
Abstract
Background
Intellectual disability (ID) is a phenotypically and genetically heterogeneous disorder.
Methods
In this study, genome wide SNP microarray and whole exome sequencing are used for the variant identification in eight Pakistani families with ID. Beside ID, most of the affected individuals had speech delay, facial dysmorphism and impaired cognitive abilities. Repetitive behavior was observed in MRID143, while seizures were reported in affected individuals belonging to MRID137 and MRID175.
Results
In two families (MRID137b and MRID175), we identified variants in the genes CCS and ELFN1, which have not previously been reported to cause ID. In four families, variants were identified in ARX, C5orf42, GNE and METTL4. A copy number variation (CNV) was identified in IL1RAPL1 gene in MRID165.
Conclusion
These findings expand the existing knowledge of variants and genes implicated in autosomal recessive and X linked ID.
Similar content being viewed by others

Introduction
Intellectual disability (ID), is a genetically heterogeneous neurodevelopmental disorder. ID is characterized by the marked reduction in individual’s intellectual capacity which is reflected in the form of intelligence quotient less than 70, and defects in adaptive behavior with an early age of onset [1]. The worldwide prevalence of ID is 1–3%, with more affected males than females [2, 3]. ID frequently co-exists with other conditions like autism, epilepsy, schizophrenia, attention deficit hyperactivity disorder or depression [4,5,6].
The earlier research on genetic causes of ID focused on X-linked intellectual disability and it helped in the identification of more than 100 disease causing genes. X-linked ID accounts for 10% of male ID cases which indicates involvement of autosomal gene defects in majority of the cases [7]. Additionally, copy number variants (CNVs) have been identified in patients/families with ID [8]. Next-generation sequencing (NGS) technology has created a paradigm shift in the genetic diagnosis of common and rare diseases. Application of NGS also led to a dramatic increase in disease gene identification in familial as well as sporadic ID cases [9, 10]. More than 700 genes have been discovered so far, in which a mutation can either cause ID or ID associated disorders [9, 11,12,13,14]. The present study aims to identify the pathogenic mutations in eight Pakistani families with ID.
Material and methods
Family recruitment and DNA extraction
The study was approved by the Bio Ethics Committee (BEC) of Quaid-i-Azam University, Islamabad, Pakistan and the institutional research ethics board of Centre for Addiction and Mental Health, Toronto, Canada. Consanguineous families with two or more ID patients were ascertained from various regions of Pakistan. Medical history was taken, and pedigrees were drawn after interviewing parents of affected individuals. Peripheral blood was withdrawn after taking an informed written consent from the respective parents of the affected individuals. DNA was extracted by using standard phenol–chloroform method.
Homozygosity mapping and CNV analysis
Genome wide SNP microarray was performed on all available affected and normal individuals of the eight families (n = 41) by using Illumina’s Infinium Human CoreExome-24v1.3 kit (551,004 fixed markers, including ~ 284,000 SNPs (~ 1 marker every 6 Kb)), according to the manufacturer’s protocol and the Illumina GenomeStudio platform (Illumina CoreExome) was used for data processing. The SNP data was exported into PLINK format for analysis with HomozygosityMapper [15] to identify homozygous by descent (HBD) regions. For each HBD region, genotype tables were checked to confirm homozygosity and haploidentity. False positive data was excluded from downstream analysis. In case of X-Chromosome, HBD regions were identified by manual curation on genotype data and regions spanning more than 1 Mb were selected. CNV analysis was performed by using Illumina GenomeStudio cnvPartition plugin to identify likely pathogenic homozygous or heterozygous CNVs. For validation of CNV, qPCR was performed by using primers designed from within the CNV and its flanking region. Breakpoints were identified by designing overlapping primers from the flanking regions of CNV and the amplified DNA fragment was subjected to Sanger sequencing.
