
Abstract
Background
Intellectual disability is characterized by impairments in adaptive behavior and cognitive functioning manifested during the developmental period. Since disabilities are heterogeneous, variant analysis can help us confirm and accurately diagnose children with intellectual disabilities. Some papers reported that bi-allelic variants of the NSUN2 gene caused a group of neurological disorders, including non-syndromic autosomal recessive intellectual disability (NS-ARID), Dubowitz syndrome, and familial restrictive cardiomyopathy 1 (RCM1). We report on a consanguineous family with three siblings diagnosed with intellectual disability.
Case presentation
The 7-year-old female was referred to Ali-Asghar hospital, Zahedan, Iran, with clinical manifestations comprising moderate intellectual disability, ptosis, long face, and short stature. Chromosome banding, metabolic testing, and magnetic resonance imaging examinations revealed no abnormalities. Accordingly, other affected siblings born of the same parents were considered. Whole-exome sequencing (WES) was conducted on the sufferer to consider NS-ARID variants. Findings identified a variant with uncertain significance (NM_017755.6: c.593 T > G) in the NSUN2 gene in the proband. This variant was confirmed through Sanger sequencing of the affected and unaffected family members. Besides, the computational results showed that the L198R exchange could change the interaction between wild-type and other residues in the protein. The affected patients with NS-ARID had similar clinical characteristics and genetic abnormalities.
Conclusion
Taken together, we described the variant in three Iranian siblings; further expanding of the other variants involved in the disease will be evident by using high-throughput sequencing technologies.
Similar content being viewed by others

Background
Intellectual disability (ID) is classified as a neurodevelopmental impairment with remarkable cognitive and adaptive functioning deficits before age 18 and an IQ score of less than 70% [1]. There are various degrees of ID categorized according to their level of intelligence quotient: mild, moderate, severe, and profound. Sufferers with intense ID may manifest signs such as decreased nasal bridge, seizures, poor or absent speech, delayed psychomotor development, self-mutilating behavior, global developmental delay, generalized hypotonia, epileptic spasms, and abnormal facial features within the first two years of life. Identifying children with mild ID may not be achieved until early school age [2]. The prevalence of ID is estimated at between 1–3% in diverse populations worldwide [3]. Respiratory illnesses are one of the major leading causes of demise in these sufferers [4].
Moreover, it is genetically a heterogeneous disorder that follows different inheritance models. Also, many essential genes are involved in the etiology of the disease [5]. Next-generation sequencing technology helps us determine a precise genetic diagnosis in children with ID [6].
Some research has found a link between NSUN2 gene variants and ID disease [7,8,9]. The NSUN2 (NOP2/Sun RNA methyltransferase 2) gene, also known as MISU, is located at 5p15 and spans 33 kb, comprising 19 exons. The gene transcribes a 2.1 kb mRNA molecule expressed in brain tissue and generates a methyltransferase enzyme. Cytosine at positions 34 and 48 of tRNA precursors is methylated to 5-methylcytosine (m5C). This modification can promote tRNA stability and mRNA export [8, 10, 11]. Enzymatic failure of the enzyme leads to Dubowitz syndrome, non-syndromic autosomal recessive intellectual disability (NS-ARID), and familial restrictive cardiomyopathy 1 (RCM1) [9]. ARID is characterized by impairments in adaptive behavior and cognitive functioning that manifest during the developmental period [12]. As a result of using whole-exome sequencing, our research found a novel homozygous missense variant in an Iranian patient diagnosed with ARID from a large consanguineous family.
Case presentation
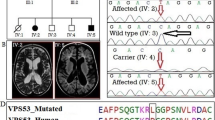
This family was racially Baloch, with five generations, including 25 members (Fig. 1a). This study considered three affected siblings born of the same parents (Fig. 1b–d). They all had normal hearing and vision.

