
Abstract
Background
Hemodynamic regulation is a substantial part of the physiological integrity of the human body. It is based on the delivery of proper blood perfusion to every organ. Five primary vasoactive substances are nearly located throughout the human body, either released from the endothelium, prostanoids, nitric oxide (NO), and endothelin-1 (ET-1); or considered as hormones, bradykinin (BK) and natriuretic peptides (NPs).
Main body
The circulating mediators are in synchronization with the renin–angiotensin system (RAS) during the pathogenesis of the main vital organs, heart, kidney, lung, liver, and brain. The RAS system has been an extensive therapeutic approach for cardiovascular and renal diseases for decades, but more recently became a crucial regulator of hemodynamics in other organs after the actions of its components were detected in other organs. All the mentioned disorders here begin with the initiation of abnormal imbalance between vasoactive mediators which causes vascular dysfunction and histopathological situations that may induce oxidative stress which exaggerates the disorder if there is no clinical intervention.
Conclusion
We will review the currently identified signaling pathways and the possible relationships between those compounds elucidating how they interfere with serious diseases including cardiovascular diseases (CVDs), chronic kidney disease (CKD), pulmonary arterial hypertension (PAH), portal hypertension (PHT), and Alzheimer's disease (AD). Thus, this updated review summarizes years of work that aims to define the contribution of each mediator in both normal and pathological states, besides the drugs based on their activity and their places in either preclinical or clinical trials.
Similar content being viewed by others


Background
To function properly, multiple tissues maintain constant blood flow during changes in blood pressure (BP) for appropriate organ perfusion. The endothelial cells that line such blood vessels secrete vasoactive substances to maintain functionality in response to abnormalities such as inflammatory mediators [1]. These compensating mechanisms are known as “Autoregulation.” Although the exact comprehensive steps of these processes are not completely defined, some hypotheses were introduced including myogenic and metabolic mechanisms that immensely involve vasoconstriction in response to high BP or preserving the vasodilating substances [2]. Among the first compounds proposed to influence autoregulation are prostaglandins (PGs) [3] and nitric oxide (NO) [4].
Regular vasoactivity can be exemplified by endothelin-1 (ET-1) which mediates both vascular smooth muscle (VSM) contraction by binding to endothelin A (ETA) receptor, and relaxation by binding to endothelin B (ETB) receptor. Another example is the ratio between prostacyclin (PGI2) and thromboxane A2 (TXA2) which regulates the balance between relaxation and constriction, respectively, which should be maintained in susceptible populations including patients with CVDs, as higher TXA2 concentrations could lead to drastic complications [5]. Prostaglandin E2 (PGE2) mediates VSM relaxation by binding to EP2 and EP4 and vasoconstriction by binding to EP1 and EP3 [6]. CVDs and cognitive impairment are two examples where natriuretic peptides (NPs) do not just serve as a therapeutic target, but also as biomarkers as we will explain, later.
Classical RAS system has an overall vasoconstrictive action, mainly, because of angiotensin II through binding to angiotensin II receptor type 1 (AT1R). On the other hand, angiotensin (1–7), a key player of the alternative system, generates NO by binding to Mas-related G-protein-coupled receptor type D (MrgD) [7].
This review does not cover all vasoactive mediators, but rather some of the endothelial and hormonal ones. This article is divided into two parts: first, it covers the mechanisms of action of the regulatory mediators including those that compose the RAS system in normal states. Second, it elaborates on their roles and the possible relationship between each other in either chronic diseases or treatments.
The pathophysiologies of the selected disorders are not explicitly discussed; for instance, genetic and environmental factors, but rather only the relevant ones associated with the mentioned mediators.
Main text
Vasoactive endothelium secretions
Prostanoids
"Prostanoids" is a term that covers both prostaglandins (PGs) and thromboxane A2 (TXA2), which also falls under the category of eicosanoids, which are lipid hormone-like compounds derived from arachidonic acid [8]. Those substances are produced by the constitutive cyclooxygenase-1 (COX-1) from the arachidonic acid and are present in most cells in normal physiology; therefore, COX-1 enzyme is considered a homeostatic regulator. Nevertheless, cyclooxygenase-2 (COX-2) is mostly induced by abnormal stimuli such as tumor promoters, and cytokines; but it also helps in the PGs generation function of COX-1 [8].
Typically, there are five main classes of G-protein-coupled receptors of prostanoids (Dp, Fp, Ip, Ep, Tp) to which PGD2, PGF2α, PGI2, PGE, and TXA2 bind, respectively [6]. Figure 1 illustrates the synthesis pathway and functions of prostanoids.

Synthesis and Functions of Prostanoids. Normally, the constitutive isoforms of COX enzymes are produced by the action of phospholipase A2 on membrane phospholipids; but in inflammatory diseases, phospholipase A2 is triggered to produce inducible COX-2. Prostaglandin H2 is the precursor of the five main types of prostanoids which generates them upon the catalysis of specific isomerase (synthase) enzymes for each type [6, 8]. GIT indicates gastrointestinal tract. VSM, vascular smooth muscle
Moreover, prostanoids are crucial for the modulation of pro- and anti-inflammatory responses depending on the status of the abnormality [9].
In kidneys, PGE2 and PGI2 have an anti-hypertensive effect through vasodilation. This effect is opposed by administering non-steroidal anti-inflammatory drugs (NSAIDs) [10].
In addition to being a potent platelet activator, TXA2 regulates BP, for that it was found, although mice deficient TXA2 maintained thrombopoiesis, there was a hemostatic irregularity due to the shift of arachidonic acid into the production of vasodilating prostaglandins, PGE2, PGD2, PGF2α, and PGI2, but it saved those mice from the drop of blood pressure compared to the TP deleted ones, which means other PGs have an affinity to TP receptor, adapting to each situation [11]. Accordingly, TXA2 antagonism and receptor blockade are still under investigation because of their efficient vasodilation over acetylsalicylic acid [12].
Endothelin-1 (ET-1)
There are mainly 3 types of endothelin polypeptides; (Endothelin-1, -2, and -3). Endothelin-1 (ET-1) is the most potent vasoconstrictor and is located in the endothelial cells of smooth muscles and myocardiocytes; thus, it has a major role in the pathogenesis of atherosclerosis and hypertension [19]. Endothelin-2 (ET-2) is found in intestinal epithelial cells, while Endothelin-3 (ET-3) is expressed in vascular endothelial cells and intestinal epithelial cells [19]. After successfully cloning the structure of preproendothelin-1, the precursor of ET-1, expressed in bovine carotid artery cells, which has the same length as in humans, it was postulated that Ang II and vasopressin were among the potent inducers of the parent protein [20].
There are two types of endothelin receptors, Endothelin A (ETA) and Endothelin B (ETB); that mediate vasoconstriction and vasodilation, respectively; since the stimulation of ETB receptor activates NO and prostacyclin resulting in vasodilation of VSM [21]. Binding to ETA receptor activates phospholipase C, resulting in increased cellular Ca2+ levels causing muscle contraction. ETA receptor is present in vascular smooth muscles. There are two types of ETB, ETB-1 that can be found in endothelial cells; while, ETB-2, like ETA, is located in the same areas producing the same effects upon binding. Hence, ET-1 is involved in chronic diseases including, but not limited to, hypertension, hepatic cirrhosis, chronic kidney disease, and pulmonary hypertension [22]. Endothelin receptor blockade drugs are either selective for ETA receptors such as ambrisentan and avosentan; or non-selective including bosentan and macitentan.
It is noteworthy that the ETA receptor has the highest affinity to ET-1, while all three types of ET bind similarly to ETB [23]. Both endothelin converting enzyme-1 (ECE-1) and -2 (ECE-2) preferably cleave Big ET-1 more than Big ET-2 and—3, generating ET-1 [24]. Also, ECE-1 is the major producer of ET-1 [23].
The contradictory coupling of NO and ET-1 which can be witnessed as low NO level usually becomes associated with elevated ET-1 release, and shear stress is controversial; nevertheless, this coupling is used as an effective therapeutic approach for several disorders [25].
Nitric Oxide (NO)
NO was identified in the early 1980s as (endothelium-derived relaxing factor) released as a result of the vasodilating effect of acetylcholine [13].
NO is, generally, produced from l-arginine amino acid by nitric oxide synthase (NOS), which is present in either a constitutive isoform or an inducible (iNOS) one. The constitutive NOS can be found in neurons, neuronal NOS (nNOS); or in the epithelium, epithelial NOS (eNOS), and platelets particularly in cardiomyocytes and airway cells. Constitutive NOS can be activated by substances, for example, acetylcholine and bradykinin (BK) under physiological conditions; meanwhile, iNOS is mainly stimulated by an abnormality such as infection [14].
NO activates guanylyl cyclase that catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine-monophosphate (cGMP), a regulator of platelet aggregation and contraction of cardiac and vascular smooth muscles. cGMP activates protein kinases releasing Ca2+ into the blood causing vascular vasodilation [15]. NO has a higher affinity to stimulate the soluble type of guanylyl cyclase (sGC) in preference to the membrane-bound one; thus, upon desensitization of sGC, the cellular responses to cGMP decline as a feedback inhibition of phosphodiesterases, namely phosphodiesterase 5 (PDE-5) counter effects; hence, tadalafil, an inhibitor of PDE-5, is approved to treat PAH [15,16,17]. Inhibition of the eNOS enzyme, caused by asymmetric-dimethyl-arginine (ADMA), is associated with detrimental disorders including cardiovascular, kidney disease [18], and pulmonary hypertension; consequently, its plasma level could be used as an assessment biomarker.
Vasoactive hormones
Natriuretic peptides (NPs)
The three substantial peptides are (1) Atrial natriuretic peptides (ANP), which by name, are secreted from the atria. (2) Brain natriuretic peptides (BNP) were detected in the brain of laboratory porcine but in humans to a little extent, but were identified to be secreted largely by ventricular myocytes in humans. (3) C-type natriuretic peptide (CNP), which has a more potent venous effect than an arterial one [26].
BNP is synthesized by its precursor, ProBNP, by proteolytic enzymes (corin and furin) along with the N-terminal prohormone of brain natriuretic peptide (NT-proBNP) [27].
The primary stimulus of the two peptides is the increased vascular resistance; however, the secretion of NT-proBNP could be induced by Ang II and endothelin, which means their plasma concentration can indicate cardiac disorders such as heart failure, coronary artery syndrome; particularly NT-proBNP due to its longer half-life [27].
Similarly, the precursor of ANP, proANP, is cleaved by corin to form the active molecule, NT-proANP which is elevated along with NT-proBNP in atrial fibrillation [28] and systemic sclerosis [29]. Interestingly, ANP regulates macrophage-mediated NO release as macrophages express iNOS, which suggests its role in reversing the deleterious effects of NO as an inflammatory factor [30].
A few years after discovering and upon the investigations of the ECEs, a new divalent cations metalloendopeptidase (NEP 24.11), which later became known as Neprilysin (Nep). This peptidase enzyme is responsible for the degradation and inactivation of the three natriuretic peptides besides, bradykinin, and endothelins; specifically, ET-1 [31, 32].
ANP and BNP exhibit their activities by binding to guanylyl cyclase which leads to vasodilation by activation of the cGMP signaling pathway, which also results in producing NO; consequently, feedback regulations of such peptides [33].
Nevertheless, CNP receptor does not follow that signaling pathway but has almost similar affinity for ANP and BNP considering the overall plasma concentration of CNP is quite low, and since the receptor was found to induce water excretion and increased perfusion in kidneys, it was proposed that its main function is wash out the NPs, although it induces relaxation of VSMs and cardiac myocytes by upregulation of NOS [34]. Washing out of ANP and BNP from plasma is either by excretion by natriuretic peptide C receptor in kidneys or by degradation by the insulin-degrading enzyme (IDE) and Nep [35].
