PL:TL ratio. The homogenization procedure enables mechanical tuning of emulsome size. As previously mentioned, smaller particles can form to some extent when the preparation is extruded through filters with smaller pores or given a longer sonication duration. The path of clearance in the body may depend on the formulation's diameter. For instance, it has been noted that macrophages are capable of clearing particles larger than 200 nm from the circulation quickly. As a result, larger nanoparticles are more suited for systems that target inflammatory areas like tumors, whereas smaller nanoparticles are better suited for systems that target macrophages [66, 67].
Drug encapsulation
During the emulsome synthesis process, when the lipid components assemble, drugs are loaded into the emulsomes. The lipophilic medicines disintegrate due to their nature, become trapped in the innermost core, and intercalate inside the phospholipid bilayers. The high level of multilamellarity increases the rate of drug encapsulation, as was previously shown. Only the watery area between the phospholipid bilayers can trap hydrophilic molecules. Moreover, amphiphilic compounds are incorporated into the phospholipid layers preferentially [68]. Curcumin emulsion formulation study recently demonstrated the high lipophilic chemical loading capacity of emulsomes [69]. The compound's solubility in water, which was 11 ng/ml, was increased up to 10,000 times, or to 0.11 mg/ml, a concentration that substantially improved the lipophilic compound's usage in biomedicine [70]. Emulsomes are unique in that they have a high loading capacity for lipophilic chemicals. Yet, because multilamellar phospholipids can hold a sizable amount of water between the bilayers, they can also be utilized to distribute water-soluble components. Assuming that emulsomes are made up of an average of 1–3 bilayers, their trapped aqueous volume is thought to be between 1.1 and 0.6 L per mol of phospholipid; it is a lot more than just one small unilamellar vesicle’s aqueous volume, which is between 0.3 and 0.2 L per mol of phospholipid. Thus, the findings indicate that emulsomes should be regarded as a nanocarrier rather than a drug delivery system (DDS) for exclusively hydrophobic molecules when it comes to delivery of both hydrophilic and hydrophobic molecules simultaneously [63, 69].
Stability of emulsomes
In terms of nanoparticle-mediated DDSs, stability refers to the nanocarrier's capacity to sustain throughout time its biophysical properties, such as size, zeta potential, and drug retention. Due to their large absolute zeta potential values, emulsomes are consequently anticipated to be physically stable, decreasing the likelihood of coalescence [71]. Emulsomes are more stable in suspensions than other lipid-based formulations like liposomes, which is a discovery that may be very helpful in clinical practice [72]. Emulsomes are created by combining two essential elements: phospholipid is walled around the lipidic core to provide vesicular steric stability. It is possible to create an emulsomal formulation that is pharmaceutically stable without the need of an extra solubilizer or surfactant. PEGylation of the surface of emulsomes will therefore help to increase steric stabilization and extend circulation duration [73, 74].
The physicochemical properties of the lipids used, as well as the storage temperature, are significant factors affecting the stability of the nanocarrier [75]. Zeta potential is a crucial component of stability; measuring zeta potential in electrostatically stabilized vesicles is essential for comprehending how dispersion and aggregation processes work and is unquestionably a crucial requirement for research on the storage stability of these vesicles. A change in the lipid type enhanced the absolute value of zeta potential, whereas the fats used in emulsomes—trilaurin, tristearin, and compritol ATO 888—offer a substantial negative value of zeta potential. Increased input energy during sonication results in a decrease in particle size and zeta potential to produce stable and tightly packed emulsomes [73, 76].
Advanced modifications in drug delivery through emulsomes
Emulsomes are a promising method of medication administration that combines the advantages of emulsions with liposomes. They are more durable and stable than conventional emulsions because they consist of an oil droplet core and a lipid bilayer membrane that stabilizes the emulsion. Emulsomes have drawn attention because of their capacity to directly target the central nervous system while bypassing the blood–brain barrier [70, 77].