Whole exome sequencing and variant prioritization
Whole exome sequencing (WES) was performed in The Centre of Applied Genomics (TCAG), Canada sequencing facility. A single ID patient from each family was used for WES, except for families MRID126, MRID137 and MRID170, for which two affected individuals were selected per family due to possible intra-familial heterogeneity. Paired-end WES was performed on NovaSeq 6000 platform (Illumina, San Diego, CA, USA) using SureSelectXT2 Target enrichment System (Agilent Technologies, Santa Clara, CA, USA). The sequencing data alignment, variant calling and annotation were done by using a previously described pipeline [12]. Golden Helix genome browser was used to check coverage of all coding sequences within HBD regions mapped in our families. Variant prioritization was done based on allele frequency < 0.01 in public databases, such as Genome Aggregation Database (gnomAD). Non-synonymous variants and indels in exonic and splice region were selected and further filtered based on the pathogenicity prediction by various mutation prediction tools. Variants falling either in the HBD regions or in known ID causing/associated genes were given priority. Co-segregation studies were performed for potential variants in parents and phenotypically normal members of the family by Sanger sequencing.
Results
Twenty-two ID patients, belonging to eight families, were recruited from different provinces of Pakistan. Pedigree analysis suggested the possible involvement of autosomal recessive and X linked ID in five and two families, respectively (Fig. 1; Table 1). The ages of patients ranged from 8 to 23 years at the time of recruitment and they mostly exhibited moderate to severe ID with or without dysmorphic features (Table 1), except three members of MRID143 family who presented with mild ID and bilateral strabismus. Seizures were experienced by all affected members of family MRID137 and just one affected member (MRID175-2) of family MRID175. In addition, ID patients from MRID137, MRID143 and MRID170 presented with some degree of facial dysmorphism (Table 1). A female individual MRID137-6 has muscular hypotonia, but complete blood tests revealed normal creatine kinase (CK) level (below the reference range of 167 U/L) and microcystic hypochromic anemia. Three affected male individuals of MRID149 family have testicular dysgenesis but external genitals were of normal size.

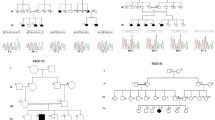
Pedigrees and haplotypes of Pakistani families (A). Segregation results of the identified variants are shown under the symbols of individuals available for this study. The arrow represents individuals selected for WES. Selected MRI images of patients MRID137-6 and MR149-4 (B)
Brain imaging (Without contrast) from a female individual MRID137-6 (Fig. 1B) and a male individual MRID149-4 (Fig. 1B) revealed the presence of mildly prominent sulci in the parietal regions. Whereas, subtle hyperintense lesions in periventricular white matter were only noted in MRID137-6 individual (Fig. 1B). In both patients, ventricles are normal in size and are in accordance with the patient’s age.
Genome wide SNP genotyping and homozygosity mapping identified 23 HBD regions in eight families (Table 2). In MRID137, analysis of genotype data of all available family members could not detect a single homozygous region over 1 Mb in size. Further analysis of the genotype data obtained from all available members of MRID137 family detected the presence of intrafamilial heterogeneity, as members were haploidentical within the respective loop but otherwise between the two loops (MRID137a and MRID137b). But reanalysis, after splitting the pedigree into two loops (MRID137a: 1,3,4,5; MRID137b: 2,6,7) identified two HBD regions each in loop (Table 2).
Later WES was applied to identify pathogenic variants in the eight ID families which led to the identification of novel and known variants in seven of the families (Table 3). The identified variants were also present in the HBD regions mapped in the respective families. However, no pathogenic variant was identified in MRID126 family despite carrying out WES of two (MRID126-3 and MRID126-4) affected individuals. CNV analysis did not identify any likely pathogenic gains or losses in this family.
WES of one affected individual from the two separated loops of MRID137 identified a recurrent missense variant in GNE (c.466T>C; p.(Tyr156His)), in MRID137a, and a homozygous missense variant, c.350G>C; p.(Gly117Ala), in CCS in MRID137b. In the case of MRID143, WES data analysis revealed a novel homozygous non-sense variant, c.8611C>T; p.(Gln2871*), in exon 42 of C5orf42, which is known to cause Joubert syndrome and Oral-facial-digital syndrome VI [16, 17].
