
Abstract
Background
During the interpretation of genome sequencing data, some types of secondary findings are identified that are located in genes that do not appear to be related to the causes of the primary disease. Although these are not the primary targets for evaluation, they have a high risk for some diseases different from the primary disease. Therefore, they can be vital for preventing and intervention from such disease.
Results
Here, we analyzed secondary findings obtained from WES in 6 families with FCHL disease who had an autosomal-dominant pattern based on their pedigrees. These finding are found in CDKAL1, ITGA2, FAM111A, WNK4, PTGIS, SCN10, TBX20, DCHS1, ANK2 and ABCA1 genes.
Conclusions
Secondary findings are very important and must be considered different variants from sequencing results in a diagnostic setting. Although we have considered these variants as secondary findings, some of them may be related to the primary disease.
Similar content being viewed by others

Background
Secondary or incidental findings are described as pathogenic (P) or more likely pathogenic variants with a phenotype unrelated to the primary disease [1]. These data are mainly obtained in families that undergo whole-genome or whole-exome sequencing (WGS/WES) due to an affected family member. These findings can be used for the early diagnosis of a new disease in the family before its symptoms appear, thus preventing mortality from latent diseases and leading to increased life expectancy [2].
Familial combined hyperlipidemia (FCHL) is a common familial lipoprotein disorder associated with premature cardiovascular disease (CVD) [3]. The frequency of FCHL in the general population is 1–3%, and in patients with a previous history of myocardial infarction (MI), it is 20–38% [4]. This disease is a heterogeneous disorder caused by a defect in the metabolism of chylomicrons, a defect in lipolysis or a decrease in fatty acid incorporation by adipose tissue, overproduction of very low-density lipoprotein (VLDL) and Apo B100's overexpression in individuals with normal BMI [5]. FCHL can be accompanied by other metabolic disorders such as obesity, hypertension, non-alcoholic fatty liver disease (NAFLD), metabolic syndrome (MS), insulin resistance (IR) and type 2 diabetes mellitus (T2D) [6].
Although FCHL is a complex disease, there are families with several affected members who may follow a monogenic pattern of inheritance. Several genes might contribute to the FCHL phenotype, including the upstream transcription factor 1 gene (USF1)1, paraoxonases (PON1), protocadherin-related 15(PCDH150), Forkhead box C2(FOXC2), low-density lipoprotein receptor (LDLR), glycosylphosphatidylinositol-anchored HDL binding protein 1(GPHDLBP1) and angiopoietin-like 3 (ANGPTL3) [7, 8]. FCHL diagnosis is therefore challenging because of a multifactorial pattern of inheritance with genetic and environmental factors involvement, but in some families there is a monogenic pattern of inheritance, and in these families with a specified pedigree and several members of affected individual, WES can be used for FCHL diagnosis to detect the major pathogenic variants associated with the disease [11]. However, in addition to the major pathogenic variants, there may be other important variants unrelated to early disease and indicate a risk of other diseases in the family. Therefore, after analyzing the WES results for main variants in related genes [12, 13], we decided to evaluate the secondary findings obtained in 6 different families with an autosomal-dominant inheritance of FCHL based on their pedigrees.
Methods
Study population
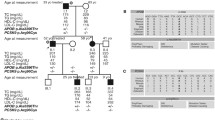
This study was performed on six families with a reoccurring and unusual constellation of juvenile-onset FCHL and early-onset coronary heart disease. All of the six families in this study had an autosomal-dominant pattern of inheritance based on their pedigrees. FCHL diagnosis was confirmed in accordance with the 2016 ESC/EAS (European Society of Cardiology/ European Atherosclerosis Society) Guidelines for dyslipidemias. The study was approved by the local university ethics committee. Besides, all participants in the study signed a consent form. Based on Fig. 1, we selected one member from each family for evaluation by WES) they are marked with an arrow in the pedigrees shown in Fig. 1. After evaluating family members and collecting clinical and laboratory data, genomic DNA was extracted from 5 ml of blood containing EDTA anticoagulant.