There has been a lot of interest lately in improving the emulsome formulation modification for intranasal delivery. Researchers have demonstrated their findings based on multiple number of methods to improve emulsome stability, drug loading capacity, and bioavailability. These improvements include the addition of targeted moieties, the insertion of novel lipid components, and compositional optimization of the formulation [75]. The utilization of hybrid lipid components, such as solid lipids and surfactants, to increase the structural stability and drug loading capability of emulsomes is one of the most promising changes. These substances can lengthen the duration that emulsomes stay in the nasal cavity and enhance the rate at which medicines pass through the nasal mucosa. Using targeting ligands to boost the specificity, drug delivery, such as antibodies, peptides, or aptamers, is another key change. Targeted emulsomes have the ability to bind specifically to cell surface receptors, increasing drug accumulation at the target region and lowering systemic adverse effects. The addition of mucoadhesive polymers or viscosity-enhancing substances has also been investigated as a modification to improve the adherence of emulsomes to the nasal mucosa and boost the bioavailability of the medicine being administered [73, 78].
For intranasal drug administration, innovative improvements to emulsome formulation hold great promise for increasing drug delivery effectiveness and lowering systemic adverse effects. We may anticipate additional developments in this area in the future since these alterations will continue to be a focus of research in the field of drug delivery. Intranasal emulsome formulations have been gaining attention in recent years due to their potential as a drug administering system for the treatment of various diseases [79]. Emulsomes are lipid-based vesicular structures that can encapsulate both hydrophilic and hydrophobic drugs, resulting in improved drug solubility, bioavailability, and sustained release. Recent advancements in intranasal emulsome formulations include the use of novel lipid materials such as ethosomes, transferosomes, and lipid nanoparticles. These materials can enhance the stability and effectiveness of the emulsomes and also offer advantages such as biocompatibility, biodegradability, and targeted delivery. Moreover, the development of emulsomes with mucoadhesive properties has enabled improved nasal retention and prolonged drug release. These advancements have opened up new possibilities for the use of intranasal emulsomes in the treatment of various diseases, in addition to neurological disorders, respiratory diseases, and cancer [68, 79].
A nasal insert is a small, solid, or semisolid dosage form that is designed to be inserted into the nasal cavity. Emulsomes are a type of lipid-based nanoparticle that can be used to encapsulate drugs and improve their bioavailability. When incorporated into a nasal insert, emulsomes can improve the absorption of the drug through the nasal mucosa, increasing its therapeutic effectiveness [80]. The emulsome structure provides a protective environment for the drug and allows for sustained release, reducing the frequency of administration [81]. Additionally, emulsomes can be engineered to target specific cells or tissues, allowing for surface modifications and targeted drug delivery. Overall, the use of emulsome-containing nasal inserts is a promising approach for improving drug delivery and efficacy through the nasal route [80, 82].
Conclusion
Emulsomes are lipid-based medication delivery systems that have benefits over other nanotechnology-based carriers. Hydrophobic and hydrophilic medicines can both be encapsulated by emulsomes, offering stability, and controlled release. They can increase drug solubility and bioavailability and have great biocompatibility. Emulsomes administered intranasally could successfully increase drug absorption, lessen systemic toxicity, and boost patient compliance. Emulsomes can avoid multidrug resistance, have a longer drug release time (up to 24 h), and have better targeting capability. The disadvantages associated with the typical vesicular system, such as a higher incidence of drug leakage and a propensity for agglomeration, could be effectively overcome by emulsomes, which can accommodate the lipophilic medicines well and have consistently shown effective therapeutics results at the targeted site of action. They show promise in the treatment of cancer, respiratory illnesses, and neurological conditions. Emulsomes present a promising route for drug development in a number of therapeutic areas, but further study is required to optimize their composition and dose.
Availability of data and materials
Not applicable.