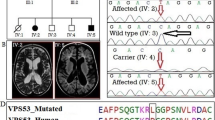
In two families (MRID149 and MRID165), deletions were identified in the genes located on the X chromosome. Two homozygous regions of 17.9 Mb and 30.8 Mb were detected on X-Chromosome in MRID149 family (Table 2). WES identified a homozygous 9 bp deletion (c.1449-1_1456delGGCTCTTTT) in ARX which is predicted to disrupt the splice acceptor site and removes first 8 bp of terminal exon, exon 5 (Fig. 2). This is predicted to result in a frameshift mutation with premature termination of protein synthesis (p.(Arg483Serfs*46)). In the second X linked family (MRID165), an approximately 0.3 Mb deletion (Fig. 3A) was identified, which was also present within the mapped HBD region of 21.5 Mb (Table 2). This genomic deletion was initially detected by cnvPartition and was then confirmed by PCR using two sets of primers located within and around the deleted region, respectively. The presence of a single band (greater than 2 kb) on agarose gel confirmed homozygous deletion in two affected male individuals (MRID165-2 and MRID165-3), but two healthy male individuals (MRID165-4 and MRID165-5) showed a single band of 1500 bp (Fig. 3B; Additional file 1: Fig. S1). However, a female affected individual (MRID165-1) and her healthy mother (MRID165-6) have two bands indicating the presence of the deletion on a single X chromosome (Fig. 3). Real time PCR also confirmed that MRID165-2 and MRID165-3 are hemizygous for the deletion while MRID165-1 is heterozygous (Fig. 3C). Sanger sequencing of deletion flanking region from an affected male individual (MRID165-2) identified a 0.29 Mb (hg19: chrX: 29,473,026–29,763,127) deletion which spans exon 6 of IL1RAPL1 gene (Fig. 3D). The deleted region is flanked by GATC repeat on both sides (Fig. 3D). IL1RAPL1 deletion is expected to remove exon 6 which encodes a 25 amino acid sequence in the extracellular domain of IL1RAPL1 protein.

Co-segregation analysis of the variants. Electropherograms of normal, carrier and affected individuals are shown

IL1RAPL1 deletion mapping performed in MRID165 family. A The cytogenetic location of deletion and deleted part of the gene is shown to highlight the strategy used for the confirmation of deletion. B Agarose gel shows the bands of products obtained with primers designed from deleted (above 2 kb) and intact (1.5 kb) regions of gene. Two affected male individuals MRID165-2 and MRID165-3 show a single band with primers designed from the deletion flanking region, but two healthy male individuals (MRID165-4, MRID165-5) show a single band of 1.5 kb with primers designed from the deleted region. Affected female individual MRID165-1 and her mother (MRID165-6) had both bands indicating heterozygous deletion on X chromosome. C Quantitative real time PCR analysis of IL1RAPL1 deleted region with multiple primers also confirmed the agarose gel findings. D Sanger sequencing results showing genomic co-ordinates (hg19) of the deleted region
In MRID170, a novel homozygous non-sense variant, c.250C>T (p.Arg84*) was identified in a known ID gene, METTL4 (Fig. 2). In MRID175 family, three potential variants c.149C>A (p.(Pro50His), c.1172T>A (p.(Val391Asp)) and c.535delC (p. Leu179Cysfs*136) were identified in ELFN1, ABCB5 and C7orf50 genes, respectively (Fig. 2). These three variants were present in the homozygous state and lie in a 22.1 Mb HBD region mapped on chromosome 7 (Table 2). The exploration of Allen brain atlas revealed the expression of ELFN1 and C7orf50 in different regions of brain. The c.149C>A change in ELFN1 resulted in the substitution of an evolutionarily conserved amino acid in a region upstream to Leucine rich repeat (LRR). The functional data available for ELFN1 suggests its role in synaptic transmission by trans regulation of mGLUR7, whose mutations are already known to cause ASD, ID, microcephaly, hypotonia and seizures [18,19,20,21].
Discussion
Next generation sequencing technology has revolutionized the process of disease gene discovery in intellectual disability. Here, we report eight families with ID and we used homozygosity mapping and whole exome sequencing to identify plausible novel disease causing mutations in ELFN1 (p.(Pro50His)), CCS (p.(Gly117Ala)), C5orf42 (p.(Gln2871*)), ARX (p.(Arg483Serfs*46)), METTL4 (p.(Arg84*)), IL1RAPL1 (intragenic 0.29 Mb deletion) in five families and a recurrent mutation in GNE (p.(Tyr156His)) in one family.