a Pedigree of the family. The family members with ID phenotype are shown at Pedigree (in black) with segregation of the p.(Lue198Arg) variant in the NSUN2 gene. Squares represent males; circles stand for females; ± , heterozygote for p.(Lue198Arg) variant; -/- homozygous for p.(Lue198Arg) variant. Clinical features of the individual V-6 showing ptosis, broad nasal bridge, and long face. b–d Photographs of individual 4–6
Patient V:4
The patient is a 16-year-old male referred to Ali-Asghar hospital, Zahedan, Iran, due to ID. A physician and a genetic counselor thoroughly examined the patient's clinical features and family history. He was the first child (first pregnancy) born by vaginal delivery at 40 weeks gestation. His mother had a normal pregnancy with no fetal decelerations or bleeding. After birth, he experienced severe developmental delay and could only walk with assistance at the age of four. On examination, he showed severe ID retardation, speech delay, seizures, a long face, broad nose, and short stature. Additionally, he was never capable of living independently. A group of medical laboratory assessments, including cytogenetic analysis, metabolic testing, magnetic resonance imaging, and array comparative genome hybridization, were performed on the sufferer. Nonetheless, there were no abnormal signs (Fig. 1b).
Patient V:5
A 14-year-old female showed a range of clinical features, including moderate mental retardation, ptosis, beaked nose, long face, poor speech, seizure, and short stature (Fig. 1c). The girl presented with moderate developmental delay and could only walk at two years old. Her birth weight was 2100 g (-2SD), and her length and head circumference at birth were 45 cm (-3SD) and 33 cm (-2SD), respectively. Furthermore, her karyotype analysis showed a standard 46, XX without any chromosomal abnormalities.
Patient V:6
The proband in this family is a 7-year-old female with a similar phenotype to her older sister V:5, but she had just the ptosis phenotype without the beaked nose (Fig. 1d). She could only walk at 26 months, and her speech was slow. She was born through a cesarean (C-section) delivery earlier than the spontaneous delivery course.
Molecular analyses
The bioinformatics analyses were narrowed down to a novel homozygous missense variant (NM_017755.6:c.593 T > G) in exon 6 of the NSUN2 gene (NP_060225.4; NG_028215.1), which can create a missense variant in the protein sequence (p. Leu198Arg) (Fig. 2). The variant was categorized according to the American College of Medical Genetics and Genomics (ACMG) as potential pathogenic (PM1, PM2, PP1, and PP3) [13] after manual adjustment for the clinical interpretation using the InterVar database. In other words, the variant is located in a conserved region (PP3) and a well-established functional domain (PM1), was not reported in the gnomAD exomes and genomes and Iranome projects (PM2), and was predicted to be pathogenic based upon nine in silico prediction tools (PP3). Furthermore, Sanger's results confirmed the homozygote variant in the three affected siblings and the heterozygote variant in their normal parents (PP1). Furthermore, the variant was perfectly co-segregated through other family members (Fig. 2). According to Jarvik's report (2016), the probability of observed cosegregation for three affected siblings who carried the same variant is 1/16. Thus, these data demonstrated an ACMG-AMP evidence level of pathogenic moderate [14]. In addition, all carriers of the p. Leu198Arg variant were clinically normal without any abnormalities.

Sanger sequence analysis of the NSUN2 gene revealing the c.593 T > G variant
Computational analysis
Following the creation of protein modeling through the SWISS-MODEL server, a crystal structure of the YebU (methyltransferase from E. coli) with PDB ID c2frxD was the best template for the model, which has a sequence identity of 30% with the NSUN2 protein. Moreover, the structure was validated by the Ramachandran plot calculations. Phyre2's results predicted that the variant is located in the disordered regions, is significant in the protein's function, and has phenotypic effects (Fig. 3a). Substituting an arginine residue (mutant) can also considerably alter the interactions between the wild type and other residues (Fig. 3b). The output of the I-Mutant2.0 indicated that the variant decreased the protein stability.