Abbreviations
- BCS:
-
Biopharmaceutical classification system
- AmB:
-
Amphotericin B
- AZT:
-
Azidothymidine
- MTX:
-
Methotrexate
- BNB:
-
Blood-nerve barrier
- o/w:
-
Oil in water
- DTE%:
-
Drug targeting efficiency
- DTP%:
-
Direct transport
- VNP:
-
Vinpocetine
- BBB:
-
Blood brain barrier
- LUV:
-
Large unilamellar vesicles
- MLV:
-
Multi lamellar vesicles
- TEM:
-
Transmission electron microscopy
References
Gill V, Nanda A (2021) Emulsomes a lipid bases drug delivery system. World J Pharm Res 10(12):113–129
Eita AS, Makky AMA, Anter A, Khalil IA (2022) Repurposing of atorvastatin emulsomes as a topical antifungal agent. Drug Deliv 29(1):3414–3431
Afreen U, Shailaja AK (2020) Pharmacosomes and emulsomes: an emerging novel vesicular drug delivery system. Glob J Anesth Pain Med 3(4):287–297
Islek Z, Ucisik MH, Keskin E, Sucu BO, Gomes-Alves AG, Tomás AM, Guzel M, Sahin F (2022) Antileishmanial activity of BNIPdaoct-and BNIPdanon-loaded emulsomes on leishmania infantum parasites. Front Nanotechnol 3(101):773741
Jadhav SM (2012) Novel vesicular system: an overview. J Appli Pharm Sci 02(01):193–202
Gupta S, Vyas SP (2007) Development and characterization of amphotericin B bearing emulsomes for passive and active macrophage targeting. J Drug Target 15:206–217
Amselem S, Yogev A, Zawoznik E (1994) Emulsomes, a novel drug delivery technology. Proc Int Symp Controll Releas Bioact Mater 21:1368–1369
Pal A, Gupta S, Jaiswal A (2012) Development and evaluation of tripalmitin emulsomes for the treatment of experimental visceral leishmaniasis. J Liposome Res 22:62–71
Choudhury H, Zakaria NFB, Tilang PAB, Tzeyung AS, Pandey M, Chatterjee B, Alhakamy NA, Bhattamishra SK, Kesharwani P, Gorain B (2019) Formulation development and evaluation of rotigotine mucoadhesive nanoemulsion for intranasal delivery. J Drug Deliv Sci Technol 54:101301
Yilmaz EN, Bay S, Ozturk G, Ucisik MH (2020) Neuroprotective effects of curcumin-loaded emulsomes in a laser axotomy-induced cns injury model. Int J Nanomed 15:9211–9229. https://doi.org/10.2147/IJN.S272931
Rizk SA, Elsheikh MA, Elnaggar YSR, Abdallah OY (2021) Novel bioemulsomes for baicalin oral lymphatic targeting: development, optimization and pharmacokinetics. Nanomedicine 16(22):1983–1998
Amselem, Shimon, Zawoznik E, Yogev A, Friedman, D (2018) Emulsomes, a new type of lipid assembly. In Handbook of Nonmedical Applications of Liposomes: Volume III: From Design to Microreactors, CRC Press, pp 209–223
Erdő F, Bors LA, Farkas D (2018) Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res Bull 143:155–170
Ghode PS, Ghode DP (2020) Applications perspectives of emulsomes drug delivery system. Int J Med Pharm Sci. https://doi.org/10.31782/IJMPS.2020.10101
Maher S, Casettari L, Illum L (2019) Transmucosal absorption enhancers in the drug delivery field. Pharmaceutics 11(7):339. https://doi.org/10.3390/pharmaceutics11070339
Aswathy N, Vidhya M, Saranya R, Sreelakshmy R, Sreeja N (2013) Emulsomes: a novel liposomal formulation for sustained drug delivery. Int Res J Pharm 3:192–196
Hadel A, El-Enin A, Mostafa ER, Ahmed FM, Naguib AI, Abdelgawad A, Ghoneim MM, Abdou ME (2022) Assessment of nasal-brain-targeting efficiency of new developed mucoadhesive emulsomes encapsulating an anti-migraine drug for effective treatment of one of the major psychiatric disorders symptoms. MDPI; Pharmaceutics 14:410