In MRID137a, a variant in GNE was co-segregating with disease phenotype (Fig. 2). GNE encodes for a bifunctional enzyme, UDP-N-acetylglucosamine 2-epimerase/N-acetyl mannosamine kinase that catalyzes a rate-limiting step in sialic acid biosynthesis. A mutation in this gene has already been reported to cause ARID in another Pakistani family [12]. The variant we identified, p.(Tyr156His), is already reported to cause Hereditary Inclusion Body Myopathy (HIBM) [22]. The clinical features indicative of HIBM, such as muscle weakness or atrophy, were not present in our family. The clinical feature of seizures in our family is suggestive of sialuria which is an autosomal dominant disorder. Harripaul et al., 2018 and our finding anticipate previously unreported autosomal recessive form of sialuria with ID as an additional clinical phenotype. Moreover our finding adds into the phenotypic variability of the p.(Tyr156His) GNE mutation.
MRID137b family has a missense variant, p.(Gly117Ala), in CCS which encodes a copper chaperon of superoxide dismutase. CCS is a 274 amino acid long cytosolic protein that is involved in delivery of copper to SOD1 via protein–protein interaction mediated by domain II, a highly homologous (SOD1 like) domain of CCS [23, 24]. A missense mutation, p.Arg163Trp, in CCS domain II along with a homozygous mutation (p.Tyr366*) in SLC33A1 gene is reported to cause severe muscular hypotonia, hypoglycemia, pericardial effusion, developmental regression, epilepsy, congenital cataracts, bilateral hearing loss, developmental delay, cerebral palsy and very low serum Cu and ceruloplasmin levels in a Turkish patient [25]. Clinical phenotype such as muscular hypotonia, seizures, anemia and speech impairment are overlapping between Turkish patient and MRID137-6. It is presumed that clinical features of congenital cataract, hearing loss and developmental regression in Turkish patient were attributed to a nonsense mutation in SLC33A1 gene [25]. The ceruloplasmin levels in our patients are normal (33.2 mg/dL Reference: 16-45 mg/dL) but reduced in the Turkish patient. The ceruloplasmin secretion is reduced in patients with SLC33A1 mutations [26], therefore we can conclude that probably CCS mutation alone has no effect on ceruloplasmin level in our patient. Immunoprecipitation studies showed that the p.Arg163Trp mutation in CCS disrupts its binding affinity to SOD1 [25]. The variant identified in MRID137b is also localized in domain II of CCS protein which may lower the binding capacity to SOD1, probably resulting in the disease phenotype in this family.
Family MRID143 and the X-linked MRID149 carried nonsense and splice site variant in C5orf42 and ARX gene respectively. C5orf42, also known as CPLANE1, encodes a cilia transition zone protein which plays an important role in ciliogenesis and mitotic progression [27, 28]. Disease causing mutations in this gene are known to cause Joubert syndrome 17 and Oral-Facial-Digital (OFD) syndrome VI [16, 17]. The presence of strabismus (Table 1) in patients of MRID143 family supports the involvement of C5orf42 gene. The protein encoded by ARX (Aristaless-related homeobox) gene plays a pivotal role in tangential migration and differentiation of GABAergic and cholinergic neurons [29, 30]. Disease causing mutations in ARX are known to causes ID with or without ASD.