The impact of the novel variant on the model of NSUN2 protein. a The amino acid sequence of NSUN2 is colored based on mutational sensitivity using Phyre2 investigator. The server predicts the discrimination between disease-causing variants with high and low (neutral variants) sensitivity. b The novel variant creates a beta-sheet structure between mutant and other residues in the enlarged image. c the schematic structure of conservation in the different regions. The purple and green colors show a highly and lowly conserved region, respectively. Lue198 is located in a conserved middle area. The B-factors provide information about the atomic displacement and dynamic parameters in the protein's crystal structure. The image shows that wild-type residue is located in a functional and structural region. d The graph shows the conservation of multiple sequence alignments from multiple species. The overall height of each symbol in that position presents the relative frequency of each nucleotide in the sequence. e The original (Leucine) and the mutant (Arginine) amino acid schematic structure
On the other hand, the pathogenicity of the novel variant (p. Lue198Arg) was evaluated by multiple in silico prediction tools. Nine of them (Polyphen-2, CADD, REVEL, ClinPred, DANN, MutPred, Provean (SCR_005762), MutationTaster (SCR_010777), and SIFT (SCR_012813)) expected it to be pathogenic (Table 1). Multiple sequence alignment by the WebLogo and ConSurf server revealed that the leucine is highly conserved among different species (Fig. 3d). In the novel variant, arginine was replaced with leucine (Fig. 3e). The HOPE results anticipated that the arginine (mutant residue) could change the main secondary structure of the wild-type protein from α-helices to B-sheets. As a result, the variant might disrupt significant interactions with different targets.
Methods
Three affected individuals were in one generation, from parents with a consanguineous relationship. The affected family members' clinical features and IQ scores were assessed by the psychiatrist and genetic counselor at Ali-Asghar hospital. The study was approved by the ethics committee of Shahid Sadoughi University of Medical Sciences (IR.SSU.MEDICINE.REC.1399.199). Before the study, written informed consent forms for publishing and participating were obtained from all family members. According to the manufacturer's instructions, DNA extraction was performed on peripheral blood from leukocytes using the QIAamp DNA Mini kit (Qiagen). Library preparation and sequencing were performed on DNA from the proband (V-6) by the SureSelect Human All Exon V6 kit (Agilent Technologies, CA) and HiSeq4000 machine sequencer (The coverage and sensitivity of the method were 100X and > 99%). We performed the quality analysis of the raw data (FASTQ file) from Hiseq4000 using IlluQC.pl and FastQC software. Next, the UCSC hg19/GRCh37 reference sequence was aligned with BWA mem (version 0.7.17-r1188). [15]. For post alignment, variant calling, and annotation, GATK (version 4.1.9.0) [16], Picard-tools [17], BCF tools [18], SAM tools [19], HaplotypeCaller [20], and ANNOVAR software [21] were applied, respectively. In our filtering strategy, we applied a list of the related panels of ID on VCF files. Then, the exonic, non-synonymous, non-benign variants were selected from the filtered files. The shortlisted variants' pathogenicity was assessed using the Varsome, InterVar, and ClinVar databases. Furthermore, we filtered out the common variants specific to the Iranian population by comparing them with the Iranome database, a catalog of genomic variations from 800 exomes from individuals belonging to eight ethnic groups [22]. The reported phenotypes of the selected variants were reviewed in the OMIM, Genecards, Malacard, and NCBI databases. We performed Sanger sequencing to confirm the variant in other family members (III-2, III-8, IV-4, IV-5, IV-6, IV-9, IV-10, IV-11, V-4, V-5, V-6). Primers F 5′-CTTGAACTGAGGTTACGG-3′ and R 5′-TTTGTTTGAGTACTACTGACG-3′ designed by Gene runner software were applied (version 6.5.52). Subsequently, we performed the polymerase chain reaction (PCR) according to the aforementioned kit. The PCR products were sequenced using the BigDye™ Terminator v3.1 Cycle Sequencing Kit and the Applied Biosystems 3700 (Thermo Fisher). The results were analyzed with Finch TV and Chromas software.