Schwarz C, Mehnert W, Lucks JS, Muller RH (1994) Solid lipid nanoparticles (SLNs) for controlled drug delivery: I. production, characterization and sterilization. J Control Releas 30:83–96
Amselem (1996) Solid fat nanoemulsions as drug delivery vehicles. US Patent 5576016. 19 November 1996
Kumar R, Nirmala KHLS (2013) Emulsomes: an emerging vesicular drug delivery system. J Drug Deliv Ther 3(6):133–142
Jain S, Jain V, Mahajan SC (2014) Lipid based vesicular drug delivery systems. Adv Pharm. https://doi.org/10.1155/2014/574673
Srinivas L, Manikanta V, Jaswitha M (2019) Protein and peptide drug delivery: a brief review. Res J Pharm and Tech 12(3):1369–1382. https://doi.org/10.5958/0974-360X.2019.00230.0
Haque S, Md S, Fazil M, Kumar M, Sahni JK, Ali J, Baboota S (2012) Venlafaxine loaded chitosan NPs for brain targeting: pharmacokinetic and pharmacodynamic evaluation. Carbohydr Polym 89(1):72–79
Illum L (2000) Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci 11(1):1–8
Aldawsari HM, Ahmed OAA, Alhakamy NA, Neamatallah T, Fahmy UA, Badr-Eldin SM (2021) Lipidic nano-sized emulsomes potentiates the cytotoxic and apoptotic effects of raloxifene hydrochloride in MCF-7 human breast cancer cells: factorial analysis and in vitro anti-tumor activity assessment. Pharmaceutics 24 13(6):783. https://doi.org/10.3390/pharmaceutics13060783
Nasr M, Mansour S, Mortada ND, Elshamy AA (2008) Vesicular aceclofenac systems: a comparative study between liposomes and niosomes. J Microencapsul 25(7):499–512. https://doi.org/10.1080/02652040802055411
Lowell GH, Kaminski RW, VanCott TC, Slike B, Kersey K, Zawoznik E, Loomis-Price L, Redfield RSG, Amselem S, Birx LD (1997) Proteosomes, emulsomes, and cholera toxin B improve nasal immunogenicity of human immunodeficiency virus gp160 in mice: induction of serum, intestinal, vaginal, and lung IgA and IgG. J Infect Dis 175:292–301. https://doi.org/10.1093/infdis/175.2.292
Pouton CW (2006) Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci 29:278–287
Gursoy RN, Benita S (2004) Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 58:173–182
Amalnath S, Ganesh NS, Chandy V (2021) A well explained review on: emulsomes as vesicular drugdelivery system. World J Pharm Pharm Sci. https://doi.org/10.20959/wjpps202110-15432
Mehanna MM, Mneimneh AT (2021) Formulation and applications of lipid-based nanovehicles: spotlight on self-emulsifying systems. Adv Pharm Bull 11(1):56–67. https://doi.org/10.34172/apb.2021.006
Briuglia ML, Rotella C, McFarlane A (2015) Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv Transl Res 5:231–242
Lofts A, Abu-Hijleh F, Rigg N, Mishra RK, Hoare T (2022) Using the intranasal route to administer drugs to treat neurological and psychiatric illnesses: rationale, successes, and future needs. CNS Drugs 36:739–770
Bourganis V, Kammona O, Alexopoulos A, Kiparissides C (2018) Recent advances in carrier mediated nose-to-brain delivery of pharmaceutics. Eur J Pharm Biopharm 128:337–362. https://doi.org/10.1016/j.ejpb.2018.05.009
Alam MI, Beg S, Samad A, Baboota S, Kohli K, Ali J, Ahuja A, Akbar M (2010) Strategy for effective brain drug delivery. Eur J Pharm Sci 40:385–403