NPs cause natriuresis by inhibition of renal Na channels, and inhibition of renin release which leads to diminished aldosterone levels, counteracting Ang II [36]. As compensatory mediators, all NPs are increased in case of heart failure, but only BNP is used as a diagnostic marker for the detection of cardiac impairment. Taking into account such positive effects, nesiritide, a recombinant BNP, was approved for acutely decompensated heart failure, but with no beneficial result in the overall mortality status [37].
Bradykinin (BK)
BK is a potent vasodilating kinin, metabolized by kininase-1 and kininase-2, the latter is also called angiotensin-converting enzyme (ACE) [38], meaning that the ACE inhibitory drugs (angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs)) elevate plasma kinin levels. Figure 2 shows the synthesis of BK, which contributes to angioedema and bronchoconstriction. The effects of BK are not fully understood, but the pain stimulation it causes could be correlated with PGs, and the vasodilation may result from the release of NO by binding to B2 receptor. BK has a high affinity for B2; while Des-Arg9-BK, an active metabolite of BK, predominantly binds to B1 receptor. Both receptors generate inflammation, but binding to the B2 receptor can, also, lead to positive actions by releasing NO and PGI2 [40].

The Correlation between renin–angiotensin system (RAS) and the kallikrein–kinin system (KKS). The interrelationship between the two systems can be found in Kallikrein which catalyzes the production of renin from its precursor (prorenin), and after the activation of the RAS, ACE-2 enzyme metabolizes bradykinin active metabolite, Des‐Arg9‐BK. C1 inhibitor protein inhibits bradykinin formation, directly by s suppressing the kininogen pathway; and indirectly by inactivating the kallikrein synthesis pathway from FXIIa factor [38, 39]
As an autacoid, there is a modulation between BK and other inflammatory mediators like NO in chronic diseases, and its solo inflammatory manifestations are witnessed in angioedema disorders. The primary receptors of BK are the G-protein-coupled receptors B1 & B2. B2 is physiologically constitutive but also involved in the acute phase of inflammation of somatic and visceral pain because it is more abundant than the B1 receptor [41].
Both receptors are located throughout the human body; particularly, in the thalamus and hypothalamus. Notably, BK is a primary inducer of COX-2-mediated inflammation as a result of binding to B2 receptor by long-term conversion of arachidonic acid to PGE2 [42].
There are several types of angioedema since there are mediators other than BK as histamine, but BK is currently corresponding to three types of the disease which are hereditary, acquired, and renin–angiotensin-system blocker-induced angioedema [43]. All of those types result from dysregulation of BK metabolism. In brief, the first two types are mainly associated with the deficiency of the C1 inhibitor (C1-INH) protein, a restrictor of activation of C1 protein of the complement system and a modulator of clotting formation. C1-INH downregulates the catalytic effect of the conversion of prekallikrein to BK as Fig. 2 illustrates; consequently, both types are associated with elevated plasma levels of BK [44]. The hereditary type is primarily either due to deficiency of gene expression of C1-INH or dysfunction of the gene which became prominently known to be mediated by BK [45]. The acquired type is due to auto-antibody response against C1-INH, which is correlated with the Kallikrein–kinin system (KKS); thus, when the infusion of C1-INH fails in the treatment of such case, kallikrein and BK receptor antagonists are considered [46].
The third type results as a rare side effect in case of long-term administration of RAS blocking agents which leads to higher plasma concentrations of BK that bind to B2 receptor leading to severe inflammation and increased capillary permeability which leads to angioedema; hence, drug withdrawal may stop the symptoms [43].
Renin–Angiotensin system (RAS)
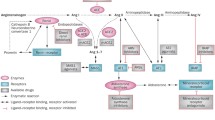
RAS has a prominent role in the hemodynamics of vital organs, especially in chronic disorders. Briefly, the RAS system activation begins essentially from renin, a proteolytic enzyme that converts its substrate, angiotensinogen, into angiotensin 1 (Ang I), which is converted later by angiotensin-converting enzyme 1 into Ang II. Furthermore, angiotensin-converting enzyme 2 (ACE-2) catalyzes the conversion of Ang II to Ang (1–7). ACE-2 may also convert (Ang I) to Ang (1–9), which is then converted to Ang (1–7) by ACE and Nep [39] as Fig. 2 elucidates.
Ang II to some extent and Ang III, an active metabolite of Ang II with the same effects, to a higher one, induce aldosterone release from zona glomerulosa of the adrenal cortex that aids in elevating BP, mainly, as a consequence of Na and water retention by acting on Na–K pumps. Consequently, aldosterone has been considered a substantial addition to the RAS, to which could be referred as renin–angiotensin–aldosterone system (RAAS) [47].
The higher extent of ACE-2 cleavage of Ang II to Ang (1–7) than converting ANG I to Ang (1–9) by binding to Mas-related G protein-coupled receptor type D (MrgD) elevates eNOS by protein kinase B signaling [7], which overall, mediates therapeutic benefits such as vasodilation, anti-inflammatory, and antifibrotic actions [48], which may explain why ARBs are more effective than ACEIs to some proportion in certain cases.
However, it was recently established that Ang (1–7) is mostly produced by direct cleavage of Ang I by neprilysin, and by less extent by the other enzymes in both murine and human kidneys in normal state and chronic kidney disease (CKD) and by Ang II pathway as alternative synthesis pathways [49].
The counterpart interactions between Ang II and NPs have been known for years which was witnessed upon the inhibition of Ang II by quinapril and candesartan inducing the activity of ANP in a preclinical study [50].
Although renin is mostly generated by the granular juxtaglomerular cells of the renal arterioles, the expression of the mRNA of renin was detected in the heart in severe conditions such as severe reduction in Na+ levels [51].
As ACE degrades the production of BK, hampering the enzyme by ACEIs would elevate BK levels; however, ARBs have a lower incidence of cough as a side effect than ACEIs by blocking BK production [52, 53]. As NOS can be stimulated by BK, administrating anti-renin–angiotensin–aldosterone drugs could increase the expression of NOS gene restricting oxidative stress.
Although there were some doubts about the direct influence of the RAAS on endothelin production, it was found that upon the expression of Ang II gene, endothelins elevate along with the upregulation of ETA receptors and downregulation of ETB [54]. Moreover, the liberation of NO and prostacyclin by the activation of ETB receptors was proved to be beneficial in female rats in renal damage modeling [55]. Moreover, ETB regulates BP by helping in the excretion of Na and water, while it is downregulated as in the case of depleted salt intake as a result of increased Ang II release; thus, administration of ARBs elevates the expression of ETB receptors in cardiovascular disorders [54].
It was found that Ang II stimulate catecholamines in pithed rats by binding to angiotensin II receptor type 1 (AT1R), even with the use of ganglionic blockade in a dose-dependent manner, but its sympathetic mediated effects were suppressed by initiating candesartan [56].
Cardiovascular diseases (CVDs)
Hypertension is one of the most prominent cardiovascular risk factors that predispose to cardiac complications such as coronary artery disease and heart failure (HF), which have significant mortality rates in 2019 [57].
Although the RAAS system is a major key element in CVDs, it is often normal in black people while the elevated Na+ levels were the primary cause besides low excretion of potassium even in normal intake which suggests the beneficial role of K+ as concluded in a recent study [58], which is why ACEIs and ARBs are not at the first line for their treatment of hypertension [59]. Even an updated study stated that administration of angiotensin receptor-neprilysin inhibitor (ARNI) (Entresto®) which combines a Nep inhibitor, sacubitril, and an ARB, valsartan, for heart failure, resulted in low but acceptable adherence with Africans compared with other participants; meanwhile, a previous initiation of ACEIs/ ARBs can improve the adherence issue, yet that solution needs further assessment [60].
Among the endothelial dysfunction mechanisms of aldosterone associated with CVDs is the alteration of COX-2 metabolites, specifically elevating the ratio of TXA2/ PGI2; although, aldosterone and renin may be at normal levels, especially in blacks [61], considering the detrimental effects of TXA2 in such disorders, which leads to the proposition of treatment with COX-2 inhibitors [5, 62].
Prostacyclins show cardioprotective action by inhibition of platelet aggregation besides relaxation of the systemic vascular endothelium as they are produced by not only COX-1 but also by constitutive isoforms of COX-2 enzyme which were found to regulate the homeostasis of the kidneys by regulating renin release and renal blood perfusion [63].
PGI2 produced by COX-2 counteracts thrombosis mediated by TXA2 which may lead to thrombosis risk upon initiating coxibs, selective COX-2 inhibitors, since TXA2 is produced mostly by COX-1 enzyme. Because of other side effects such as abdominal bleeding of the non-selective COX inhibitors, both types of NSAIDs should be used only if necessary with precaution in CVD, taking into account the exacerbations of NSAIDs, particularly with patients who have a history of CVD [64].
PGE2 has controversial cardiac effects; for that reason, much effort was made to develop receptor agonists of the four PE2 receptors that are described here [65]. EP3 and EP4 are almost the most remarkable receptors to attenuate inflammatory cytokines in ischemic heart models.
Peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear receptor family that has multiple roles in organism’s biological processes whose activity is exhibited by binding to ligands namely prostaglandin PGJ2, a dehydrated form of PGD2 [66]. One of the crucial roles of PPARγ agonism is anti-inflammatory which appeared markedly in using the antidiabetic thiazolidinediones by inhibiting cytokines as well as improving contractile function by lowering insulin resistance in the process of glucose metabolism [67]. However, this class was taken out of the market in EU countries and the FDA black-boxed the category, specifically rosiglitazone, due to its association with myocardial infarction and possible heart failure due to overactivation of NPs and collagens causing cardiac hypertrophy [67].
Many cases have been reported regarding the exaggerated angioedema associated with bradykinin-releasing drugs namely ACEI, ARBs, and Dipeptidyl peptidase-4 inhibitors; including an old female patient who was diagnosed with acquired angioedema and stopped using sitagliptin and irbesartan, but owing to interrupted follow-up and taking ramipril, even icatibant, selective BK B2 antagonist, and tracheostomy were unsuccessful which led to her death [68].
Despite the main indication of endothelin antagonists is pulmonary arterial hypertension and that endothelin influence is, perhaps, lower than Ang II and sympathetic adrenergic vasoconstriction, ET-1 receptor blockers could only be used after assessment in hypertension, particularly in resistant cases [69]. The imbalance between NO and ET-1 could be a risk factor for essential hypertension. Moreover, certain concomitant gene polymorphisms of ACE and eNOS were added to the currently known risk factors of coronary artery disease [70].
To take advantage of NPs, omapatrilat was developed based on Nep and ACE inhibition showing positive effects in the atrium in heart failure with reduced ejection fraction (HFrEF) due to ANP release [71], and higher efficacy than enalapril as in OCTAVE trial in hypertensive patients and those with concomitant heart failure; however, it resulted in more cases of angioedema than enalapril [72] and its approval was denied. Entresto is a newer drug that was approved for HFrEF (NYHA class II to IV) in 2015 [73] and became recently indicated in HF with preserved ejection fraction and left ventricular hypertrophy [74]; besides having a greater efficacy in lowering BP in comparison with ARBs [75] and ACEIs [76]. According to the PIONEER-HF clinical trial of 881 patients diagnosed with HF, only one non-black patient experienced angioedema with Entresto; moreover, other side effects, for example, hypotension, hyperkalemia, and renal dysfunction were higher during Entresto administration compared to enalapril in all races, even though the blacks had an overall lower risk ratio of side effects than non-blacks [77].
Exploiting the alternative pathway of the RAS, a novel nonpeptide analog of Ang (1–7), AVE0991, was found to decrease the mass of collagen mass of the fibrotic hearts of rats improving cardiac functions protecting them from high BP [78] reducing oxidative stress-mediated enzymes [79].
Chronic kidney disease (CKD)
The protection activities of PGE2 in kidneys were witnessed when BK [80] and noradrenaline [81] were infused in animal models which resulted in induction of diuresis to wash out the vasopressors; however, other PGs such as PGF2α may not be induced in such extent of the PGE2 due to lower abundance and potency [80, 82].
PGs contribute to renal functions, particularly, PGE2 which increases glomerular blood flow by afferent vasodilation which aids the filtration function of the kidney; due to the expression of both COX-1 and -2 and phospholipase A2 in endothelial and mesangial cells [83, 84].
