
Abstract
Background
Sepsis reaction is a response to an infection composed of genetic elements. This research aims to better understand how sepsis affects the molecular pathways in whole blood samples.
Methods
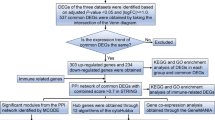
Whole blood samples from healthy controls (n = 18), sepsis nonsurvivors (n = 9), and sepsis survivors (n = 26) were retrieved from the gene expression omnibus (GEO) collection of the national center for biotechnology information (NCBI) (accession number GSE54514). The NCBI's GEO2R program was used to determine differential expression, and the ingenuity pathway analysis (IPA) software was utilized to do a pathway analysis.
Results
In sepsis patients, 2672 genes were substantially differently expressed (p value 0.05). One thousand three hundred four genes were overexpressed, and one thousand three hundred sixty-eight were under-expressed. The inhibition of ARE-mediated mRNA degradation pathway and the Pl3K/AKT signaling spliceosomal cycle were the most significant canonical pathways identified by ingenuity pathway analysis (IPA). The IPA upstream analysis predicted the ESR1, SIRT1, and PTPRR proteins, and the drugs filgrastim and fluticasone were top transcriptional regulators.
Conclusions
The inhibition of ARE-mediated mRNA degradation pathway and the Pl3K/AKT signaling spliceosomal cycle were highlighted as essential pathways of inflammation by IPA, indicating widespread cancer owing to sepsis. Our data imply that sepsis considerably influences gene pathways in whole blood samples, pointing to possible targets for sepsis treatment.
Similar content being viewed by others


Background
A dysregulated host response to infection causes sepsis, a life-threatening organ failure that affects over 19 million individuals yearly, killing ~ 11 million persons [1,2,3,4]. General management measures such as supportive care, source control, and antibiotics are still used to treat sepsis. Despite numerous clinical trials, there is no sepsis-specific drug beneficial in clinical practice [5]. A complete list of gene names and their abbreviations are delivered in the Additional file 1.
According to two consensus publications, the lack of tools to reliably diagnose sepsis at the molecular level and significant human heterogeneity in the sepsis syndrome are the main reasons for the persistent failure of proposed sepsis therapeutics [5, 6]. Clinical severity ratings are used to classify the risk of sepsis blood lactate levels, such as Acute Physiology, Age and Chronic Health Evaluation (APACHE) or Sequential Organ Failure Assessment (SOFA). These ratings estimate the overall severity of the illness, and they are incapable of quantifying the patient's response severity [7].
There are various advantages to having a molecular definition of the seriousness of the sepsis host response. The clinical outcomes can be improved by improving sepsis diagnosis accuracy by better matching resources with patients; more accurate diagnosis predictions would allow for more information about the efficiency of interventions and patient preferences. Clinical trials can be boosted by better molecular phenotyping of sepsis patients through enhancing medication, intervention, patient selection, and calculating observed-to-expected mortality ratios [5, 6, 8]. Since molecular biomarkers are a lineal quantitative indicator of the host response dysregulations, they may be used to help construct a quantitative diagnosis of sepsis versus non-septic critical illness [9, 10]. Overall, a quantitative sepsis test might be valuable for doctors if used as a fast assay.
This research aimed to figure out which molecular pathways are changed in sepsis. IPA (Ingenuity Pathway Analysis) is a web-based software program that identifies new targets within biological systems and is used to examine gene expression data from whole blood of sepsis patients versus healthy controls.
Methods
Data collection
In the present work, the investigated microarray dataset was obtained from (NCBI) the National Center for Biotechnology Information's (GEO) Gene expression Omnibus repository (accession number GSE54514). The dataset included gene expression data of whole blood samples collected for five days for healthy controls (n = 18), sepsis nonsurvivors (n = 9), and sepsis survivors (n = 26) [11]. Sepsis was described as the occurrence of at least two of four clinical criteria in addition to a proven bacterial infection. These clinical criteria are the change in white blood cell count, abnormally rapid breathing or if mechanical ventilation is required, abnormal rapid heart rate, and the presence of hypothermia or fever [11]. Whole blood samples collected from patients were preserved in PAXgene tubes, then RNA extraction was carried out in batches. Illumina Sentrix was used to profile gene expression for the extracted RNA.