In-silico structural modeling and protein stability
As the 3D structure of the NSUN2 protein has not been reported in the protein data bank (PDB), the SWISS-MODEL online server (https://swissmodel.expasy.org) was applied to build the 3D structure of the protein (UniProt ID: Q08J23). This server is a computational tool for building protein models using the homology of protein structures and energy minimization. Afterward, Phyre2 investigator (Protein Homology/analogY Recognition Engine) (PHYRE2 Protein Fold Recognition Server (https://ic.ac.uk)) was used to assess the effect of the amino acid variant in the top model [23]. Also, we applied the I-TASSER server (Iterative Threading Assembly Refinement) to predict the impact of a missense variant (p. Lue198Arg) on the structure and function of the protein (https://zhanglab.ccmb.med.umich.edu/I-TASSER) [24]. The ConSurf server was used to predict the conserved and dynamic areas in the NSUN2 protein [25]. Highly and lowly conserved areas are colored purple and green, respectively. Following the collection of amino acid sequences from the UniProt database (http://www.uniprot.org), the WebLogo server (http://weblogo.threeplusone.com/create.cgi) was applied to determine the conservation among different species of multiple amino acid sequence alignments. The I-Mutant2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html) measured the changes in free energy (ΔΔG) for each variant in the protein sequences and was used to predict the impact of the variant on the protein stability [26]. Furthermore, HOPE as an online service (https://www3.cmbi.umcn.nl/hope) was used for analyzing the structural assessment of a missense variant in the protein sequence [27].
Discussion
In the present study, we found a novel homozygous missense variant in the NSUN2 gene NM_020919.3:c.593 T > G in the three siblings diagnosed with autosomal recessive intellectual disability (ARID) from a large consanguineous family. This form of the disease has a heterogeneous molecular basis, caused by variants in many genes, such as DDX3X, NHS, WDR45, MECP2, and DYRK1A genes [5]. Several studies have reported that variants in the NSUN gene are linked to ARID in countries with frequent parental consanguinity. Abbasi-Moheb et al. (2012) reported a homozygous transition in the NSUN2 gene, associated with the ID in a consanguineous Iranian family [7]. In another study, variant analysis of candidate genes by homozygosity mapping showed that NSUN2-flanking STR markers might be a powerful strategy for ARID diagnosis in medical genetic labs [28].
The NSUN gene encodes the methyltransferase, which is responsible for modifying tRNAs by methylation. Improper functioning of this enzyme may lead to the absence of tRNA-Leu (CAA) in the cytoplasm and, consequently, could cause some alterations in translational efficiency or fidelity, which might manifest some changes in the tissue-specific protein expression in ID patients. The protein was strongly conserved from bacteria to humans, and it was the first SUN-domain-containing protein characterized by invertebrates [29]. The NSUN2 gene expression analysis in the fetal brain has shown that NSUN2 is expressed in the early stages of brain development. Therefore, it is conceivable that the absence of NSUN2 protein could result in proteomic shifts in the affected individuals during brain development and may involve human neurocognitive development [30, 31].
NSUN2 variant-associated clinical features in the studied family included broad nasal bridge, severe and moderate intellectual disability, developmental delay, long face, short stature, and ptosis. As Abbasi-Moheb et al. previously reported, there is a range of overlapped phenotypes among individuals with NSUN2 variants; these clinical features include moderate to severe ID and facial dysmorphisms [7]. Recently, some papers reported that Nsun2 (− / −) knockout mice had a remarkable reduction in their size; these results might explain the short stature observed in several affected individuals [8]. Compared to the clinical symptoms of present cases related to other intellectual disabilities, molecularly-confirmed patients are reported in Table 2. Our bioinformatics results predicted that the wild-type residue (leucine) rather than the mutant residue (arginine) at p. L198R substitution is more significant, hydrophilic, and its charge changes from NEUTRAL to POSITIVE. So, hydrophobic interactions will be lost either in the core of the protein or on the surface. Besides, the results obtained from the HOPE software demonstrated that the p. L198R, which is located in a SUN-domain, is necessary for the protein's main activity. The variant of the residue may reduce the activity of methyltransferase, which could cause the disease (Fig. 3).
Using next-generation sequencing (NGS), we can identify rare and heterogeneous diseases like ARID, which have irreparable consequences for health care [6]. Moreover, due to the high rate of consanguineous marriages in Iran, especially in the Baloch ethnicity, considering the NGS panels, including the NSUN2 gene for individuals with ID, could shed light on the way of pediatricians and geneticists in confirming a molecular diagnosis of ID and probably distinguishing this disease from others [32].
Conclusion
In conclusion, this paper described a novel variant in the NSUN2 gene in three Iranian siblings. High-throughput sequencing technologies might be promising to expand further our knowledge of the potential pathogenic variants involved in heterogeneous disease. Also, according to the results from Sanger sequencing, this variant was co-segregated in this family, and the in silico analyses predicted its high level of pathogenicity. In this regard, this study's results are consistent with the results of previous publications and emphasize that analysis of the different variations of NSUN2, as a genetic test, might play a key role in ARID diagnosis.