Pardeshi C, Vijaysing B, Shailendra V (2013) Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood–brain barrier: an excellent platform for brain targeting. Expert Opin Drug Deliv 10(7):957–972. https://doi.org/10.1517/17425247.2013.790887
Weerasuriya A, Mizisin AP (2011) The blood-nerve barrier: structure and functional significance. Methods Mol Biol 686:149–173
Sezer AD (ed) (2014) Application of nanotechnology in drug delivery. IntechOpen, Croatia
Mistry GA, Kjems ZS, Randel J, Howard J, Stolnik AK, Illum SL (2009) Effect of physicochemical properties on intranasal nanoparticle transit into murine olfactory epithelium. J Drug Target 17:543–552
El-Zaafarany GM, Soliman ME, Mansour S, Awad SAG (2016) Identifying lipidic emulsomes for improved oxcarbazepine brain targeting: In vitro and rat in vivo studies. Int J Pharm 503:127–140. https://doi.org/10.1016/j.ijpharm.2016.02.038
Aldawsari HM, Badr-Eldin SM, Assiri NY, Alhakamy NA, Privitera A, Caraci F, Caruso G (2022) Surface-tailoring of emulsomes for boosting brain delivery of vinpocetine via intranasal route: in vitro optimization and in vivo pharmacokinetic assessment. Drug Deliv 29(1):2671–2684. https://doi.org/10.1080/10717544.2022.2110996
Sopyan I, Kurniawansyah I, Gozali D (2020) A review: a novel of efforts to enhance liposome stability as drug delivery approach. Syst Rev Pharm. https://doi.org/10.31838/srp.2020.6.85
Shringarpure M, Gharat S, Momin M, Omri A (2021) Management of epileptic disorders using nanotechnology-based strategies for nose-to-brain drug delivery. Expert Opin Drug Deliv 18(2):169–185
Costa C, Moreira JN, Amaral MH, Sousa Lobo JM, Silva AC (2019) Nose-to-brain delivery of lipid-based nanosystems for epileptic seizures and anxiety crisis. J Control Releas 295:187–200. https://doi.org/10.1016/j.jconrel.2018.12.049
El-Zaafarany GM, Soliman ME, Mansour S, Awad GAS (2016) Identifying lipidic emulsomes for improved oxcarbazepine brain targeting: In vitro and rat in vivo studies. Int J Pharm 503(1–2):127–140. https://doi.org/10.1016/j.ijpharm.2016.02.038
Inoue D, Furubayashi T, Tanaka A, Sakane T, Sugano K (2020) Effect of cerebrospinal fluid circulation on nose-to-brain direct delivery and distribution of caffeine in rats. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.0c00495
Vyas PS, Subhedar R, Jain S (2006) Development and characterization of emulsomes for sustained and targeted delivery of an antiviral agent to liver. JPP 58(3):321–326. https://doi.org/10.1211/jpp.58.3.0005
Jaiswal M, Dudhe R, Sharma PK (2015) Nanoemulsion: an advanced mode of drug delivery system. 3 Biotech 2:123–127. https://doi.org/10.1007/s13205-014-0214-0
Zhang H (2017) Thin-film hydration followed by extrusion method for liposome preparation. Methods Mol Biol 1522:17–22. https://doi.org/10.1007/978-1-4939-6591-5_2
Govender T, Stolnik S, Garnett MC, Illum L, Davis SS (1999) PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water-soluble drug. J Control Releas 57(2):171–185. https://doi.org/10.1016/s0168-3659(98)00116-3
Shi NQ, Qi XR (2018) Preparation of drug liposomes by reverse-phase evaporation. In: Lu WL, Qi XR (eds) Liposome-based drug delivery systems biomaterial engineering. Springer, Berlin Heidelberg
Ong S, Chitneni M, Lee KS, Ming LC, Yuen K (2016) Evaluation of extrusion technique for nanosizing liposomes. Pharmaceutics 8(4):36. https://doi.org/10.3390/pharmaceutics8040036