The discovery of genes that are differently expressed
The NCBI's GEO2R software generated a list of 14,703 genes that were expressed differently between sepsis and non-sepsis blood samples. Microsoft Excel was used to sort and process the 14,703 genes. After applying stringent cutoff criteria, the list of differentially expressed genes was limited down to 2672 genes after applying stringent cutoff criteria (p value 0.05 and absolute fold change between − 0.1 and 0.1).
Pathway analysis using IPA
The differentially expressed gene list was entered into IPA software (QIAGEN, Hilden, Germany), which employed the program's 'core analysis' feature to analyze the data regards upstream regulators and canonical pathways.
Results
DE genes (differently expressed genes) and upstream regulators by IPA
Figure 1 (A) Graphical summary and (B) Different shapes represent the molecular class of the protein. Inhibition and activation are shown by blue and orange, respectively. A solid line denotes a direct relationship, a dashed line denotes an indirect interaction, and a dotted line denotes machine-based learning inferred association. Multiple gene IDs in the dataset are represented by a single gene or molecule in the Global Molecular Network, indicated by an asterisk.

A Graphical summary and B Different shapes represent the molecular class of the protein. Blue and orange indicate inhibition and activation. A solid line indicates a direct interaction, a dashed line indicates an indirect interaction, and a dotted line indicates inferred correlation from machine-based learning. An asterisk indicates that multiple identifiers in the dataset file map to a single gene or chemical in the Global Molecular Network
Figure 2 Upstream regulators: filgrastim and the estrogen receptor 1 (ESR1). (A) Chemical drug filgrastim is predicted to be activated in sepsis with p value = 1.36 × 10–7 and Z-score = 4.170. (ESR1 is predicted to be activated in sepsis with p value = 1.55 × 10–8 and Z-score = 4.064). Different forms represent the molecular class of the protein. Red and green show up-regulation and down-regulation, respectively. Inhibition and activation are indicated by blue and orange, respectively, in predicted relationships. A solid line denotes a direct relationship, a dashed line denotes an indirect interaction, and a dotted line denotes machine-based learning inferred association.

Upstream regulators (A) filgrastim and (B) ESR1. Chemical drug filgrastim is predicted to be activated in sepsis with p-value = 1.36 x 10–7 and Z-score 4.170. (BESR1 is predicted to be activated in sepsis with P-value = 1.55 x 10–8 and Z-score = 4.064. Different shapes represent the molecular class of the protein. Red and green indicate down-regulation and up-regulation, respectively, while blue and orange indicate inhibition and activation, respectively. A solid line indicates a direct interaction, a dashed line indicates an indirect interaction, and a dotted line indicates inferred correlation from machine-based learning. An asterisk indicates that multiple identifiers in the dataset file map to a single gene or chemical in the Global Molecular Network
Figure 3 Regulatory effect genes PTPRR and EPO are predicted during sepsis. Different forms represent the molecular class of the protein. Up-regulation and down-regulation are shown in red and green, respectively. Inhibition and activation are indicated by blue and orange, respectively. A solid line denotes a direct relationship, a dashed line denotes an indirect interaction, and a dotted line denotes machine-based learning inferred association.