Availability of data and materials
The data to support the findings in the study is available on request from the corresponding author.
Abbreviations
- ID:
-
Intellectual disability
- NS-ARID:
-
Autosomal recessive non-syndromic intellectual disability
- RCM1:
-
Restrictive cardiomyopathy 1
- WES:
-
Whole-exome sequencing
- NSUN2:
-
NOP2/Sun RNA methyltransferase 2
- m5C:
-
5-Methylcytosine
- SD:
-
Standard deviation
- PDB:
-
Protein data bank
- OMIM:
-
Online mendelian inheritance in man
- NGS:
-
Next-generation sequencing.
- I-TASSER:
-
Iterative threading assembly refinement
- Phyre2:
-
Protein homology/analogy recognition engine
- gnomAD:
-
Genome aggregation database
References
Shree A, Shukla P (2016) Intellectual disability: definition, classification, causes and characteristics. Learn Commun 7(1):9
Patel Dr, Apple R, Kanungo S, Akkal A. Intellectual disability: definitions, evaluation and principles of treatment. Pediatric Med. 2018;1.
Mckenzie K, Milton M, Smith G, Ouellette-Kuntz H (2016) Systematic review of the prevalence and incidence of intellectual disabilities: current trends and issues. Curr Dev Disord Rep 3(2):104–115
Glover G, Williams R, Heslop P, Oyinlola J, Grey J (2017) Mortality in people with intellectual disabilities In England. J Intellect Disabil Res 61(1):62–74
Ilyas M, Mir A, Efthymiou S, Houlden H. The genetics of intellectual disability: advancing technology and gene editing. F1000research. 2020;9.
Bruel Al, Vitobello A, Mau‐Them Ft, Nambot S, Sorlin A, Denommé‐Pichon As, Et Al. Next‐generation sequencing approaches and challenges in the diagnosis of developmental anomalies and intellectual disability. Clin Genet. 2020.
Abbasi-Moheb L, Mertel S, Gonsior M, Nouri-Vahid L, Kahrizi K, Cirak S et al (2012) Mutations in Nsun2 Cause autosomal-recessive intellectual disability. Am J Hum Genet 90(5):847–855
Khan M, Rafiq M, Noor A, Hussain S, Flores JV, Rupp V et al (2012) Mutation In Nsun2, which encodes an Rna methyltransferase, causes autosomal-recessive intellectual disability. Am J Hum Genet. 90(5):856–63
Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S et al (2012) Whole exome sequencing identifies a splicing mutation in Nsun2 As a cause of a dubowitz-like syndrome. J Med Genet. 49(6):380–5
Shinoda S, Kitagawa S, Nakagawa S, Wei F-Y, Tomizawa K, Araki K et al (2019) Mammalian Nsun2 Introduces 5-methylcytidines into mitochondrial trnas. Nucleic Acids Res 47(16):8734–8745
Yang X, Yang Y, Sun B-F, Chen Y-S, Xu J-W, Lai W-Y et al (2017) 5-methylcytosine promotes Mrna export—Nsun2 as the methyltransferase and Alyref as An M5c reader. Cell Res 27(5):606–625
Hill WD, Davies G, Liewald D, Payton A, Mcneil C, Whalley L et al (2016) Examining non-syndromic autosomal recessive intellectual disability (Ns-Arid) genes for an enriched association with intelligence differences. Intelligence. 54:80–9
Li Q, Wang K (2017) Intervar: clinical interpretation of genetic variants by the 2015 Acmg-Amp guidelines. Am J Hum Genet 100(2):267–280
Gp J, Bl B (2016) Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet 98(6):1077–1081
Houtgast EJ, Sima V-M, Bertels K, Al-Ars Z (2018) Hardware acceleration of bwa-mem genomic short read mapping for longer read lengths. Comput Biol Chem. 75:54–64
DoValle Íf, Giampieri E, Simonetti G, Padella A, Manfrini M, Ferrari A, et al. Optimized pipeline of mutect and gatk tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. Bmc Bioinformatics. 2016;17(12):341.