Batzri S, Korn ED (1973) Single bilayer liposomes prepared without sonication. Biochim Biophys Acta 298:1015–1019
Szoka F, Papahadjopoulos D (1978) Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci U S A 75:4194–4198
Batzri S, Korn DE (1973) Single bilayer liposomes prepared without sonication. Biochim Biophys Acta (BBA)-Biomembr 298(4):1015–1019. https://doi.org/10.1016/0005-2736(73)90408-2
Mendez R, Banerjee S (2017) Sonication-based basic protocol for liposome synthesis. In: Bhattacharya S (ed) Lipidomics methods in molecular biology, vol 1609. Humana Press, New York. https://doi.org/10.1007/978-1-4939-6996-8_21
Betzer O, Shilo M, Opochinsky R (2017) The effect of nanoparticle size on the ability to cross the blood-brain barrier: an in vivo study. Nanomedicine (Lond) 12:1533–1546
Zhang HJ, Zhu BJ (1999) A novel method to prepare liposomes containing amikacin. J Microencapsul 16(4):511–516. https://doi.org/10.1080/026520499288951
Stano P, Bufali S, Pisano C, Bucci F, Barbarino SM, Carminati M, Luisi P, PL, (2004) Novel camptothecin analogue (Gimatecan)-containing liposomes prepared by the ethanol injection method. J Liposome Res 14:87–109. https://doi.org/10.1081/LPR-120039794
Wagner A, Vorauer-Uhl K (2011) Liposome technology for industrial purposes. J Drug Deliv. https://doi.org/10.1155/2011/591325
Catala A (ed) (2019) Liposomes: advances and perspectives. IntechOpen, United Kingdom
Kanásová M, Nesměrák K (2017) Systematic review of liposomes’ characterization methods. Monatshefte für Chemie: Chem Month. https://doi.org/10.1007/s00706-017-1994-9
Ucisik MH, Küpcü S, Debreczeny M, Schuster B, Sleytr UB (2013) S-layer coated emulsomes as potential nanocarriers. Small 9:2895–2904. https://doi.org/10.1002/smll.201203116
Pratishtha SS, Gupta S (2020) Nose to brain drug delivery for the treatment of epilepsy. Nanoformulations Human Health: Chall Approaches. https://doi.org/10.1007/978-3-030-41858-8_8
Sonvico CF, Buttini A, Colombo F, Pescina G, Guterres SS, Pohlmann RS, Nicoli A, Sara, (2018) Surface-modified nanocarriers for nose-to-brain delivery: from bioadhesion to targeting. Pharmaceutics 10(1):34. https://doi.org/10.3390/pharmaceutics10010034
Kaminaga Y, Nagatsu A, Akiyama T, Sugimoto N, Yamazaki T, Maitani T, Mizukami H (2003) Production of unnatural glucosides of curcumin with drastically enhanced water solubility by cell suspension cultures of Catharanthus roseus. FEBS Lett 555(2):311–316
Paliwal R, Paliwal SR, MishraN MA, Vyas SP (2009) Engineered chylomicron mimicking carrier emulsome for lymph targeted oral delivery of methotrexate. Int. J Pharmaceut 380(1–2):181–188
Narayan R, Singh M, Ranjan OP, Nayak Y, Garg S, Shavi GV, Nayak UY (2016) Development of risperidone liposomes for brain targeting through intranasal route. Life Sci 163:38–45. https://doi.org/10.1016/j.lfs.2016.08.033
Ucisik MH, Kupcu S, Schuster B, Sleytr UB (2013) Characterization of CurcuEmulsomes: nanoformulation for enhanced solubility and delivery of curcumin. J Nanobiotechnol 11(1):37
Ucisik MH, Sleytr UB, Schuster B (2015) Emulsomes meet S-layer proteins: an emerging targeted drug delivery system. Curr Pharm Biotechnol 16(4):392–405. https://doi.org/10.2174/138920101604150218112656
Zhou X, Chen Z (2015) Preparation and performance evaluation of emulsomes as a drug delivery system for silybin. Arch Pharm Res 38(12):2193–2200. https://doi.org/10.1007/s12272-015-0630-7