Regulatory effect genes PTPRR and EPO are predicted during sepsis. Different shapes represent the molecular class of the protein. Red and green indicate down-regulation and up-regulation, respectively, while blue and orange indicate inhibition and activation, respectively. A solid line indicates a direct interaction, a dashed line indicates an indirect interaction, and a dotted line indicates inferred correlation from machine-based learning. An asterisk indicates that multiple identifiers in the dataset file map to a single gene or chemical in the global molecular network
Figure 4 Regulatory effect genes EIF4G2 and NFKB2 are predicted during sepsis. Different forms represent the molecular class of the protein. Up-regulation and down-regulation are shown in red and green, respectively. Inhibition and activation are indicated by blue and orange, respectively. A solid line denotes a direct relationship, a dashed line denotes an indirect interaction, and a dotted line denotes machine-based learning inferred association.

Regulatory effect genes EIF4G2 and NFKB2 are predicted during sepsis. Different shapes represent the molecular class of the protein. Red and green indicate down-regulation and up-regulation, respectively, while blue and orange indicate inhibition and activation, respectively. A solid line indicates a direct interaction, a dashed line indicates an indirect interaction, and a dotted line indicates inferred correlation from machine-based learning. An asterisk indicates that multiple identifiers in the dataset file map to a single gene or chemical in the global molecular network
Upstream regulators
Results showed that (ESR1, SIRT1, and PTPRR) proteins, filgrastim, and fluticasone drugs were among the top 20 regulators predicted by IPA (Table 1). Figure 2 depicts the data in Table 1 and emphasizes the major upstream regulators' anticipated activation state as reported by IPA. The ESR1 protein and medication filgrastim are the most active upstream regulator in sepsis. As shown in Fig. 2,ESR1 is among the essential proteins in IPA's graphical summary results.
The most significant regulatory effects are networks EIF4G2 and NFKB2. They are important regulatory underlying factors in sepsis pathogenesis identified by IPA. Figures 3, 4, and Table 4 show that the most significantly differentially expressed protein-coding genes during sepsis exposure were the CLEC1B, PPBP, HBE1, SNX10, and H3P6 genes.
Biological pathways that have been enriched by IPA
The inhibition of ARE-Mediated mRNA Degradation Pathway and the Pl3K/AKT Signaling Spliceosomal Cycle were essential canonical pathways (Table 2).
The relationship between sepsis and other illnesses
The differently expressed genes in sepsis are linked to cancer and organ harm, among other illnesses (Table 3).
Discussion
We applied the IPA tool to find important molecular pathways and regulatory networks in the context of septic disease. Consequently, we carried out upstream regulators analysis, revealing that estrogen receptor 1 (ESR1) and filgrastim drug are the most significant DE regulators during sepsis. ESR1 gene was not revealed to be associated with sepsis before. Recent studies showed that the ESR1 gene was associated with other diseases such as osteoporosis [12], coronary artery disease [13], Parkinson's disease [14], ovarian cancer [15], multiple myeloma [16], and finally, Alzheimer's disease [17].
Consequently, we carried out upstream regulators analysis that revealed the estrogen receptor 1 (ESR1) and filgrastim drug. Filgrastim (NEUPOGEN®) is a humanized granulocyte colony-stimulating factor used to treat and prevent neutropenia [18]. Filgrastim is the best agent for patients with severe sepsis/septic shock [19]. A recent report showed that Filgrastim is useful in postoperative patients at risk of sepsis. It results in improved production and function of neutrophils. It also appeared to counter the regulatory process of hyperactivation of pro-inflammatory processes [20]. Such inflammatory processes might agree with our results that filgrastim modulating many inflammatory genes, such as IL-10, is a crucial anti-inflammatory cytokine that was overexpressed during sepsis and was targeted by filgrastim as his upstream regulator discovered by IPA.
IL-1RA increased in 10 patients in the previous study [20], was increased in our dataset, and targeted by filgrastim. Interestingly, some cytokines were associated with sepsis; IL-8, IL-6, and IL-10 are practical sepsis biomarkers. The same report showed that Filgrastim increased the expression of these markers, and authors observed deficiency in the IL-23-IL-17 dual genes associated with sepsis [21].