Normally, Ang II causes vasoconstriction of the efferent renal artery to promote the filtration function, but upon its overproduction, intraglomerular pressure rises and the GFR elevates followed by dysfunction of glomerular barrier allowing Na and water retention and glucose reabsorption, this can be observed in type 2 diabetes mellitus [85].
This explains an important drug interaction between afferent arteriolar vasoconstrictors (i.e., NSAIDs, COX-2 inhibitors, and cyclosporine) and efferent vasodilators (i.e., ACEIs and ARBs) that can cause a significant decline in renal function [86] as Fig. 3 shows.

How drugs alter renal perfusion, especially if co-administrated. Prostaglandins (PGs) cover the endothelium of afferent arteriole causing its vasodilation, while angiotensin-2 (Ang II) binds to angiotensin II receptor type 1 (AT1R) located in the efferent arteriole resulting in vasoconstriction. Non-steroidal anti-inflammatory drugs (NSAIDs), cyclooxygenase-2 (COX-2) angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), Calcium channel blockers (CCBs) [86, 87]. Figure created with biorender.com
Ang II was found to cause direct apoptosis to the podocytes, the third layer forming cells of the glomerular filtration barrier, by upregulation of Na+ /H+ exchanger type 1 (NHE-1) protein, an intracellular pH regulator which is correlated with reactive oxygen species (ROS) production, which was prevented by administration of olmesartan and amiloride, potassium-sparing diuretic and NHE-1 inhibitor [88]. The same pathogenic pathway of Ang II occurs in proximal tubules as the peptide activates NHE-3 through the same complicated signaling pathway which includes protein kinase C. The inhibition of NHE-3 could be a new antihypertensive approach [89]. After its induction by Ang II, aldosterone advances podocyte injury by mitochondrial damage by enhancement of ROS signaling which is demonstrated as the elevation of ROS biomarkers in in vivo and in vitro studies [90]. Abnormal glomerular disorders and decreased GFR may cause CKD, the end-stage renal disease, which is one of the complications of diabetes mellitus.
Another pathogenic factor of CKD is reduced NO production following eNOS inhibition by ADMA [91]. Inhaled NO was proved effective in the prevention of AKI risk during cardiopulmonary bypass surgery besides enhanced pulmonary perfusion and general anti-inflammatory properties that led to a better response, particularly for those with PAH [92]. The same positive results with nitroglycerin in cardiopulmonary bypass surgery due to maintenance of renal and cerebral perfusion [93].
Since ETB is underregulated and considering its aspects, ETA selective antagonism should theoretically provide more beneficiaries than the non-selective ones to support NO generation; however, multiple clinical trials were developed to assess the role of ET antagonists including the ASCEND trial of avosentan, and it was concluded that despite the improved albuminuria, other adverse events such as heart failure and fluid retention have restricted the use of such drugs except for considering risk–benefit ratio [94].
Due to their proven beneficial effects, ACEIs and ARBs are included in the first lines for diabetic nephropathy and CKD according to the latest guidelines [95]. Aliskiren, a renin inhibitor drug, hinders the expression of angiotensin II receptor type 2 and ACE-2 enzyme [96]; thus, aliskiren is restricted to cases characterized by overactive RAAS system such as hypertension and not used in renal failure due to its overall detrimental implications [97]. Therefore, combination therapy of ACEIs or ARBs with ET antagonists can be beneficial at low doses [98].
Because there is a strong link between renal and cardiovascular pathology, in a follow-up study of 239 CKD patients stage 4–5, a directly proportional relationship between NT-proBNP and edema was detected. Furthermore, 48 participants had cardiovascular adverse events and 23 of them died; confirming the effectiveness of the peptide as an active diagnostic marker of HF and CKD [99]. Owing to the satisfactory impacts of NPs such as improved GFR and lowering renal BP, ARNI was studied in cardiorenal syndrome (CRS)-induced Sprague–Dawley rats which proposed promising results in cardiorenal preservation by maintaining left ventricular ejection fraction and kidney functions saving the animal life using a high dose (100 mg/kg/day) which promotes its indication in HF associated with CKD [100].
B2 receptor of bradykinin was found in mesangial cells of rats contributing to their contractile function as binding to B2 receptor, specifically, stimulates inositol trisphosphate and Ca2+ to release PGE2, serving its physiologic role [101]. More recently, it was found that BK has also a proliferative function by inhibiting transforming growth factor-β1 (TGF-β1) inflammatory signaling inducing apoptosis to slow renal fibrosis [102].
Pulmonary arterial hypertension (PAH)
PAH is a rare but life-threatening condition characterized by increased pulmonary vascular resistance (PVR) and blood pressure (≥ 25Hgmm). It is the first group of the five categories of pulmonary hypertension (PH) as classified by the World Health Organization, which can be associated with hypertension, heart failure, and frequently a bad prognosis [103]. PAH could be either due to an abnormal genetic or an idiopathic factor. PH can be associated with right-sided HF, a rare case but frequent only as a consequence of left-side HF (group 2 PH). PAH may manifest in association with pulmonary vascular damage (sclerosis) and chronic inflammatory lung diseases [104]. Principally, the pathogenesis includes atypical vasoconstriction and exaggerated inflammatory response.
Upon initiation of proinflammatory mediators, hypoxia, and deterioration of the vascular function of cardiopulmonary vasculature, NT-proBNP is released as a biomarker for the failure of the right [103]. Based on that and due to the intervention of the RAAS system in the pathogenesis of HFrEF, ACEIs and ARBs proved to lower mortality rates in pre- (pulmonary vascular dysfunction) and post-capillary PH (left-sided HF) [105] besides normalizing filling pressure followed by relaxing the pulmonary artery [106]. Indeed, the same goes for ARNI considering the additional advantage of ANP and BNP preservation which helps to lower pulmonary pressure in patients with HF including blacks [107].
As ET-1 induces Ca2+ pulmonary contraction, Ca channel blockers were used in the early management of the disease along with anticoagulants for HF associated with PAH. Newer agents were approved to treat PAH itself rather than HF such as NO derivatives and PGs analogs which are two of the principal treatment options for PAH.
Recombinant human ACE-2, GSK2586881, is also a novel agent which showed significant improvements in inflammatory biomarkers of PAH and limiting oxidative stress with no concerning safety precautions in a clinical trial [108]. In another small trial, it was proved to be safe upon intravenous administration by healthy subjects; yet, no improvements in the hypoxic state after the exercise test [109].
From investigations of PAH in newborns, persistent pulmonary hypertension of the newborn, endothelial cells were able to generate NO endogenously during gestation and had high tolerability to exogenous NO donor, for example, Na nitroprusside and inhaled NO in animals, and later, when it was combined with oxygenation, neonates had notable improvements; mainly because inhaled NO and prostacyclin have a local pulmonary effect rather than vascular systemic effect [110, 111]. Nonetheless, restricted doses are still required to avoid the risks, particularly NO as it can be metabolized to nitrogen dioxide which causes lung damage in a such susceptible population [111]. Since the pathogenesis of PAH includes impairment of the NO-sGC-cGMP pathway, a new oral drug, riociguat, was approved to target that approach by maintaining the sensitization of sGC for the NO to bind to its receptor. It showed the potential to reduce vascular resistance and NT-proBNP levels [112] with no serious adverse drug events compared to the control group [113].
PGs play a crucial role in the respiration process, as PGI2 is produced after fetus delivery to aid airway vasodilation and pass oxygenated blood to other organs [111]. Therefore, it is not surprising that Velvis et al. assumed that COX inhibition suppressed the reduction in PVR along with the aforementioned balance between PGI2 and TXA2 [114] which should, also, be considered to prevent thrombotic pulmonary hypertension.
Epoprostenol, the first powerful prostacyclin analog, was approved for PAH with multiple benefits including vasodilatory effects and inhibition of platelet aggregation [115]. Newer analogs were approved for PAH, iloprost and treprostinil.
Iloprost normalized ejection fraction and right ventricular function in chronic PAH rat model similar to clinical cases which used a dose-dependent aspect [116].
Treprostinil had similar efficacy with improvements in cardiopulmonary signs and symptoms of PAH [117] along with a decreased risk of sepsis which resulted in remarkable cost-effective treatment when it was tested against epoprostenol [118]. Additionally, inhaled therapy of treprostinil showed a 15% reduction in NT-proBNP against a 46% increase in the placebo group upon its testing in a randomized trial of efficacy in PH in association with interstitial lung disease besides less worsening of the case against the placebo group of patients [119].
A small cohort study concluded that initiating selexipag, a new oral PGI2 agonist, is more efficient than beraprost sodium, the first oral prostacyclin analog, and epoprostenol for children with PAH [120] due to chemical stability, longer half-life, and its conversion into an active metabolite upon its degradation.
Tadalafil showed better oxygen saturation than sildenafil upon drug transition in children and young adults [121]; not to mention the one-time daily dose. A small study of older adult patients showed that due to adverse effects that patients on sildenafil experienced, the transition to tadalafil was successful, despite considerable intolerability to the transition and switching back to sildenafil [122].
Vascular injury of PAH results from autoimmune sclerosis and is characterized by the initiation of proinflammatory cytokines including TGF-β1 and tumor necrosis factor α (TNF- α); while macrophages initiate fibrosis [123]. Again, endothelin overexpression following low production of NO and PGs composes a critical marker of the disease [124]. ET-1 was found to be in increased amounts in PH cases, specifically groups 1 and 3, compared to normal lungs [125] increasing capillary permeability and vascular pressure which may lead to fluid retention causing edema. Nonetheless, ET-1 was anticipated to be an anti-apoptotic agent, normally, by binding to ETA receptors in vivo and in vitro, which supports the functionality of non-selective ET-1 antagonists more than the selective ones in PAH [126].
Considering endothelin receptor blockers, Zhao Q. et al. concluded in a meta-analysis that both ambrisentan and bosentan showed similar therapeutic results [127]. Ambrisentan also showed higher potential when combined with tadalafil against monotherapy of each drug on its own [128]; nevertheless, its combination with riociguat showed no promising outcomes in an open-label study [129]. Indeed, other factors such as the long duration of action and having an active metabolite, as with macitentan, increase the efficacy against ambrisentan [130].
B9972, a bradykinin analog and a B2 agonist, showed apoptotic activity to the hyperproliferative cells in HF-associated PH which is followed by reduction in pulmonary pressure and right heart hypertrophy [131]; while nearly the same opposite results occurred on investigating bradykinin antagonist, B9430 [132], due to the bradykinin-activated eNOS pathway which induces PGI2 synthesis that enhances vasorelaxation effects.
Portal hypertension (PHT)
PHT is a syndrome resulting from a constant abnormal increase in BP within the portal system mostly by cirrhosis, the end stage of fibrosis, with predisposing factors such as ascites and variceal bleeding. High-output heart failure, a sporadic type of HF characterized by a high mortality rate, is mostly associated with cirrhosis due to decreased peripheral resistance and increased cardiac output [133]. While intrahepatic circulation is impacted by high plasma levels of vasoconstrictors and low vasodilators, splanchnic circulation is the opposite, and reversing the hemodynamics defines the therapeutic approach. PHT involves norepinephrine, Ang II as vasoconstrictors; and BK and PGs as vasodilators; but ET-1 and NO compose the most prominent vasoactive substances.
Liver sinusoidal endothelial cells (LSECs) and hepatic stellate cells (HSCs) are two major types of cells that play a vital role in homeostasis and vasoregulation. HSCs are fibrosis-forming cells present in between LSECs and hepatocytes. Their contractile state is regulated by Et-1 which is produced, normally, in lower amounts by LSECs [134].
HSCs exaggerate the secretion of ET-1 [134], which is, also, regulated by proinflammatory mediators including TGF-β1 [135] and TNF-α [136] as well as platelet-derived growth factor and Ang II [135]. Spleen is also a primary site of ET-1 production in cirrhotic patients [137]. iNOS is more abundant than eNOS in the hepatic Kupffer cells, macrophage-like cells, and modulated by nuclear factor-κB (NF-κB) which also stimulates TGF-β1 and TNF-α.