The second X-Linked family, MRID165, harbored deletion in IL1RAPL1 (Interleukin 1 Receptor Accessory Protein Like 1) gene which is known to cause ID with or without co-morbid features. IL1RAPL1 regulates dendritic synapse formation and stabilization [31,32,33,34]. Intragenic deletions of IL1RAPL1 have been identified in ID patients. Deletion of exon 6 was reported in two families with moderate ID, language delay, facial dysmorphism, autistic and aggressive behavior. Δex6 functional analysis revealed that the mutant IL1RAPL1 is unstable and mislocalized within dendrites [31]. Deletion of exon 3–7 has been reported to cause autistic behavior and ID [35], deletion of exon 2–5 has been reported in ID with hyperactivity [36], deletion of exon 3–5 has been reported to cause microcephaly, dysmorphic features and ID [37, 38]. IL1RAPL1 pericentric inversions characterized in ID affected patients suggest that this region is prone to non-homologous recombination events [39,40,41]. In our family (MRID165), deletion of exon 6 and flanking intronic regions is associated with moderate ID, poor speech development and impaired cognition, but all three patients have normal head circumference. Additionally, we could not identify any difference in the phenotypic presentation of male (hemizygous for IL1RAP1 deletion) and female (heterozygous for IL1RAP1 deletion) individuals of this family (Fig. 1). Phenotypic expression in carrier XLID females has been reported in previous studies and almost 13 genes are known to give equivalent or more expression in carrier females [42]. A de novo frameshift mutation, p.Ile367Serfs*6, in IL1RAPL1 has been reported in a girl with ASD [35]. It is suspected that the functional consequence of the deletion in our family will be same as reported earlier [31]. We anticipate that ID phenotype in the female patient, heterozygous for IL1RAPL1 deletion, is probably due to X chromosome inactivation.
MRID170 carried a nonsense variant in METTL4 genes which co-segregate with ID phenotype (Fig. 2). METTL4 is a member of MT-A70 domain containing adenine methyltransferases [43]. It performs the function of N6-methyladenine (6 mA) modification in DNA which is responsible for epigenetic silencing [44, 45]. Mettl4 KO mice studies showed anatomical defects including craniofacial dysmorphism and anophthalmia in KO mice as compared to wild type controls [46]. A disease-causing nonsense variant, p.(Cys196*), in this gene has already been identified to cause ARID in a family with Pakistani descent [13]. Furthermore, mutations in other methyltransferase-like gene, METTL5 and METTL23, have also been implicated in ID [47, 48].
Family MRID175 has three rare variants including a frameshift variant in C7orf50, and missense variants in ABCB5 and ELFN1 genes. Extracellular leucine rich repeat and fibronectin Type III domain containing 1 (ELFN1) expression is evident in a subset of interneurons in cortex and hippocampus and is localized mostly to excitatory post-synaptic sites where it acts as a trans-regulator of mGluR7 [19, 49]. Efln1 knock out (KO) mice exhibits seizures and hyperactivity, and similar neurological phenotype was observed in mGluR7 deficient mice suggesting a close functional relationship between the two proteins [19, 50, 51]. Heterozygous missense variants, p.Ala481Val, p.Arg650Cys, p.Asp678Asn, p.Arg691Trp, in ELFN1 have been identified in a Japanese cohort diagnosed with epilepsy, autism spectrum disorder (ASD), and attention deficit hyperactivity disorder [19]. Affected individuals of MRID175 family are homozygous for missense variant c.149C>A (p.(Pro50His)) in ELFN1 gene and presented moderate ID, poor speech and cognition deficit. A 16-year-old female individual from MRID175 currently suffers from epilepsy, but it was absent in two younger individuals of this family. Considering the earlier reports of ELFN1 involvement in neurological disorders in heterozygous state, we can conclude that homozygous variant detected in MRID175 is a likely cause of moderate ID.
Conclusion
This study supports the genetic and phenotypic heterogeneity of Mendelian forms of ID. We identified novel variants in previously reported ID causing genes i.e. ARX, C5orf42 and METTL4, a recurrent variant in GNE which is involved in causing ID and a CNV in IL1RAPL1. We also report novel missense variants in ELFN1 and CCS to be implicated in intellectual disability which are not reported previously in ID phenotype.
Availability of data and materials
Written consent forms of the participating subjects or their legal representatives, are available upon request. The raw whole-exome sequencing data are not publicly available due to privacy or ethical restrictions. Genotype and DNA sequencing data generated or analyzed within this study are available upon an email request from the corresponding author Muhammad Ansar.