The most significant regulatory effects are networks EIF4G2 and NFKB2. They are important regulatory underlying factors in sepsis pathogenesis identified by IPA as shown in Fig. 4.
Firstly, Eukaryotic Translation Initiation Factor 4 Gamma 2 (EIF4G2) was found to be associated with sepsis previously when sepsis changed the distribution of eukaryotic initiation factor 4E (eIF4E) [22]. Another report showed that Eukaryotic Translation Initiation Factor 4 Gamma 1 (EIF4G1) is a potential target for cancer treatment [23]. The same report showed that this gene, EIF4G1, was dominant in multiple cancers, for instance, cervical cancer, prostate cancer, Head and Neck Cancer, and ovarian cancer [23].
Secondly, Nuclear Factor Kappa B Subunit 2 (NFKB2), Nuclear factor-kappa B (NF-κB), is a master regulator of the inflammatory response and represents a key regulatory node in the complex inflammatory signaling network [24].
We highlighted that this gene is an essential regulator of sepsis-induced genetic expressions. Recent reports showed that sepsis provoked cardiac malfunction by NF-κB Pathway in animal models [25]. Another report signifies the role of this gene in sepsis; toll-like receptors activation provoked the nuclear factor-κB pathway, which led to the downregulation of specific sodium transporter expression during sepsis [26]. This vital gene is involved in acute inflammation [24], respiratory diseases [27], and joint diseases such as osteoarthritis [28].
We also discovered that the most significantly differentially expressed protein-coding genes during sepsis exposure were the CLEC1B, PPBP, HBE1, SNX10, and H3P6 genes, as shown in Table 4. The CLEC1B gene, CLEC1B lower expression was a prognostic factor indicating the poor outcome for hepatic cancer [29]. Another report showed that this gene was among the hub genes associated with hepatocellular carcinoma [30]. Overexpression of CLEC1B gene inhibited hepatocellular carcinoma cells' proliferation and migration [31]. PPBP gene translated to pro-platelet basic protein was associated with the diagnosis and prognosis of sepsis in an animal model; rats [32], and other diseases such as chronic, allergic aspergillosis [33] and tongue cancer [34]. PPBP gene is a biomarker in acute ischemic stroke in Type 2 Diabetes [35].
The current report has limitations. The sample size was small, and the patients were not equally distributed, which could confound the interpretation of the genetic variation. Additionally, several genes in sepsis were uncharacterized or unmapped to pathways, meaning their effects are not considered in the current analysis.
Conclusion
The current findings signify the importance of inflammation as a significant component of sepsis-induced tissue injury. Most significantly, EIF4G2 and NFKB2 are the regulatory effects networks underlying genetic and molecular pathway changes coupled with exposure to sepsis. Future lines of research should focus on validating the results of the current study in a larger population to ascertain potential therapeutic targets in the context of sepsis-induced damage.
Availability of data and materials
The current report utilized a previously published dataset for the analysis. The dataset used in this work was acquired from The National Center for Biotechnology Information’s (NCBI) Gene Expression Omnibus (GEO) depository (accession number GSE54514).