Dubey S, Vyas PS (2021) Emulsomes for lipophilic anticancer drug delivery: development, optimization and in vitro drug release kinetic study. Int J App Pharm 13(2):114–121. https://doi.org/10.22159/ijap.2021v13i2.40339
Gupta S, Vyas SP (2006) Development and characterization of Amphotericin B bearing emulsomes for passive and active macrophage targeting. J Drug Target 15:206–217
Zakaria MY, Zaki I, Alhomrani M, Alamri AS, Abdulaziz O, Abourehab MA (2022) Boosting the anti MERS-CoV activity and oral bioavailability of resveratrol via PEG-stabilized emulsomal nano-carrier: factorial design, in-vitro and in-vivo assessments. Drug Deliv 29(1):3155–3167
Ghosh A, Kaur CD, Gupta A, Saraf S (2017) Surface engineered lamivudine loaded emulsome for targeting drug delivery to lymphatic system for effective treatment of hiv. Int J Appl Pharm Biol Res 2(1):25–37
Priya S, Desai MV, Singhvi G (2023) Surface modification of lipid-based nanocarriers: a potential approach to enhance targeted drug delivery. ACS Omega 8(1):74–86. https://doi.org/10.1021/acsomega.2c05976
Zhang Y, Hong H, Cai W (2011) Tumor-targeted drug delivery with aptamers. Curr Med Chem 18(27):4185–4194. https://doi.org/10.2174/092986711797189547
El-Zaafarany SG, Mahmoud M, Cespi S, Palmieri M, Illum G, Casettari L, Awad L, Gehanne, (2018) A tailored thermosensitive PLGA-PEG-PLGA/emulsomes composite for enhanced oxcarbazepine brain delivery via the nasal route. Pharmaceutics 10(4):217. https://doi.org/10.3390/pharmaceutics10040217
Ucisik MH, Küpcü S, Breitwieser A, Gelbmann N, Schuster B, Sleytr UB (2015) S-layer fusion protein as a tool functionalizing emulsomes and Curcu Emulsomes for antibody binding and targeting. Colloids Surfac B: Biointerfaces. https://doi.org/10.1016/j.colsurfb.2015.01.055
Abdelmonema R, El-Eninb AAH, Abdelkaderc G, Abdel-Hakeem M (2023) Formulation and characterization of lamotrigine nasal insert targeted brain for enhanced epilepsy treatment. Drug Deliv 30(1):2163321
Alhakamy NA, Badr-Eldin SM, Aldawsari HM, Alfarsi A, Neamatallah T, Okbazghi SZ, Md S (2021) Fluvastatin-loaded emulsomes exhibit improved cytotoxic and apoptosis in prostate cancer cells. AAPS PharmSciTech 22(5):177
Misra SK, Pathak K (2023) Nose-to-brain targeting via nanoemulsion: significance and evidence. Colloids Interfaces 7(1):23
Acknowledgements
Not applicable
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SS prepared and wrote the initial draft of the manuscript and coordinated the incorporation of all the required data and major contributor in writing the manuscript. KK further led the checking and revising the manuscript for various aspects such as clarity, grammar and scientific content. IS and SBC have made mandatory contributions toward all the crucial editing, interpretation, provided critical feedback on the manuscript and revised the final revision. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There is no conflict of interest among the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, S., Khurana, K., Chauhan, S.B. et al. Emulsomes: new lipidic carriers for drug delivery with special mention to brain drug transport. Futur J Pharm Sci 9, 78 (2023). https://doi.org/10.1186/s43094-023-00530-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-023-00530-z