The disproportionate relationship between ADMA and eNOS provokes reactive nitrogen species (RNS) [138].
LSECs are characterized by a distinctive permeability barrier that aids in fenestration which is regulated by Vascular endothelial growth factor (VEGF). Its downregulation results in activation of HSCs to form fibrous tissues [139]. The activity of VEGF is mediated by NO which also regulates the HSCs cycle, as it induces its apoptosis via oxidative stress, meaning it is a crosstalk between both types of cells [139]. The production of VEGF which is associated with the release of iNOS and the predominance of ET-A receptors, located on HSCs, over ET-B, present on LSECs surface, are two factors that contribute to the angiogenesis and metastasis in the liver [140]. It was found that chronic non-selective antagonism of ET-1 has potential anti-cirrhotic results and is even more feasible to reach the desired portal pressure goals (< 10 mmHg) than ambrisentan by lowering intrahepatic vascular resistance; although, both drugs ameliorated the angiogenesis of portosystemic shunt and mesentery [141]. Ambrisentan is less hepatotoxic than bosentan; thus, safer in advanced liver cirrhosis; therefore, the benefit/ risk ratio should be evaluated [141].
As Ang II is released in liver injury following the stimulation of the RAS system, Ang (1–7) generates NO upon binding to MrgD through the protein kinase B (Akt) pathway [142]; while MrgD receptor agonism contributes negatively to splanchnic hemodynamics in PHT; hence, its blockade composes a new therapeutic option [142]. Meanwhile, NO is released as a response to intrahepatic vasoconstriction exaggerating portal-splanchnic circulation dynamics by vasodilation by a phenomenon called “NO paradox,” along with other vasodilators including PGE2 and PGI2 [143]. Riociguat was tested in cirrhotic rats at early and advanced stages, and the PHT was reduced by (24%) to elevate NO levels intrahepatically by the NO-sGC-cGMP pathway. Notably, this pathway is restricted to damaged hepatic sinusoids not affecting splanchnic circulation, unlike NO donors which have systemic effects [144]. For example, vasopressin and nitroglycerin combination demonstrated systemic vasoconstriction and diminished cardiac output followed by a remarkable decrease in hepatosplanchnic perfusion in normal persons, regardless of other side effects that may manifest in the long-term which were not included in this study [145]. Moreover, riociguat was effective in reducing vascular resistance and PHT in biliary cirrhotic animals when combined with tadalafil [146].
A meta-analysis of seven studies that included 1066 patients with liver cirrhosis demonstrated that ACEIs, including captopril, lisinopril, and enalapril, are potent antifibrotic agents since they decreased pathogenic markers of hepatic cirrhosis [147]. Moreover, a large retrospective cohort study of 12,327 non-alcoholic fatty liver disease patients concluded that ACEIs were more hepatoprotective than ARBs against cirrhosis complications and cancer [148].
On the other hand, studies on the overall effectiveness of ARBs were controversial because, for instance, unlike propranolol, losartan did not reduce hepatic venous pressure gradient in patients who had an episode of variceal bleeding besides arterial hypotension and altered GFR [149]. However, losartan was as effective as propranolol in lowering hepatic pressure in moderate liver disease with successful management of variceal bleeding [150]. Even upon its addition to propranolol, candesartan did not show better therapeutic outcomes than the non-selective beta-blocker monotherapy [151]; however, due to their antifibrotic effects, ARBs could be added to treat scarring tissues in advanced liver disease [152].
Entresto reduced arterial pressure and the intervention of macrophages in a PHT animal model; yet, not NF-κB and iNOS, but only sacubitril not valsartan diminished PHT [153]. The effects of ARNI on NO and ET-1 are complicated and require further clarification.
Regardless of the reduction in cardiac output and heart rate in PHT, a new study confirmed that the use of non-selective beta-blocker in cirrhotic patients including those who had surgery of trans-jugular intrahepatic portosystemic shunt had an additional advantage of a dramatically decreased magnitude of Ang II [154], which favors Ang (1–7) thus normalizing the porto-mesenteric circulation. This gives another reason to use ACEIs and ARBs since both peptides are upregulated in liver cirrhosis as an add-on [155].
AVE0991 was found to decrease PHT in rat models but not arterial pressure without affecting systemic vasodilation, either by NO-mediated MasR binding or interference with AT1R signaling [156]. It is not confirmed if it has anti-cirrhotic impacts in the long-term use.
Due to the proven positive effects of ameliorating PAH, tadalafil, macitentan, and selexipag were used as a triple combination therapy to improve the stress test of a progressive portopulmonary hypertension, PAH in association with PHT, in a very recent and infrequent case [157]. Selexipag improved the escalated dyspnea status from WHO functional class III to class I.
Gao JH et al. tested the role of a combination of celecoxib and octreotide, a somatostatin analog used in the management of variceal bleeding, in PHT-induced rats by thioacetamide, since the combination demonstrated an anti-angiogenic effect in the case of hepatocellular induced carcinoma in an animal model, owing to the presence of intra- and extra-hepatic angiogenesis by in vivo suppression of COX-2/ PGE2/EP-2/somatostatin receptor-2 signaling paradigm [158]. Later, they proposed that the same combination may be used as a prophylaxis for PHT due to the restriction of hepatocyte apoptosis which would ameliorate the disease [159]. More investigations regarding the combination need to be made considering cardiac and renal side events of COX-2 inhibitors.
Alzheimer's disease (AD)
As the most common cause of dementia, AD is an umbrella term that covers memory loss and cognitive impairment that interfere with the patient’s daily life. Amyloid β (Aβ) protein and tau are the two principal proteins of the pathogenesis of AD that form 10–15 years in advance before the diagnosis which makes it a slowly progressive disease [160]. Normally, tau proteins stabilize the microtubules, giving cerebral neurons internal support; meanwhile, in AD, tau proteins lose such ability and form neurofibrillary tangles upon aggregation that may induce stress response [161].
Amyloid precursor protein (APP) is cut into fragments of Aβ upon its adherence to the neuronal membrane of the cerebral cortex by β-amyloid–converting enzymes, α- and γ-secretases. These fragments form clusters of plaques surrounded by astrocytes, star-shaped cells that regulate neuronal cells, and microglia which activate iNOS [162]. The genetic mutations besides apolipoprotein E alleles will not be discussed here. iNOS was identified in such areas in the pathogenesis of AD and other neurodegenerative diseases such as Parkinson's disease and stroke, stimulating RNS; hence, it was found that iNOS-deficient mice were protected from AD by prohibiting Aβ accumulation and tau deactivation [163].
The three types of NOS are present in the brain. There is a proportional relationship between the provocation of NO by nNOS, present in the cortex of the hippocampus which maintains memory, and the binding of glutamate to n-methyl-D-aspartate (NMDA). This is clear in ischemic states as upregulated NMDA abnormally elevates NO concentrations that bind to superoxide anion forming RNS intermediate, peroxynitrite, which is distinguished by cytotoxicity [162]. Therefore, NMDA blockade by memantine is a treatment step that slows down AD progression. According to the cholinergic hypothesis, acetylcholine is an important factor for memory preservation due to its proportional relationship with NO-cGMP signaling, thus inhibiting its degrading enzyme, esterase, is considered another important approach [164]. In this manner, sildenafil was effective in memory restoration in preclinical studies [164]. Jiansong Fang et al. developed a computational study on AD patients and concluded that sildenafil is neuroprotective against neuronal tau formation [165]. Tadalafil also exhibited neuroprotection against oxidative proteins [166].
ETA receptors are located in pericytes, present in brain capillaries to support its blood barrier; while ETB receptors are widely expressed in astrocytes upon which activation by ET-1 binding [167, 168]. This attributes to the disease immensely by vasoconstriction and stimulation of proinflammatory response. Therefore, both receptors antagonism proved to reduce APPs in a murine model by improving the disruption of cerebral edema during traumatic brain injury in animal models [169].
ECE-1 was among the suggested enzymes that cleave Aβ, as upon its inhibition by phosphoramidon in hamsters, accumulations of the plaques were noticed suggesting its beneficial role [170]; thus, its genetic mutation increases Aβ plaques [168]. Although other studies propose as it generates ET-1 [171] and corresponds to Aβ protein, ECE-1 is a double-edged element in AD pathophysiology.
It is noteworthy that all the components of the RAS system are expressed in the brain, but at different degrees, for instance, renin and angiotensinogen are present in inferior amounts, unlike ACE, which is also another strongly proposed degrading enzyme of Aβ [172]; thus, its inhibition by ACEIs causes excessive amyloidosis. Nonetheless, angiotensin-blocking agents could be beneficial, especially ARBs, since ACEIs prevent Ang (1–7) and Ang (1–9) formation and such peptides bind to AT4 receptor which is known for facilitating memory and learning promotion. Like ACE, Nep also degrades Aβ in the cerebral cortex to an even higher extent than other enzymes, ECEs and IDE, since it is almost non-selectively depose oligomeric and monomeric forms of Aβ [173] and both Aβ1–40 and Aβ1–42 isoforms, which correlate with the neurodegenerative impacts of ET-1 if produced in higher concentrations making Nep preservation a potential AD target.
To determine the comparative effects of ACEIs and ARBs on AD patients with mild cognitive impairment, a retrospective control study showed that ARBs were associated with a lower risk of AD than ACEIs [174] and remarkable improvement of AD patients with physical disability than ACEIs [175]. This could also be explained by the fact that Ang III induces Mitogen-activated protein kinase (MAPK) enzyme [176], activating abnormal astrocyte growth through binding to AT1R which is blocked by those medications. In addition to the concept of the negative influence of ACEIs in AD. Moreover, AVE0991 reversed neuroinflammation and maintained the blood–brain barrier (BBB) in the Sprague–Dawley rats model by targeting Ang (1–7)/ Mas mechanism, which can be an advantageous add-on post-surgery drug for clinically susceptible patients [177].
Taken together, this is perhaps why valsartan/ sacubitril caused no effect on cognition when it was compared to valsartan monotherapy in the PERSPECTIVE study [178] and the same results against enalapril in PARADIGM-HF trial among patients with HF [179]; however, in a very recent large cohort study, there was a smaller mortality incidence, and neurocognitive diagnosis, among those who administered Entresto than those who took ACEIs/ ARBs, 25.8% vs. 31.2%, respectively [180]. Before initiation of valsartan/ sacubitril in case of cognitive impairment in future clinical trials, it should be noted that NPs such as BNP [181] and NT-proBNP [182] are robustly elevated in deteriorated cognitive examinations.
Since both COX-1 and -2 isoforms are expressed in microglia during the progression of AD [183] and considering the correlation between phospholipase A2 and Aβ formation besides arachidonic acid-derived MAPK relationship with phosphorylation of tau; in addition to the upregulation of PGE2, PGD2, and TXA2 in AD, the inhibitory effect of NSAIDs on inflammatory mediators such as interleukins, cytokines, and PGs was considered as a clinical option as a conventional therapy during AD [184]; but because most PGs play constitutive roles, the overall influence of NSAIDs was negligible in ADAPT trial of naproxen and celecoxib [185]. However, after considering the BBB permeability of naproxen, it was studied computationally, suggesting that it may have an anti-aggregation effect against Aβ [186]; and again in-vivo animal study in combination with rivastigmine, acetylcholine esterase inhibitor, but the positive synergistic effect was limited to neurogenesis and neuronal preservation with no cognitive ability enhancement [187].