Abbreviations
- IPA:
-
Ingenuity pathway analysis
- DE:
-
Differentially expressed
- GEO:
-
Gene expression omnibus
- NCBI:
-
National centre for biotechnology information
References
Prescott HC, Angus DC (2018) Enhancing recovery from sepsis: a review. JAMA 319(1):62–75
Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C et al (2021) Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med 47(11):1181–1247
Fleischmann-Struzek C, Mellhammar L, Rose N, Cassini A, Rudd KE, Schlattmann P et al (2020) Incidence and mortality of hospital- and ICU-treated sepsis: results from an updated and expanded systematic review and meta-analysis. Intensive Care Med 46(8):1552–1562
Olwal CO, Nganyewo NN, Tapela K, Djomkam Zune AL, Owoicho O, Bediako Y et al (2021) Parallels in sepsis and COVID-19 conditions: implications for managing severe COVID-19. Front Immunol 12:602848
Opal SM, Dellinger RP, Vincent JL, Masur H, Angus DC (2014) The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C?*. Crit Care Med 42(7):1714–1721
Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T et al (2015) Sepsis: a roadmap for future research. Lancet Infect Dis 15(5):581–614
Shankar-Hari M, Phillips GS, Levy ML, Seymour CW, Liu VX, Deutschman CS et al (2016) Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315(8):775–787
Sweeney TE, Perumal TM, Henao R, Nichols M, Howrylak JA, Choi AM et al (2018) A community approach to mortality prediction in sepsis via gene expression analysis. Nat Commun 9(1):694
Abraham E (2016) New Definitions for sepsis and septic shock: continuing evolution but with much still to be done. JAMA 315(8):757–759
Bermejo-Martin JF, Tamayo E, Andaluz-Ojeda D, Martin-Fernandez M, Almansa R (2017) Characterizing systemic immune dysfunction syndrome to fill in the gaps of SEPSIS-2 and SEPSIS-3 definitions. Chest 151(2):518–519
Parnell GP, Tang BM, Nalos M, Armstrong NJ, Huang SJ, Booth DR et al (2013) Identifying key regulatory genes in the whole blood of septic patients to monitor underlying immune dysfunctions. Shock 40(3):166–174
Krela-Kazmierczak I, Skrzypczak-Zielinska M, Kaczmarek-Rys M, Michalak M, Szymczak-Tomczak A, Hryhorowicz ST et al (2019) ESR1 gene variants are predictive of osteoporosis in female patients with crohn’s disease. J Clin Med 8(9):1306
Sumi MP, Guru SA, Mir R, Masroor M, Bhat MA, Girish MP et al (2019) Clinical Importance of estrogen receptor 1 (ESR1) gene polymorphisms and their expression patterns in coronary artery disease patients: a study from India. Indian J Clin Biochem 34(2):133–142
Retraction Note to: Genetic POLYMORPHISMS in VDR, ESR1 and ESR2 genes may contribute to susceptibility to Parkinson's disease: a meta-analysis. Mol Biol Rep. 2015;42(10):1457
Gong G, Lin T, Yuan Y (2020) Integrated analysis of gene expression and DNA methylation profiles in ovarian cancer. J Ovarian Res 13(1):30
Cui Y, Wang F, Zhang D, Huang J, Yang Y, Xu J et al (2022) Estrogen responsive gene MAST4 regulates myeloma bone disease. J Bone Miner Res 37:711
Ma SL, Tang NL, Tam CW, Lui VW, Lau ES, Zhang YP et al (2009) Polymorphisms of the estrogen receptor alpha (ESR1) gene and the risk of Alzheimer’s disease in a southern Chinese community. Int Psychogeriatr 21(5):977–986
Dale DC, Crawford J, Klippel Z, Reiner M, Osslund T, Fan E et al (2018) A systematic literature review of the efficacy, effectiveness, and safety of filgrastim. Support Care Cancer 26(1):7–20
Wunderink R, Leeper K Jr, Schein R, Nelson S, DeBoisblanc B, Fotheringham N et al (2001) Filgrastim in patients with pneumonia and severe sepsis or septic shock. Chest 119(2):523–529
Weiss M, Gross-Weege W, Harms B, Schneider EM (1996) Filgrastim (RHG-CSF) related modulation of the inflammatory response in patients at risk of sepsis or with sepsis. Cytokine 8(3):260–265
de Araujo OR, Salomao R, Brunialti MKC, da Silva DCB, Senerchia AA, de Moraes Costa Carlesse FA, et al. Cytokine kinetics in febrile neutropenic children: insights on the usefulness as sepsis biomarkers, influence of filgrastim, and behavior of the IL-23/IL-17 pathway. Mediat Inflamm. 2017;2017:8291316.