Abbreviations
- ID:
-
Intellectual disability
- SNP:
-
Single nucleotide polymorphism
- CCS:
-
Copper chaperon of superoxide dismutase
- SOD1:
-
Superoxide dismutase 1
- ELFN1:
-
Extracellular leucine rich repeat and fibronectin Type III domain containing 1
- GNE:
-
UDP-N-acetylglucosamine 2-epimerase/N-acetyl mannosamine kinase
- ABCB5:
-
ATP-Binding Cassette, Subfamily B, Member 5
- ARX:
-
Aristaless-related homeobox
- CPLANE1 :
-
Ciliogenesis and Planar Polarity Effector 1
- IL1RAPL1 :
-
Interleukin 1 Receptor Accessory Protein Like 1
- METTL4:
-
Methyltransferase-Like 4
- METTL5 :
-
Methyltransferase-Like 5
- METTL23 :
-
Methyltransferase-Like 23
- OFD:
-
Oral-Facial-Digital Syndrome
- mGluR7:
-
Metabotropic Glutamate Receptor 7
- 6 mA:
-
N6-methyladenine
- HIBM:
-
Hereditary Inclusion Body Myopathy
- ASD:
-
Autism Spectrum Disorder
- LRR:
-
Leucine rich repeat
- CNV:
-
Copy Number Variant
- NGS:
-
Next Generation Sequencing
- DNA:
-
Deoxyribonucleic acid
- HBD:
-
Homozygous by descent
- PCR:
-
Polymerase chain reaction
- WES:
-
Whole exome sequencing
- gnomAD:
-
Genome aggregation database
References
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. American Psychiatric Association; 2013.
Bhasin TK, Brocksen S, Achven RN, Van Naarden BK. Morbidity and mortality weekly report: prevalence of four developmental disabilities among children aged 8 years—Metropolitan Atlanta Developmental Disabilities Surveillance Program, 1996 and 2000. MMWR Surveill Summ. 2006;55:1–10.
Leonard H, Wen X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev. 2002;8:117–34.
Simonoff E, Pickles A, Wood N, Gringras P, Chadwick O. ADHD symptoms in children with mild intellectual disability. J Am Acad Child Adolesc Psychiatry. 2007;46(5):591–600.
Anagnostou E, Zwaigenbaum L, Szatmari P, Fombonne E, Fernandez BA, Woodbury-Smith M, et al. Autism spectrum disorder: advances in evidence-based practice. CMAJ. 2014;186:509–19.
Robertson J, Hatton C, Emerson E, Baines S. Prevalence of epilepsy among people with intellectual disabilities: a systematic review. Seizure. 2015;29:46–62.
Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–87.
Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med. 2012;367(14):1321–31.
Hu H, Kahrizi K, Musante L, Fattahi Z, Herwig R, Hosseini M, et al. Genetics of intellectual disability in consanguineous families. Mol Psychiatry. 2019;24(7):1027–39.
Grozeva D, Carss K, Spasic-Boskovic O, Tejada MI, Gecz J, Shaw M, et al. Targeted next-generation sequencing analysis of 1,000 individuals with intellectual disability. Hum Mutat. 2015;36(12):1197–204.
Vissers LELM, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Geneti. 2016;17:9–18.
Harripaul R, Vasli N, Mikhailov A, Rafiq MA, Mittal K, Windpassinger C, et al. Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry. 2018;23(4):973–84.
Riazuddin S, Hussain M, Razzaq A, Iqbal Z, Shahzad M, Polla DL, et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol Psychiatry. 2017;22(11):1604–14.
Santos-Cortez RLP, Khan V, Khan FS, Mughal ZN, Chakchouk I, Lee K, et al. Novel candidate genes and variants underlying autosomal recessive neurodevelopmental disorders with intellectual disability. Hum Genet. 2018;137(9):735–52.
Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(SUPPL. 2):W593–9.
Srour M, Schwartzentruber J, Hamdan FF, Ospina LH, Patry L, Labuda D, et al. Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am J Hum Genet. 2012;90(4):693–700.
Bonnard C, Shboul M, Tonekaboni SH, Ng AYJ, Tohari S, Ghosh K, et al. Novel mutations in the ciliopathy-associated gene CPLANE1 (C5orf42) cause OFD syndrome type VI rather than Joubert syndrome. Eur J Med Genet. 2018;61(10):585–95.
Gai X, Xie HM, Perin JC, Takahashi N, Murphy K, Wenocur AS, et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol Psychiatry. 2012;17(4):402–11.
Tomioka NH, Yasuda H, Miyamoto H, Hatayama M, Morimura N, Matsumoto Y, et al. Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nat Commun. 2014;5:4501.