Upon examining BK receptors, loss of occludin (zonula occludens), which contributes to the structural integrity of the BBB, was observed which facilitated more permeability for cytokine release and leukocyte adhering substances upon activation of B1 receptor by Des‐Arg9‐BK [188]. Notably, the same effect was noted upon injection of a high dose of kallikrein, but with mRNA of B2 receptor stimulation and eNOS; while in lower dose elevated iNOS and B1 expression as well as BBB hyperpermeability with downregulation of zonula occludens in both cases [189]. Nevertheless, a study of the corresponding relationship between B2 receptor and nerve growth factor (NGF), a neuroprotective peptide, showed that in neurodegenerative disease, B2 receptor is underexpressed in favor of BK and B1 receptor which diminishes the effect of NGF, confirmed by B2 selective antagonism, by icatibant, that reversed the rescue effect of NGF [190]. Furthermore, an in vivo mouse study concluded that elevated BK levels may worsen AD, in addition to the possibility of contribution of the peripheral BK to the disease due to delocalization of Aβ [191].
Conclusion
The human body compensates for the needs of each organ by stimulating vasoactive substances which play a crucial role in organ functionality. The imbalance between those mentioned mediators can deteriorate already critical statuses as seen in the CRS and portopulmonary hypertension which requires rapid intervention of combination therapies of mediator-based drugs besides the indicated one as those defects are multifactorial. KKS and the alternative pathway of the RAS are promising targets since they intervene in several disorders. Despite the possibility of an indirect relationship with certain pathogeneses, they can pave new paths for more effective therapeutics than conventional treatments to achieve better therapeutic goals.
Accordingly, and considering what this article discussed, future clinical trials should, mainly, focus on three aspects. First, the potential candidates such as AVE0991, since it demonstrated competence in restoring the normal state in several diseases of animal models, but its efficacy and safety profiles need to be determined in human trials; also, the long-term use of the ARNI drug in chronic diseases. Second, the NO-sGC-cGMP signaling pathway should be reviewed as it requires more drug candidates to be tested. Finally, the theoretical hypotheses of utilizing NSAIDs, including COX-2 inhibitors, in such pathologies, take into account their detrimental side effects.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- ACE-2:
-
Angiotensin-converting enzyme 2
- ACEIs:
-
Angiotensin-converting enzyme inhibitors
- AD:
-
Alzheimer's disease
- ADMA:
-
Asymmetric-dimethyl-arginine
- Ang I:
-
Angiotensin 1
- Ang(1–7):
-
Angiotensin (1–7)
- ANP:
-
Atrial natriuretic peptides
- ARBs:
-
Angiotensin II receptor blockers
- ARNI:
-
Angiotensin receptor-neprilysin inhibitor
- AT1R:
-
Angiotensin II receptor type 1
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid β
- BBB:
-
Blood–brain barrier
- BK:
-
Bradykinin
- BNP:
-
Brain natriuretic peptides
- BP:
-
Blood pressure
- CNP:
-
C-type natriuretic peptide
- C1-INH:
-
C1 inhibitor
- CKD:
-
Chronic kidney disease
- cGMP:
-
Cyclic guanosine-monophosphate
- COX-1:
-
Cyclooxygenase-1
- COX-2:
-
Cyclooxygenase-2
- CRS:
-
Cardiorenal syndrome
- CVDs:
-
Cardiovascular diseases
- ET-1:
-
Endothelin-1
- ET-2:
-
Endothelin-2
- ET-3:
-
Endothelin-3
- ETA:
-
Endothelin A
- ETB:
-
Endothelin B
- eNOS:
-
Epithelial nitric oxide synthase
- GFR:
-
Glomerular filtration rate
- GTP:
-
Guanosine triphosphate
- HF:
-
Heart failure
- HfrEF:
-
Heart failure with reduced ejection fraction
- HSCs:
-
Hepatic stellate cells
- IDE:
-
Insulin-degrading enzyme
- iNOS:
-
Inducible nitric oxide synthase
- KKS:
-
Kallikrein–kinin system
- LSECs:
-
Liver sinusoidal endothelial cells
- MAPK:
-
Mitogen-activated protein kinase
- MrgD:
-
Mas-related G protein-coupled receptor type D
- Nep:
-
Neprilysin
- NF-κB:
-
Nuclear factor-κB
- NGF:
-
Nerve growth factor
- nNOS:
-
Neuronal nitric oxide synthase
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NHE:
-
Na+ /H+ exchanger
- NMDA:
-
N-methyl-D-aspartate
- NPs:
-
Natriuretic peptides
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- NT-proBNP:
-
N-terminal prohormone of brain natriuretic peptide
- PAH:
-
Pulmonary arterial hypertension
- PDE-5:
-
Phosphodiesterase-5
- PGs:
-
Prostaglandins
- PH:
-
Pulmonary hypertension
- PHT:
-
Portal hypertension
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PVR:
-
Pulmonary vascular resistance
- RAAS:
-
Renin–angiotensin–aldosterone system
- RAS:
-
Renin–angiotensin system
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- sGC:
-
Soluble type of guanylyl cyclase
- TGF-β1:
-
Transforming growth factor-β1
- TNF-α:
-
Tumor necrosis factor α
- TXA2 :
-
Thromboxane A2
- VEGF:
-
Vascular endothelial growth factor
- VSM:
-
Vascular smooth muscle
References
Vaughan CJ, Delanty N (2000) Hypertensive emergencies. The Lancet 356(9227):411–417
Carlson BE, Arciero JC, Secomb TW (2008) Theoretical model of blood flow autoregulation: roles of myogenic, shear-dependent, and metabolic responses. Am J Physiol Heart Circulat Physiol 295(4):H1572–H1579
Dole WP (1987) Autoregulation of the coronary circulation. Prog Cardiovasc Dis 29(4):293–323
Dautzenberg M, Keilhoff G, Just A (2011) Modulation of the myogenic response in renal blood flow autoregulation by NO depends on endothelial nitric oxide synthase (eNOS), but not neuronal or inducible NOS. J Physiol 589(19):4731–4744
Xavier FE, Aras-López R, Arroyo-Villa I, Del Campo L, Salaices M, Rossoni LV, Ferrer M, Balfagón G (2008) Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane A2 and prostacyclin. Br J Pharmacol 154(6):1225–1235
Local Hormones 1: histamine and the biologically active lipids. In: Rang HP (ed) Rang and Dale’s Pharmacology, 9th edn. Elsevier, London.
Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM (2007) Angiotensin-(1–7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 49(1):185–192
Smyth EM, FitzGerald GA (2012) The eicosanoids: prostaglandins, thromboxanes, eukotrienes, & related compounds. In: Katzung BG (ed) Basic & clinical pharmacology, 12th edn. McGraw Hill Companies, Inc. New York.
Tilley SL, Coffman TM, Koller BH (2001) Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Investig 108(1):15–23
Epstein M (2002) Non-steroidal anti-inflammatory drugs and the continuum of renal dysfunction. J Hyperten 20(6):S17–S23
Yu IS, Lin SR, Huang CC, Tseng HY, Huang PH, Shi GY, Wu HL, Tang CL, Chu PH, Wang LH, Wu KK, Lin SW (2004) TXAS-deleted mice exhibit normal thrombopoiesis, defective hemostasis, and resistance to arachidonate-induced death. Blood 104(1):135–142
Ozen G, Aljesri K, Abdelazeem H, Norel X, Turkyılmaz G, Turkyılmaz S, Topal G (2021) Comparative study on the effect of aspirin, TP receptor antagonist and TxA2 synthase inhibitor on the vascular tone of human saphenous vein and internal mammary artery. Life Sci 286:120073
Linder L, Kiowski W, Bühler FR, Lüscher TF (1990) Indirect evidence for release of endothelium-derived relaxing factor in human forearm circulation in vivo. Blunted response in essential hypertension. Circulation 81(6):1762–1767
Blumenthal DK (2018) Pharmacodynamics: molecular mechanisms of drug action. In: Brunton LL (ed) Goodman & Gilman’s the pharmacological basis of therapeutics, 13th edn. McGraw Hill Companies, Inc. New York
Kreisel W, Lazaro A, Trebicka J, Perdekamp MG, Schmitt-Graeff A, Deibert P (2021) Cyclic gmp in liver cirrhosis—role in pathophysiology of portal hypertension and therapeutic implications. Int J Mol Sci 22(19):10372
Bellamy TC, Wood J, Goodwin DA, Garthwaite J (2000) Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc Natl Acad Sci 97(6):2928–2933
Falk JA, Philip KJ, Schwarz ER (2010) The emergence of oral tadalafil as a once-daily treatment for pulmonary arterial hypertension. Vascular Health Risk Manag 6:273
Singh J, Lee Y, Kellum JA (2022) A new perspective on NO pathway in sepsis and ADMA lowering as a potential therapeutic approach. Crit Care 26(1):1–8
Kedzierski RM, Yanagisawa M (2001) Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol 41(1):851–876
Imai T, Hirata Y, Emori, T, Yanagisawa M, Masaki T, Marumo F (1992) Induction of endothelin-1 gene by angiotensin and vasopressin in endothelial cells. Hypertension, 19(6_pt_2), 753–757
Kuc RE, Carlebur M, Maguire JJ, Yang P, Long L, Toshner M, Morrell NW, Davenport AP (2014) Modulation of endothelin receptors in the failing right ventricle of the heart and vasculature of the lung in human pulmonary arterial hypertension. Life Sci 118(2):391–396
Stow LR, Jacobs ME, Wingo CS, Cain BD (2011) Endothelin-1 gene regulation. FASEB J 25(1):16–28
Kawanabe Y, Nauli SM (2011) Endothelin. Cell Mol Life Sci 68:195–203
Turner AJ, Murphy LJ (1996) Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol 51(2):91–102
Rapoport RM (2014) Nitric oxide inhibition of endothelin-1 release in the vasculature: in vivo relevance of in vitro findings. Hypertension 64(5):908–914
Vanderheyden M, Bartunek J, Goethals M (2004) Brain and other natriuretic peptides: molecular aspects. Eur J Heart Fail 6(3):261–268
Hall C (2005) NT-ProBNP: the mechanism behind the marker. J Cardiac Fail 11(5):S81–S83
Buettner P, Schumacher K, Dinov B, Zeynalova S, Sommer P, Bollmann A, Husser D, Hindricks G, Kornej J (2018) Role of NT-proANP and NT-proBNP in patients with atrial fibrillation: association with atrial fibrillation progression phenotypes. Heart Rhythm 15(8):1132–1137
Romaniello A, Rubattu S, Vaiarello V, Gigante A, Volpe M, Rosato E (2021) Circulating NT-proANP level is a predictor of mortality for systemic sclerosis: a retrospective study of an Italian cohort. Expert Rev Clin Immunol 17(6):661–666
Najenson AC, Courreges AP, Perazzo JC, Rubio MF, Vatta MS, Bianciotti LG (2018) Atrial natriuretic peptide reduces inflammation and enhances apoptosis in rat acute pancreatitis. Acta Physiol 222(3):e12992
Opgenorth TJ, Wu-Wong JR, Shiosaki K (1992) Endothelin-converting enzymes. FASEB J 6(9):2653–2659
Campbell DJ (2018) Neprilysin inhibitors and bradykinin. Front Med 5:257
Madhani M, Scotland RS, MacAllister RJ, Hobbs AJ (2003) Vascular natriuretic peptide receptor-linked particulate guanylate cyclases are modulated by nitric oxide–cyclic GMP signalling. Br J Pharmacol 139(7):1289–1296
Zhou H, Murthy KS (2003) Identification of the G protein-activating sequence of the single-transmembrane natriuretic peptide receptor C (NPR-C). Am J Physiol Cell Physiol 284(5):C1255–C1261
Ichiki T, Burnett JC Jr (2017) Atrial natriuretic peptide-old but new therapeutic in cardiovascular diseases. Circ J 81(7):913–919
Drugs affecting major organ systems. In: Rang HP (ed) Rang and Dale’s pharmacology, 9th edn. Elsevier, London
Yan B, Peng L, Zhao X, Chung H, Li L, Zeng L, Ong H, Wang G (2014) Nesiritide fails to reduce the mortality of patients with acute decompensated heart failure: an updated systematic review and cumulative meta-analysis. Int J Cardiol 177(2):505–509
Sugawara A, Shimada H, Otsubo Y, Kouketsu T, Suzuki S, Yokoyama A (2021) The usefulness of angiotensin-(1–7) and des-Arg9-bradykinin as novel biomarkers for metabolic syndrome. Hypertens Res 44(8):1034–1036
Bork K, Davis-Lorton M (2013) Overview of hereditary angioedema caused by C1-inhibitor deficiency: assessment and clinical management. Eur Ann Allergy Clin Immunol 45(1):7–16
Obtułowicz K (2016) Bradykinin-mediated angioedema. Polish Arch Int Med 126(1–2).