Lang CH, Frost RA (2007) Sepsis-induced suppression of skeletal muscle translation initiation mediated by tumor necrosis factor alpha. Metabolism 56(1):49–57
Jaiswal PK, Koul S, Palanisamy N, Koul HK (2019) Eukaryotic translation initiation factor 4 gamma 1 (EIF4G1): a target for cancer therapeutic intervention? Cancer Cell Int 19:224
Rius-Perez S, Perez S, Marti-Andres P, Monsalve M, Sastre J (2020) Nuclear factor kappa B signaling complexes in acute inflammation. Antioxid Redox Signal 33(3):145–165
Huang X, Zhang MZ, Liu B, Ma SY, Yin X, Guo LH (2021) Astragaloside IV attenuates polymicrobial sepsis-induced cardiac dysfunction in rats via IKK/NF-kappaB pathway. Chin J Integr Med 27(11):825–831
Li D, Zou L, Feng Y, Xu G, Gong Y, Zhao G et al (2016) Complement factor B production in renal tubular cells and its role in sodium transporter expression during polymicrobial sepsis. Crit Care Med 44(5):e289–e299
Tang X, Sun L, Wang G, Chen B, Luo F (2018) RUNX1: a regulator of NF-kB signaling in pulmonary diseases. Curr Protein Pept Sci 19(2):172–178
Chin KY (2016) The spice for joint inflammation: anti-inflammatory role of curcumin in treating osteoarthritis. Drug Des Devel Ther 10:3029–3042
Hu K, Wang ZM, Li JN, Zhang S, Xiao ZF, Tao YM (2018) CLEC1B expression and PD-L1 expression predict clinical outcome in hepatocellular carcinoma with tumor hemorrhage. Transl Oncol 11(2):552–558
Wang S, Song Z, Tan B, Zhang J, Zhang J, Liu S (2021) Identification and validation of hub genes associated with hepatocellular carcinoma via integrated bioinformatics analysis. Front Oncol 11:614531
Zhang G, Su L, Lv X, Yang Q (2021) A novel tumor doubling time-related immune gene signature for prognosis prediction in hepatocellular carcinoma. Cancer Cell Int 21(1):522
Jiao J, Gao M, Zhang H, Wang N, Xiao Z, Liu K et al (2014) Identification of potential biomarkers by serum proteomics analysis in rats with sepsis. Shock 42(1):75–81
Smith NL, Bromley MJ, Denning DW, Simpson A, Bowyer P (2015) Elevated levels of the neutrophil chemoattractant pro-platelet basic protein in macrophages from individuals with chronic and allergic aspergillosis. J Infect Dis 211(4):651–660
Sun W, Qiu Z, Huang W, Cao M (2018) Gene expression profiles and proteinprotein interaction networks during tongue carcinogenesis in the tumor microenvironment. Mol Med Rep 17(1):165–171
Moin ASM, Nandakumar M, Al-Qaissi A, Sathyapalan T, Atkin SL, Butler AE (2021) Potential biomarkers to predict acute ischemic stroke in type 2 diabetes. Front Mol Biosci 8:744459
Acknowledgements
The authors would like to acknowledge the efforts of the University of Petra’s Faculty of Pharmacy and Medical Sciences, its dean, and its Department Head of Pharmacology and Biomedical Sciences.
Funding
This study was not funded.
Author information
Authors and Affiliations
Contributions
AAE contributed to conceptualization, and editing of manuscript, and analysis, validation and visualization. AK contributed to official analysis, IPA methodology, and writing, draft. AA contributed to visualization and final manuscript review and editing. All authors have read and approved the manuscript. ANAM has contributed to the final draft writing and to the first-round revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
RAW DATA GSE54514.top.table supplementary file 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elbakkoush, A.A., Khaleel, A., Mohamed, A.N.A. et al. Pathway analysis of sepsis-induced changes gene expression. Egypt J Med Hum Genet 23, 142 (2022). https://doi.org/10.1186/s43042-022-00352-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00352-3