Charng WL, Karaca E, Coban Akdemir Z, Gambin T, Atik MM, Gu S, et al. Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med Genomics. 2016;9(1):42–56.
Reuter MS, Tawamie H, Buchert R, Gebril OH, Froukh T, Thiel C, et al. Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiat. 2017;74(3):293–9.
Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MCV, Leoyklang P, et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. 2014;35(8):915–26.
Rothstein JD, Dykes-Hoberg M, Corson LB, Becker M, Cleveland DW, Price DL, et al. The copper chaperone CCS is abundant in neurons and astrocytes in human and rodent brain. J Neurochem. 1999;72(1):422–9.
Schmidt PJ, Rae TD, Pufahl RA, Hamma T, Strain J, O’Halloran TV, et al. Multiple protein domains contribute to the action of the copper chaperone for superoxide dismutase. J Biol Chem. 1999;274(34):23719–25.
Huppke P, Brendel C, Korenke GC, Marquardt I, Donsante A, Yi L, et al. Molecular and biochemical characterization of a unique mutation in CCS, the human copper chaperone to superoxide dismutase. Hum Mutat. 2012;33(8):1207–15.
Huppke P, Brendel C, Kalscheuer V, Korenke GC, Marquardt I, Freisinger P, et al. Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am J Hum Genet. 2012;90(1):61–8.
Hong H, Joo K, Park SM, Seo J, Kim MH, Shin EB, et al. Extraciliary roles of the ciliopathy protein JBTS17 in mitosis and neurogenesis. Ann Neurol. 2019;86(1):99–115.
Damerla RR, Cui C, Gabriel GC, Liu X, Craige B, Gibbs BC, et al. Novel Jbts17 mutant mouse model of joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum Mol Genet. 2015;24(14):3994–4005.
Colasante G, Collombat P, Raimondi V, Bonanomi D, Ferrai C, Maira M, et al. Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J Neurosci. 2008;28(42):10674–86.
Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, et al. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27(17):4786–98.
Ramos-brossier M, Montani C, Lebrun N, Gritti L, Martin C, Seminatore-nole C, et al. Novel IL1RAPL1 mutations associated with intellectual disability impair synaptogenesis. Hum Mol Genet. 2015;24(4):1106–18.
Valnegri P, Khelfaoui M, Dorseuil O, Bassani S, Lagneaux C, Gianfelice A, et al. A circadian clock in hippocampus is regulated by interaction between oligophrenin-1 and Rev-erbα. Nat Neurosci. 2011;14(10):1293–301.
Yoshida T, Yasumura M, Uemura T, Lee SJ, Ra M, Taguchi R, et al. IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphatase δ. J Neurosci. 2011;31(38):13485–99.
Pavlowsky A, Gianfelice A, Pallotto M, Zanchi A, Vara H, Khelfaoui M, et al. A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr Biol. 2010;20(2):103–15.
Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet. 2008;17(24):3965–74.
Nawara M, Klapecki J, Borg K, Jurek M, Moreno S, Tryfon J, et al. Novel mutation of IL1RAPL1 gene in a nonspecific X-linked mental retardation (MRX) family. Am J Med Genet Part A. 2008;146(24):3167–72.
Whibley AC, Plagnol V, Tarpey PS, Abidi F, Fullston T, Choma MK, et al. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am J Hum Genet. 2010;87(2):173–88.
Mignon-Ravix C, Cacciagli P, Choucair N, Popovici C, Missirian C, Milh M, et al. Intragenic rearrangements in X-linked intellectual deficiency: results of a-CGH in a series of 54 patients and identification of TRPC5 and KLHL15 as potential XLID genes. Am J Med Genet Part A. 2014;164(8):1991–7.
Laumonnier F, Ronce N, Hamel BCJ, Thomas P, Lespinasse J, Raynaud M, et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71(6):1450–5.
Leprêtre F, Delannoy V, Froguel P, Vasseur F, Montpellier C. Dissection of an inverted X(p21.3q27.1) chromosome associated with mental retardation. Cytogenet Genome Res. 2003;101(2):124–9.
Tabolacci E, Pomponi MG, Pietrobono R, Terracciano A, Chiurazzi P, Neri G. A truncating mutation in the IL1RAPL1 gene is responsible for X-linked mental retardation in the MRX21 family. Am J Med Genet. 2006;140(5):482–7.