Golias C, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A (2007) The kinin system-bradykinin: biological effects and clinical implications. Multiple role of the kinin system-bradykinin. Hippokratia 11(3):124
Terzuoli E, Meini S, Cucchi P, Catalani C, Cialdai C, Maggi CA, Giachetti A, Ziche M, Donnini S (2014) Antagonism of bradykinin B2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-kB pathway activation. PLoS ONE 9(1):e84358
Cicardi M, Zuraw BL (2018) Angioedema due to bradykinin dysregulation. J Allergy Clin Immunol 6(4):1132–1141
Schmaier AH (2002) The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. J Clin Investig 109(8):1007–1009
Patel G, Pongracic JA (2019). Hereditary and acquired angioedema. In Allergy & Asthma Proceedings (Vol. 40, No. 6)
Cicardi M, Zanichelli A (2010) Acquired angioedema. Allergy Asthma Clin Immunol 6(1):14
Fountain JH, Lappin SL (2017) Physiology, Renin angiotensin system. In: StatPearls. StatPearls Publishing, Treasure Island (FL); 2022. PMID: 29261862
Grace JA, Herath CB, Mak KY, Burrell LM, Angus PW (2012) Update on new aspects of the renin–angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci 123(4):225–239
Kaltenecker CC, Domenig O, Kopecky C, Antlanger M, Poglitsch M, Berlakovich G, Kain R, Stegbauer J, Rahman M, Hellinger R, Gruber C, Grobe N, Fajkovic H, Eskandary F, Böhmig GA, Säemann MD, Kovarik JJ (2020) Critical role of neprilysin in kidney angiotensin metabolism. Circ Res 127(5):593–606
Sata N, Tanaka Y, Suzuki S, Kamimura R, Mifune H, Nakamura K, Miyahara K, Arima T (2003) Effectiveness of angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker on atrial natriuretic peptide. Circ J 67(12):1053–1058
Kaschina E, Steckelings UM, Unger T (2018) Hypertension and the renin-angiotensin-aldosterone system. In Encyclopedia of endocrine diseases, pp 505–510. Elsevier Editora
Schwinghammer TL (2015) Hypertension. In: Wells BG (ed) Pharmacotherapy handbook, 9th edn. McGraw Hill Education, New York.
Wright SA, Bardolia C, Bankes D, Amin NS, Turgeon J (2020) Angiotensin converting enzyme (ACE) inhibitor-induced cough resulting in prescribing cascade. Clin Case Rep J 10:1368
Kittikulsuth W, Pollock JS, Pollock DM (2012) Loss of renal medullary endothelin B receptor function during salt deprivation is regulated by angiotensin II. Am J Physiol Renal Physiol 303(5):F659–F666
Kittikulsuth W, Sullivan JC, Pollock DM (2013) ET-1 actions in the kidney: evidence for sex differences. Br J Pharmacol 168(2):318–326
Dendorfer A, Thornagel A, Raasch W, Grisk O, Tempel K, Dominiak P (2002) Angiotensin II induces catecholamine release by direct ganglionic excitation. Hypertension 40(3):348–354
Cardiovascular diseases (CVDs). https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds). Accessed 23 September 2022.
Gafane-Matemane LF, Kruger R, Smith W, Mels CM, Van Rooyen JM, Mokwatsi GG, Uys AS, Brits SJ, Schutte AE (2021) Characterization of the renin-angiotensin-aldosterone system in young healthy black adults: The african prospective study on the early detection and identification of hypertension and cardiovascular disease (African-PREDICT Study). Hypertension 78(2):400–410
Bain A (2019) Use of calcium channel blockers in cardiovascular disease. Br J Cardiac Nursing 14(2):64–70
Bhatt AS, Vaduganathan M, Solomon SD, Schneeweiss S, Lauffenburger JC, Desai RJ (2022) Sacubitril/valsartan use patterns among older adults with heart failure in clinical practice: a population-based cohort study of> 25 000 Medicare beneficiaries. Eur J Heart Fail 24(9):1506–1515
Somova LI, Mufunda JJ (1992) Renin-angiotensin-aldosterone system and thromboxane A2/prostacyclin in normotensive and hypertensive black Zimbabweans. Ethnicity Disease, pp 27–34
Krum H, Aw TJ, Liew D, Haas S (2006) Blood pressure effects of COX-2 inhibitors. J Cardiovasc Pharmacol 47:S43–S48
Mitchell JA, Kirkby NS (2019) Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacol 176(8):1038–1050
Pawlosky N (2013) Cardiovascular risk: Are all NSAIDs alike? Can Pharm J 146(2):80–83
Suzuki JI, Ogawa M, Watanabe R, Takayama K, Hirata Y, Nagai R, Isobe M (2011) Roles of prostaglandin E2 in cardiovascular diseases focus on the potential use of a novel selective EP4 receptor agonist. Int Heart J 52(5):266–269
Katsumata Y, Shinmura K, Sugiura Y, Tohyama S, Matsuhashi T, Ito H, Yan X, Ito K, Yuasa S, Ieda M, Urade Y, Suematsu M, Fukuda K, Sano M (2014) Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia–reperfusion injury by activating Nrf2. Hypertension 63(1):80–87
Palee S, Chattipakorn S, Phrommintikul A, Chattipakorn N (2011) PPARγ activator, rosiglitazone: Is it beneficial or harmful to the cardiovascular system? World J Cardiol 3(5):144
Escure G, Nudel M, Terriou L, Farhat MM, Launay D, Staumont-Salle D, Hachulla GS, Sanges S (2022) Tolerance of bradykinin-releasing drugs in patients with acquired C1 inhibitor deficiency: a case series and review of the literature. Eur J Dermatol 32(1):49–55
Yuan W, Cheng G, Li B, Li Y, Lu S, Liu D, Xiao J, Zhao Z (2017) Endothelin-receptor antagonist can reduce blood pressure in patients with hypertension: a meta-analysis. Blood Press 26(3):139–149
Mokretar K, Velinov H, Postadzhiyan A, Apostolova M (2016) Association of polymorphisms in endothelial nitric oxide synthesis and renin–angiotensin–aldosterone system with developing of coronary artery disease in bulgarian patients. Genet Test Mol Biomarkers 20(2):67–73
McClean DR, Ikram H, Garlick AH, Crozier IG (2001) Effects of omapatrilat on systemic arterial function in patients with chronic heart failure. Am J Cardiol 87(5):565–569
Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E (2004) Omapatrilat and enalapril in patients with hypertension: the omapatrilat cardiovascular treatment vs enalapril (OCTAVE) trial. Am J Hypert 17(2):103–111
FDA Approves Entresto. https://hfsa.org/fda-approves-entresto. Accessed 28 Sept 2022.
Novartis Entresto® granted expanded indication in chronic heart failure by FDA. https://www.novartis.com/news/media-releases/novartis-entresto-granted-expanded-indication-chronic-heart-failure-fda. Accessed 28 Sep 2022
Rakugi H, Kario K, Yamaguchi M, Sasajima T, Gotou H, Zhang J (2022) Efficacy of sacubitril/valsartan versus olmesartan in Japanese patients with essential hypertension: a randomized, double-blind, multicenter study. Hypertens Res 45(5):824–833
Yandrapalli S, Khan MH, Rochlani Y, Aronow WS (2018) Sacubitril/valsartan in cardiovascular disease: evidence to date and place in therapy. Ther Adv Cardiovasc Dis 12(8):217–231
Berardi C, Braunwald E, Morrow DA, Mulder HS, Duffy CI, O’Brien TX, Ambrosy AP, Chakraborty H, Velazquez EJ, DeVore AD, Investigators PIONEER-HF (2020) Angiotensin-neprilysin inhibition in black Americans: data from the PIONEER-HF trial. Heart Failure 8(10):859–866
Silva MVBD, Sousa Júnior CPD, Silva HVCD, Santos VMD, Feijao FIM, Bernardino ADO, Melo JACRTD (2022) Evaluation of the cardioprotective and antihypertensive effect of AVE 0991 in normotensive and hypertensive rats. Revista da Associação Médica Brasileira
Ma Y, Huang H, Jiang J, Wu L, Lin C, Tang A, Dai G, He J, Chen Y (2016) AVE 0991 attenuates cardiac hypertrophy through reducing oxidative stress. Biochem Biophys Res Commun 474(4):621–625
McGiff JC, Terragno NA, Malik KU, Lonigro AJ (1972) Release of a prostaglandin E-like substance from canine kidney by bradykinin: comparison with eledoisin. Circ Res 31(1):36–43
McGiff JC, Crowshaw K, Terragno NA, Malik KU, Lonigro AJ (1972) Differential efect of noradrenaline and renal nerve stimulation on vascular resistance in the dog kidney and the release of a prostaglandin e-like substance. Clin Sci 42(2):223–233
Wang J, Liu M, Zhang X, Yang G, Chen L (2018) Physiological and pathophysiological implications of PGE2 and the PGE2 synthases in the kidney. Prostaglandins Other Lipid Mediat 134:1–6
Pfeilschifter J, Schalkwijk C, Briner VA, Van den Bosch H (1993). Cytokine-stimulated secretion of group II phospholipase A2 by rat mesangial cells. Its contribution to arachidonic acid release and prostaglandin synthesis by cultured rat glomerular cells. J Clin Investigat 92(5):2516–2523
Völzke A, Koch A, Zu Heringdorf DM, Huwiler A, Pfeilschifter J (2014) Sphingosine 1-phosphate (S1P) induces COX-2 expression and PGE2 formation via S1P receptor 2 in renal mesangial cells. Molecul Cell Biol Lipids, 1841(1): 11–21
Barutta F, Bellini S, Gruden G (2022) Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin Sci 136(7):493–520
Eschenhagen T (2018) Treatment of hypertension, therapy of heart failure. In: Brunton LL (ed) Goodman & Gilman’s the pharmacological basis of therapeutics, 13th edn. McGraw Hill Companies, Inc. New York.