Institute B. http://Broadinstitute.Github.Io/Picard. 19 Sept 2018.
Danecek P, Mccarthy SA (2017) Bcftools/Csq: haplotype-aware variant consequences. Bioinformatics. 33(13):2037–9
Weeks NT, Luecke GR, Editors. Performance analysis and optimization of samtools sorting. European Conference On Parallel Processing; 2016: Springer.
Eliseev A, Gibson KM, Avdeyev P, Novik D, Bendall Ml, Pérez-Losada M, et al. Evaluation of haplotype callers for next-generation sequencing of viruses. Infect Genet Evol. 2020;82:104277.
Yang H, Wang K (2015) Genomic variant annotation and prioritization with annovar and wannovar. Nat Protoc 10(10):1556–1566
Fattahi Z, Beheshtian M, Mohseni M, Poustchi H, Sellars E, Nezhadi Sh et al (2019) Iranome: a catalog of genomic variations in the iranian population. Hum Mutat 40(11):1968–1984
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10(6):845–58
Zheng W, Zhang C, Bell EW, Zhang Y (2019) I-Tasser gateway: a protein structure and function prediction server powered by Xsede. Fut Gen Comput Syst. 99:73–85
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T et al (2016) Consurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucl Acids Res 44(W1):W344–W350
Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucl Acids Res. 2005;33(Web Server Issue):W306–10.
Venselaar H, Te Beek Tah, Kuipers Rkp, Hekkelman Ml, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An E-science approach with life scientist friendly interfaces. Bmc Bioinform. 2010;11:548-.
Najmabadi H, Motazacker MM, Garshasbi M, Kahrizi K, Tzschach A, Chen W et al (2007) Homozygosity mapping in consanguineous families reveals extreme heterogeneity of non-syndromic autosomal recessive mental retardation and identifies 8 novel gene loci. Hum Genet. 121(1):43–8
Tomac V, Pušeljić S, Škrlec I, Anđelić M, Kos M, Wagner J (2017) Etiology and the genetic basis of intellectual disability in the pediatric population. Southeastern Eur Med J Seemedj 1(1):144–153
Flores JV, Cordero-Espinoza L, Oeztuerk-Winder F, Andersson-Rolf A, Selmi T, Blanco S et al (2017) Cytosine-5 Rna methylation regulates neural stem cell differentiation and motility. Stem Cell Rep. 8(1):112–24
Hussain S, Bashir Zi. The epitranscriptome in modulating spatiotemporal rna translation in neuronal post-synaptic function. Frontiers In Cellular Neuroscience. 2015;9(420).
Baralle D (2020) Ismail V’. Bmj Publishing Group Ltd, Next Generation Sequencing’as A Diagnostic Tool In Paediatrics
Sun S, Chen L, Wang Y, Wang J, Li N, Wang X. Further delineation of autosomal recessive intellectual disability syndrome caused by homozygous variant of the Nsun2 gene in a chinese pedigree. Mol Genet Genomic Med. 2020;8(12):E1518-E.
Acknowledgements
We would like to gratitude the affected individuals and their families for their participation in the study. We thank the technical operators of Ali Asghar Hospital in Zahedan.
Funding
No funding.
Author information
Authors and Affiliations
Contributions
Author's contribution to the paper as follows: MD, SMK and MT designed the study and contributed in the conception. data collection: ZM and NG performed the physical and historical examination of patients; MYM and MM analysed and interpreted the patient data. Also, MM prepared the draft manuscript. The final version of the manuscript was reviewed and the results were approved by all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for this study was obtained from the School of Medicine-Shahid Sadoughi University of Medical Science. (IR.SSU.MEDICINE.REC.1399.199). Informed consent forms to participate were obtained from all family members before the study.
Consent for publication
Written informed consent to publish this information was obtained from their parent before the submission. Because our patients were under the age of 18 years.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moudi, M., Vahidi Mehrjardi, M.Y., Kalantar, S.M. et al. Co-segregation of variant NSUN2 Lue198Arg among Iranian family with intellectual disability: a case report. Egypt J Med Hum Genet 23, 82 (2022). https://doi.org/10.1186/s43042-022-00293-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-022-00293-x