Ziats CA, Schwartz CE, Gecz J, Shaw M, Field MJ, Stevenson RE, et al. X-linked intellectual disability: phenotypic expression in carrier females. Clin Genet. 2020;97(3):418–25.
Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m6A methyltransferase. J Mol Evol. 2002;55(4):431–44.
Le XC, Zhu S, He M, Chen D, Zhang Q, Chen Y, et al. N6-methyladenine DNA modification in the human genome. Mol Cell. 2018;71(2):306–18.
Wu TP, Wang T, Seetin MG, Lai Y, Zhu S, Lin K, et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature. 2016;532(7599):329–33.
Kweon SM, Chen Y, Moon E, Kvederaviciutė K, Klimasauskas S, Feldman DE. An adversarial DNA N6-methyladenine-sensor network preserves polycomb silencing. Mol Cell. 2019;74(6):1138-1147.e6.
Bernkopf M, Webersinke G, Tongsook C, Koyani CN, Rafiq MA, Ayaz M, et al. Disruption of the methyltransferase-like 23 gene METTL23 causes mild autosomal recessive intellectual disability. Hum Mol Genet. 2014;23(15):4015–23.
Richard EM, Polla DL, Assir MZ, Contreras M, Shahzad M, Khan AA, et al. Bi-allelic variants in METTL5 cause autosomal-recessive intellectual disability and microcephaly. Am J Hum Genet. 2019;105(4):869–78.
Dunn HA, Patil DN, Cao Y, Orlandi C, Martemyanov KA. Synaptic adhesion protein ELFN1 is a selective allosteric modulator of group III metabotropic glutamate receptors in trans. Proc Natl Acad Sci USA. 2018;115(19):5022–7.
Sansig G, Bushell TJ, Clarke VRJ, Rozov A, Burnashev N, Portet C, et al. Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J Neurosci. 2001;21(22):8734–45.
Dolan J, Mitchell KJ. Mutation of Elfn1 in mice causes seizures and hyperactivity. PLoS ONE. 2013;8(11):e80491.
Acknowledgements
We are thankful to the affected individuals and their families for their volunteer participation. We are also thankful to Higher Education Commission of Pakistan for providing International Research Support Fellowship to Memoona Rasheed and Research Grant (NRPU-7099) to Muhammad Ansar.
Funding
The research was supported by Research Grant (NRPU-7099) to Muhammad Ansar by Higher Education Commission of Pakistan and International Research Support Fellowship to Memoona Rasheed by Higher education Commission of Pakistan.
Author information
Authors and Affiliations
Contributions
MR, JBV and MA contributed to the study conception and design. Identification and collection of families, data collection and analysis (genome wide genotyping, homozygosity mapping, exome data) were performed by MR, VK, RH, MAM, ZU and MZ. MS reviewed the brain images of the patients. The first draft of the manuscript was written by MR and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
All studies were performed in accordance with the Declaration of Helsinki protocols. The studies were reviewed and approved by institutional ethics boards including Bio Ethics Committee (BEC) of Quaid-i-Azam University, Islamabad, Pakistan and the institutional research ethics board of Centre for Addiction and Mental Health, Toronto, Canada. Subjects or their legal representatives participating in this study gave written informed consent to participate in this study and consented for DNA tests including exome sequencing.
Consent for publication
Subjects or their legal representatives participating in this study provided written consent for publication of the results.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Raw agarose gel electrophoresis image showing the results of IL1RAPL1 Deletion mapping in MRID165. D1 represents the PCR product (~ 2000 bp) obtained with primers flanking the deleted region while D2 represents PCR product obtained with primers located within the deleted region. Individual showing only D1 band (IV-4, IV-5) are hemizygous for deletion and individuals with D2 band (III-1, IV-3) are homozygous for normal allele. Individuals (III-2, IV-6) with both bands are heterozygous for deletion.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Rasheed, M., Khan, V., Harripaul, R. et al. Exome sequencing identifies novel and known mutations in families with intellectual disability. BMC Med Genomics 14, 211 (2021). https://doi.org/10.1186/s12920-021-01066-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01066-y