Laurent S (2017) Antihypertensive drugs. Pharmacol Res 124:116–125
Liu Y, Hitomi H, Diah S, Deguchi K, Mori H, Masaki T, Nakano D, Kobori H, Nishiyama A (2013) Roles of Na+/H+ exchanger type 1 and intracellular pH in angiotensin II-induced reactive oxygen species generation and podocyte apoptosis. J Pharmacol Sci 122(3):176–183
Li XC, Zhu D, Chen X, Zheng X, Zhao C, Zhang J, Soleimani M, Rubera I, Tauc M, Zhou X, Zhuo JL (2019) Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates Ang II (angiotensin II)-induced hypertension in mice. Hypertension 74(3):526–535
Su M, Dhoopun AR, Yuan Y, Huang S, Zhu C, Ding G, Liu B, Yang T, Zhang A (2013) Mitochondrial dysfunction is an early event in aldosterone-induced podocyte injury. Am J Physiol-Renal Physiol 305(4):F520–F531
Baylis C (2008) Nitric oxide deficiency in chronic kidney disease. Am J Physiol-Renal Physiol 294(1):F1–F9
Lei C, Berra L, Rezoagli E, Yu B, Dong H, Yu S, Hou L, Chen M, Chen W, Wang H, Zheng Q (2018) Nitric oxide decreases acute kidney injury and stage 3 chronic kidney disease after cardiac surgery. Am J Respir Crit Care Med 198(10):1279–1287
Tai YH, Wu HL, Su FW, Chang KY, Huang CH, Tsou MY, Lu CC (2019) The effect of high-dose nitroglycerin on the cerebral saturation and renal function in cardiac surgery: a propensity score analysis. J Chin Med Assoc 82(2):120–125
Smeijer JD, Kohan DE, Webb DJ, Dhaun N, Heerspink HJ (2021) Endothelin receptor antagonists for the treatment of diabetic and nondiabetic chronic kidney disease. Curr Opin Nephrol Hypertens 30(4):456–465
Flack JM, Adekola B (2020) Blood pressure and the new ACC/AHA hypertension guidelines. Trends Cardiovasc Med 30(3):160–164
96. Ding W, Li X, Wu W, He H, Li Y, Gao L, Gan L, Wang M, Ou S, Liu J (2018) Aliskiren inhibits angiotensin II/angiotensin 1–7 (Ang II/Ang1–7) signal pathway in rats with diabetic nephropathy. Chinese J Cellular Molecul Immunol 34(10):891–895
Angeli F, Reboldi G, Poltronieri C, Angeli E, De Filippo V, Crocetti A, Bartolini C, D’Ambrosio C, Verdecchia P (2014) Efficacy and safety profile of aliskiren: practical implications for clinicians. Curr Drug Saf 9(2):106–117
Zhang L, Xue S, Hou J, Chen G, Xu ZG (2020) Endothelin receptor antagonists for the treatment of diabetic nephropathy: a meta-analysis and systematic review. World J Diabetes 11(11):553
Tsai YC, Tsai HJ, Lee CS, Chiu YW, Kuo HT, Lee SC, Chen TH, Kuo MC (2018) The interaction between N-terminal pro-brain natriuretic peptide and fluid status in adverse clinical outcomes of late stages of chronic kidney disease. PLoS ONE 13(8):e0202733
Yang CC, Chen YT, Chen CH, Li YC, Shao PL, Huang TH, Chen YL, Sun CK, Yip HK (2019) The therapeutic impact of entresto on protecting against cardiorenal syndrome-associated renal damage in rats on high protein diet. Biomed Pharmacother 116:108954
Bascands JL, Pecher C, Rouaud S, Emond C, Tack JL, Bastie MJ, Burch R, Regoli D, Girolami JP (1993) Evidence for existence of two distinct bradykinin receptors on rat mesangial cells. Am J Physiol-Renal Physiol 264(3):F548–F556
Dong JI, Ding LI, Wang L, Yang Z, Wang Y, Zang Y, Cao X, Tang L (2021) Effects of bradykinin on proliferation, apoptosis, and cycle of glomerular mesangial cells via the TGF-ß1/Smad signaling pathway. Turk J Biol 45(1):17–25
Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A (2016) 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37(1):67–119
Bazan IS, Fares WH (2015) Pulmonary hypertension: diagnostic and therapeutic challenges. Therapeut Clin Risk Manage, pp 1221–1233
Lahm T, Hess E, Barón AE, Maddox TM, Plomondon ME, Choudhary G, Maron BA, Zamanian RT, Leary PJ (2021) Renin-angiotensin-aldosterone system inhibitor use and mortality in pulmonary hypertension: insights from the Veterans Affairs Clinical Assessment Reporting and Tracking Database. Chest 159(4):1586–1597
Costanzo MR, Stevenson LW, Adamson PB, Desai AS, Heywood JT, Bourge RC, Bauman J, Abraham WT (2016) Interventions linked to decreased heart failure hospitalizations during ambulatory pulmonary artery pressure monitoring. JACC Heart Failure 4(5): 333–344
Desai AS, Heywood JT, Rathman L, Abraham WT, Adamson P, Brett ME, Costanzo MR, Lamba S, Raval N, Shavelle D, Sood P, Stevenson W (2021) Early reduction in ambulatory pulmonary artery pressures after initiation of sacubitril/valsartan. Circulation Heart Failure 14(7): e008212
Hemnes AR, Rathinasabapathy A, Austin EA, Brittain EL, Carrier EJ, Chen X, Fessel JP, Fike CD, Fong P, Fortune N, Gerszten RE (2018) A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur Respirat J 51(6)
Hall DA, Hanrott K, Badorrek P, Berliner D, Budd DC, Eames R, Powley WM, Hewens D, Siederer S, Lazaar AL, Cahn A, Hohlfeld JM (2021) Effects of recombinant human angiotensin-converting enzyme 2 on response to acute hypoxia and exercise: a randomised, Placebo-Controlled Study. Pulmonary Therapy 7:487–501
Schilling T, Bergmann A (2022) Modulating the pulmonary circulation: nitric oxide and beyond. In Cohen's comprehensive thoracic anesthesia. Elsevier, pp 105–114
Ziegler JW, Ivy DD, Kinsella JP, Abman SH (1995) The role of nitric oxide, endothelin, and prostaglandins in the transition of the pulmonary circulation. Clin Perinatol 22(2):387–403
Wang L, Zhu L, Wu Y, Li Q, Liu H (2021) Riociguat therapy for pulmonary hypertension: a systematic review and meta-analysis. Ann Palliat Med 10(10):11117–11128
Zhao R, Jiang Y (2019) Influence of riociguat treatment on pulmonary arterial hypertension. Herz 44(7):637–643
Velvis H, Moore P, Heymann MA (1991) Prostaglandin inhibition prevents the fall in pulmonary vascular resistance as a result of rhythmic distension of the lungs in fetal lambs. Pediatr Res 30(1):62–68
Sitbon O, Noordegraaf AV (2017) Epoprostenol and pulmonary arterial hypertension: 20 years of clinical experience. Eur Respirat Rev 26(143)
Zeineh NS, Bachman TN, El-Haddad H, Champion HC (2014) Effects of acute intravenous iloprost on right ventricular hemodynamics in rats with chronic pulmonary hypertension. Pulmon Circulat 4(4):612–618
Humbert M, Sitbon O, Simonneau G (2004) Treatment of pulmonary arterial hypertension. N Engl J Med 351(14):1425–1436
Narine L, Hague LK, Walker JH, Vicente C, Schilz R, Desjardins O, Einarson TR, Iskedjian M (2005) Cost-minimization analysis of treprostinil vs epoprostenol as an alternate to oral therapy non-responders for the treatment of pulmonary arterial hypertension. Curr Med Res Opin 21(12):2007–2016
Waxman A, Restrepo-Jaramillo R, Thenappan T, Ravichandran A, Engel P, Bajwa A, Allen R, Feldman J, Argula R, Smith P, Rollins K (2021) Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. N Engl J Med 384(4):325–334
Takatsuki S, Nakayama T, Shimizu Y, Kawai R, Matsuura H (2022) Clinical efficacy and safety of selexipag in children and young adults with idiopathic and heritable pulmonary arterial hypertension. Cardiol Young 33(2):196–200
Sabri MR, Beheshtian E (2014) Comparison of the therapeutic and side effects of tadalafil and sildenafil in children and adolescents with pulmonary arterial hypertension. Pediatr Cardiol 35:699–704
Lichtblau M, Harzheim D, Ehlken N, Marra A, Pinado FP, Grünig E, Egenlauf B (2015) Safety and long-term efficacy of transition from sildenafil to tadalafil due to side effects in patients with pulmonary arterial hypertension. Lung 193:105–112
Coral-Alvarado P, Quintana G, Garces MF, Cepeda LA, Caminos JE, Rondon F, Iglesias-Gamarra A, Restrepo JF (2009) Potential biomarkers for detecting pulmonary arterial hypertension in patients with systemic sclerosis. Rheumatol Int 29:1017–1024
Hajra A, Safiriyu I, Balasubramanian P, Gupta R, Chowdhury S, Prasad AJ, Kumar A, Kumar D, Khan B, Bilberry RS, Sarkar A, Malik P, Aronow WS (2022) Recent advances and future prospects of treatment of pulmonary hypertension. Current Problem Cardiol 101236.
Collum SD, Amione-Guerra J, Cruz-Solbes AS, DiFrancesco A, Hernandez AM, Hanmandlu A, Youker K, Guha A, Karmouty-Quintana H (2017) Pulmonary hypertension associated with idiopathic pulmonary fibrosis: current and future perspectives. Can Respirat J
Jankov RP, Kantores C, Belcastro R, Yi M, Tanswell AK (2006) Endothelin-1 inhibits apoptosis of pulmonary arterial smooth muscle in the neonatal rat. Pediatr Res 60(3):245–251
Zhao Q, Guo N, Chen J, Parks D, Tian Z (2022) Comparative assessment of efficacy and safety of ambrisentan and bosentan in patients with pulmonary arterial hypertension: a meta-analysis. J Clin Pharm Ther 47(2):146–156
Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grünig E, Oudiz RJ (2015) Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med 373(9):834–844
Weatherald J, Thakrar MV, Varughese RA, Kularatne M, Liu J, Harper L, Kiamanesh O, Fine N, Orlikow E, Nwaroh C, Thornton C (2022) Upfront riociguat and ambrisentan combination therapy for newly diagnosed pulmonary arterial hypertension: a prospective open-label trial. J Heart Lung Transplant 41(5):563–567
Bedan M, Grimm D, Wehland M, Simonsen U, Infanger M, Krüger M (2018) A focus on macitentan in the treatment of pulmonary arterial hypertension. Basic Clin Pharmacol Toxicol 123(2):103–113
Taraseviciene-Stewart L, Scerbavicius R, Stewart JM, Gera L, Demura Y, Cool C, Kasper M, Voelkel NF (2005) Treatment of severe pulmonary hypertension: a bradykinin receptor 2 agonist B9972 causes reduction of pulmonary artery pressure and right ventricular hypertrophy. Peptides 26(8):1292–1300
Taraseviciene-Stewart L, Gera L, Hirth P, Voelkel NF, Tuder RM, Stewart JM (2002) A bradykinin antagonist and a caspase inhibitor prevent severe pulmonary hypertension in a rat model. Can J Physiol Pharmacol 80(4):269–274
Singh S, Sharma S (2022) High-output cardiac failure. In: StatPearls. StatPearls Publishing.
Rockey DC, Fouassier L, Chung JJ, Carayon A, Vallée P, Rey C, Housset C (1998) Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells. Hepatology 27(2):472–480
Pinzani M, Milani S, Franco RD, Grappone C, Caligiuri A, Gentilini A, Tosti-Guerra C, Maggi M, Failli P, Ruocco C, Gentilini P (1996) Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology 110(2):534–548
Marsden PA, Brenner BM (1992) Transcriptional regulation of the endothelin-1 gene by TNF-alpha. Am J Physiol Cell Physiol 262(4):C854–C861
Nagasue N, Dhar DK, Yamanoi A, Emi Y, Udagawa J, Yamamoto A, Tachibana M, Kubota H, Kohno H, Harada T (2000) Production and release of endothelin-1 from the gut and spleen in portal hypertension due to cirrhosis. Hepatology 31(5):1107–1114
Hu LS, George J, Wang JH (2013) Current concepts on the role of nitric oxide in portal hypertension. World J Gastroenterol: WJG 19(11):1707
Iwakiri Y, Groszmann RJ (2020) Pathophysiology of portal hypertension. In: Arias M (ed) The liver biology and pathobiology, 6th edn. Wiley Blackwell, New Jersey
Ezhilarasan D (2022) Hepatic stellate cells in the injured liver: Perspectives beyond hepatic fibrosis. J Cell Physiol 237(1):436–449
Hsu SJ, Lin TY, Wang SS, Chuang CL, Lee FY, Huang HC, Hsin IF, Lee JY, Lin HC, Lee SD (2016) Endothelin receptor blockers reduce shunting and angiogenesis in cirrhotic rats. Eur J Clin Invest 46(6):572–580
Gunarathne LS, Rajapaksha IG, Casey S, Qaradakhi T, Zulli A, Rajapaksha H, Trebicka J, Angus PW, Herath CB (2022) Mas-related G protein-coupled receptor type D antagonism improves portal hypertension in cirrhotic rats. Hepatol Commun 6(9):2523–2537
Wiest R, Groszmann RJ (2002) The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatology 35(2):478–549
Schwabl P, Brusilovskaya K, Supper P, Bauer D, Königshofer P, Riedl F, Hayden H, Fuchs CD, Stift J, Oberhuber G, Aschauer S (2018) The soluble guanylate cyclase stimulator riociguat reduces fibrogenesis and portal pressure in cirrhotic rats. Sci Rep 8(1):9372
Wisén E, Svennerholm K, Bown LS, Houltz E, Rizell M, Lundin S, Ricksten SE (2018) Vasopressin and nitroglycerin decrease portal and hepatic venous pressure and hepato-splanchnic blood flow. Acta Anaesthesiol Scand 62(7):953–961
Brusilovskaya K, Königshofer P, Lampach D, Szodl A, Supper P, Bauer D, Beer A, Stift J, Timelthaler G, Oberhuber G, Podesser BK (2020) Soluble guanylyl cyclase stimulation and phosphodiesterase-5 inhibition improve portal hypertension and reduce liver fibrosis in bile duct–ligated rats. United Eur Gastroenterol J 8(10):1174–1185
Kim G, Kim J, Lim YL, Kim MY, Baik SK (2016) Renin–angiotensin system inhibitors and fibrosis in chronic liver disease: a systematic review. Hep Intl 10:819–828
Zhang X, Wong GLH, Yip TCF, Tse YK, Liang LY, Hui VWK, Lin H, Li GL, Lai JCT, Chan HLY, Wong VWS (2022) Angiotensin-converting enzyme inhibitors prevent liver-related events in nonalcoholic fatty liver disease. Hepatology 76(2):469–482
González-Abraldes J, Albillos A, Bañares R, Del Arbol LR, Moitinho E, Rodríguez C, González M, Escorsell A, García-Pagán JC, Bosch J (2001) Randomized comparison of long-term losartan versus propranolol in lowering portal pressure in cirrhosis. Gastroenterology 121(2):382–388
Agasti AK, Mahajan AU, Phadke AY, Nathani PJ, Sawant P (2013) Comparative randomized study on efficacy of losartan versus propranolol in lowering portal pressure in decompensated chronic liver disease. J Dig Dis 14(5):266–271
Kim JH, Kim JM, Cho YZ, Na JH, Kim HS, Kim HA, Kang HW, Baik SK, Kwon SO, Cha SH, Kim YJ, Kim MY (2014) Effects of candesartan and propranolol combination therapy versus propranolol monotherapy in reducing portal hypertension. Clin Mol Hepatol 20(4):376
Sookoian S, Fernández MA, Castaño G (2005) Effects of six months losartan administration on liver fibrosis in chronic hepatitis C patients: a pilot study. World J Gastroenterol 11(48):7560
Hsu SJ, Huang HC, Chuang CL, Chang CC, Hou MC, Lee FY, Lee SD (2020) Dual angiotensin receptor and neprilysin inhibitor ameliorates portal hypertension in portal hypertensive rats. Pharmaceutics 12(4):320
Casey S, Schierwagen R, Mak KY, Klein S, Uschner F, Jansen C, Praktiknjo M, Meyer C, Thomas D, Herath C, Jones R, Trebicka J, Angus P (2019) Activation of the alternate renin-angiotensin system correlates with the clinical status in human cirrhosis and corrects post liver transplantation. J Clin Med 8(4):419
Lubel JS, Herath CB, Burrell LM, Angus PW (2008) Liver disease and the renin–angiotensin system: recent discoveries and clinical implications. J Gastroenterol Hepatol 23(9):1327–1338
Klein S, Herath CB, Schierwagen R, Grace J, Haltenhof T, Uschner FE, Strassburg CP, Sauerbruch T, Walther T, Angus PW, Trebicka J (2015) Hemodynamic effects of the non-peptidic angiotensin-(1–7) agonist AVE0991 in liver cirrhosis. PLoS ONE 10(9):e0138732
Iwahashi N, Horii M, Tamura K, Kimura K (2022) Worsening dyspnea in patients with idiopathic portal hypertension. Chest 161(4):e245–e248
Gao JH, Wen SL, Feng S, Yang WJ, Lu YY, Tong H, Liu R, Tang SH, Huang ZY, Tang YM, Yang JH (2016) Celecoxib and octreotide synergistically ameliorate portal hypertension via inhibition of angiogenesis in cirrhotic rats. Angiogenesis 19:501–511
Feng S, Tong H, Gao JH, Tang SH, Yang WJ, Wang GM, Zhou HY, Wen SL (2021) Anti-inflammation treatment for protection of hepatocytes and amelioration of hepatic fibrosis in rats. Exp Ther Med 22(5):1–11
Grimaldi A, Brighi C, Peruzzi G, Ragozzino D, Bonanni V, Limatola C, Ruocco G, Di Angelantonio S (2018) Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer’s disease in the 3xTg-AD mouse model. Cell Death Dis 9(6):685
Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14:450–464
Tang X, Li Z, Zhang W, Yao Z (2019) Nitric oxide might be an inducing factor in cognitive impairment in Alzheimer’s disease via downregulating the monocarboxylate transporter 1. Nitric Oxide 91:35–41
Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J (2005) Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med 202(9):1163–1169
Liu L, Xu H, Ding S, Wang D, Song G, Huang X (2019) Phosphodiesterase 5 inhibitors as novel agents for the treatment of Alzheimer’s disease. Brain Res Bull 153:223–231
Fang J, Zhang P, Zhou Y, Chiang CW, Tan J, Hou Y, Stauffer S, Li L, Pieper AA, Cummings J, Cheng F (2021) Endophenotype-based in silico network medicine discovery combined with insurance record data mining identifies sildenafil as a candidate drug for Alzheimer’s disease. Nature Aging 1(12):1175–1188
Al-Amin MM, Hasan SN, Alam T, Hasan AT, Hossain I, Didar RR, Alam MA, Rahman MM (2014) Tadalafil enhances working memory, and reduces hippocampal oxidative stress in both young and aged mice. Eur J Pharmacol 745:84–90
Michinaga S, Koyama Y (2019) Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int J Mol Sci 20(3):571
Sharma S, Behl T, Kumar A, Sehgal A, Singh S, Sharma N, Bhatia S, Al-Harrasi A, Bungau S (2021) Targeting endothelin in Alzheimer’s disease: a promising therapeutic approach. BioMed Research Int
Michinaga S, Inoue A, Yamamoto H, Ryu R, Inoue A, Mizuguchi H, Koyama Y (2020) Endothelin receptor antagonists alleviate blood-brain barrier disruption and cerebral edema in a mouse model of traumatic brain injury: a comparison between bosentan and ambrisentan. Neuropharmacology 175:108182
Eckman EA, Reed DK, Eckman CB (2001) Degradation of the Alzheimer’s amyloid β peptide by endothelin-converting enzyme. J Biol Chem 276(27):24540–24548
Koyama Y, Endothelin ETB (2021) Receptor-mediated astrocytic activation: pathological roles in brain disorders. Int J Mol Sci 22(9):4333
Hemming ML, Selkoe DJ (2005) Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 280(45):37644–37650
Saido TC (2013) Metabolism of amyloid β peptide and pathogenesis of Alzheimer’s disease. Proc Jpn Acad Ser B 89(7):321–339
Deng Z, Jiang J, Wang J, Pan D, Zhu Y, Li H, Zhang X, Liu X, Xu Y, Li Y, Tang Y (2022) Angiotensin receptor blockers are associated with a lower risk of progression from mild cognitive impairment to dementia. Hypertension 79(10):2159–2169
Brown JD, Smith SM, Strotmeyer ES, Kritchevsky SB, Gill TM, Blair SN, Fielding RA, Buford TW, Pahor M, Manini TM (2019) Comparative effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on response to a physical activity intervention in older adults: results from the Lifestyle Interventions and Independence for Elders Study. J Gerontol Ser A 75(5):1010–1016
Clark MA, Tran H, Nguyen C (2011) Angiotensin III stimulates ERK1/2 mitogen-activated protein kinases and astrocyte growth in cultured rat astrocytes. Neuropeptides 45(5):329–335
Mi X, Cao Y, Li Y, Li Y, Hong J, He J, Liang Y, Yang N, Liu T, Han D, Kuang C (2021) The Non-peptide Angiotensin-(1–7) Mimic AVE 0991 attenuates delayed neurocognitive recovery after laparotomy by reducing neuroinflammation and restoring blood-brain barrier integrity in aged rats. Front Aging Neurosci 13:624387
Efficacy and Safety of Sacubitril/Valsartan Compared to Valsartan on Cognitive Function in Patients With Chronic Heart Failure and Preserved Ejection Fraction. American College of Cardiology. https://www.acc.org/Latest-in-Cardiology/Clinical-Trials/2022/08/25/03/38/PERSPECTIVE. Accessed 1 Nov 2022.
Cannon JA, Shen L, Jhund PS, Kristensen SL, Købe L, Chen F, Gong J, Lefkowitz MP, Rouleau JL, Shi VC, Swedberg K (2017) Dementia-related adverse events in PARADIGM-HF and other trials in heart failure with reduced ejection fraction. Eur J Heart Fail 19(1):129–137
Grewal P, Yoo J, Myserlis EP, Skopicki HA, Kalogeropoulos A (2022) Sacubitril-valsartan and 5-year incidence of neurocognitive diagnoses. J Cardiac Fail 28(5):S11
Kerola T, Nieminen T, Hartikainen S, Sulkava R, Vuolteenaho O, Kettunen R (2010) B-type natriuretic peptide as a predictor of declining cognitive function and dementia—a cohort study of an elderly general population with a 5-year follow-up. Ann Med 42(3):207–215
Daniels LB, Laughlin GA, Kritz-Silverstein D, Clopton P, Chen WC, Maisel AS, Barrett-Connor E (2011) Elevated natriuretic peptide levels and cognitive function in community-dwelling older adults. Am J Med 124(7):670-e1
Famitafreshi H, Karimian M (2020) Prostaglandins as the agents that modulate the course of brain disorders. Degenerat Neurol Neuromuscul Disease, pp 1–13
Thomas MH, Olivier JL (2016) Arachidonic acid in Alzheimer's disease. J Neurol Neuromed 1(9)
ADAPT Research Group (2008) Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65(7):896
Sharma P, Sharma D, Sharma L (2021) Exploring NSAIDs as anti-amyloidal agents: a drug repurposing approach to treat Alzheimer’s disease. Alzheimers Dement 17:e058504
Abdel-Aal RA, Hussein OA, Elsaady RG, Abdelzaher L (2021) The potential role of naproxen in promoting rivastigmine effect against aluminum chloride-induced Alzheimer-like model in rats
Mugisho OO, Robilliard LD, Nicholson LF, Graham ES, O’Carroll SJ (2020) Bradykinin receptor-1 activation induces inflammation and increases the permeability of human brain microvascular endothelial cells. Cell Biol Int 44(1):343–351
Zhang Q, Tan J, Wan L, Chen C, Wu B, Ke X, Wu R, Ran X (2021) Increase in blood–brain barrier permeability is modulated by tissue kallikrein via activation of bradykinin B1 and B2 receptor-mediated signaling. J Inflamm Res 14:4283
Petrella C, Ciotti MT, Nisticò R, Piccinin S, Calissano P, Capsoni S, Mercanti D, Cavallaro S, Possenti R, Severini C (2020) Involvement of bradykinin receptor 2 in nerve growth factor neuroprotective activity. Cells 9(12): 2651
Singh PK, Chen ZL, Ghosh D, Strickland S, Norris EH (2020) Increased plasma bradykinin level is associated with cognitive impairment in Alzheimer’s patients. Neurobiol Dis 139:104833
Acknowledgements
The author would like to thank Dr. Bassnt El-Mokadem for her assistance in preparing this article.
Funding
Not applicable for this work.
Author information
Authors and Affiliations
Contributions
Author ASM collected all the study material, analyzed it, and prepared the complete manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable for this work.
Consent for publication
The authors declare no conflict of interest.
Competing interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mansour, A.S. Autoregulation: mediators and renin–angiotensin system in diseases and treatments. Futur J Pharm Sci 9, 30 (2023). https://doi.org/10.1186/s43094-023-00482-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-023-00482-4