pedigrees of studied pedigrees from 1 to 6, Circles indicate female family members and squares male family members; slashes indicate that the family member is deceased. Family members with FCHL disease are indicated by solid symbols and those without FCHL indicated by open symbols. FCHL, familial combined hyperlipidemia
Whole-exome sequencing
Six patients were screened using sequencing all exons of protein encoding regions and some other critical genomic regions. WES has been read 100 million times using the Illumina Sequencer. In total > 95% of the target areas with acceptable sensitivity of about 99% has been investigated. WES can be used for some variants detection in genome and these variants are including gain/loss mutations, start loss or frame shift variants, in-frame insertions or deletions, variants within the intron–exon boundary, missense variants in exons and some lateral sequences in target areas. Following this, bioinformatics analysis was performed using international databases and some bioinformatics software. At this stage, various filters are used for the raw output to reduce the data to a limited number. Subsequent identifying related mutations, secondary findings were studied, and their characteristics were determined by some international databases such as OMIM, Gene Cards and MalaCards. In the next step we used some prediction software’s such as SIFT, Mutation Taster, FATHMM and DANN for investigation and prediction of identified variants on the cell's biological function.
Results
All affected family members in this study had coronary artery diseases (CHD) events before the age of 50 years. Also they had total cholesterol > 240 mg/dl, LDL > 160 mg/dl and TG > 133 mg/dl, and according to the 2016 ESC/EAS guidelines associated with management of dyslipidemias, they had the standard criteria for FCHL. The diagnosis of genetic diseases using the WES technique provides numerous data and some of these findings are important for follow-up. Our study showed some likely pathogenic/pathogenic unrelated variants to FCHL. At least one secondary finding was identified in each family, with a disease-causing effect in some prediction software which are shown in Table 1. These findings include CDKAL1 (NM_017774: exon3: c. G116A: p. R39Q, ITGA2(NM_002203: exon4: c. A352G: p. I118V), FAM111A (NM_001312911: exon4:c.C1216T: p Q406X), WNK4(NM_032387: exon1: c.178delA: p. S60fs), PTGIS (NM_000961: exon1: c. G62A: p. R21H), SCN10(NM_001293307: exon26: c.5116_5117del: p. L1706fs), TBX20(NM_001077653: exon4: c. G577A: p. G193S), DCHS1(NM_003737: exon10: c. G4330A: p. A1444T), ANK2(NM_001148: exon38:c.C6749T: p. S2250L), ABCA1(NM_005502: exon5: c.C357A: p. D119E). The interpretation of these variants is given in Table 1.
Discussion
The American College of Medical Genetics and Genomics (ACMG) has published instructions on reporting secondary findings in clinical exome and genome sequencing [14]. Accordingly, only pathogenic or, most likely, pathogenic variants are reported as secondary findings. It is estimated that 1–4% of all individuals who have had an exome or genome sequencing have a secondary finding [15, 16]. Therefore, it can be said it is a form of screening that should be analyzed using a diagnostic approach [17]. It may provide patients with an additional clinical follow-up for a special health condition [18]. However, the interpretation of these findings has always been challenging. Researchers and physicians have never reached a similar conclusion about them. On the one hand, identifying secondary findings can be a clue for patients and physicians to prevent diseases that may be in the relatively early stages. Also, genetic variants diagnosis associated with certain diseases before the phenotype is revealed can have a psychological burden on individuals [20, 21].
To date, various studies have been performed on a considerable number of samples among different populations, and the data obtained confirm the presence of a change in the number of secondary findings between different groups. For example, Jalkh et al. have identified 4262 variants in the 59 selected genes among 280 Lebanese individuals, of which 6% were pathogenic or possibly pathogenic [22]. Xue et al. have reported a frequency of 11% of secondary findings among 179 subjects [23], and in the examination of 1,000 WES belonging to Europeans/African people, they have obtained 1.2 to 3.4% of secondary findings [24]. This difference indicates that secondary findings' evaluation depends on various factors such as study design, sequencing technology, experimental group and mainly data interpretation. In this study, secondary findings related to cardiac diseases were identified in some samples, and these findings may be important, being related to the underlying disease. One of the secondary findings was a mutation associated with mitral valve prolapse in the DCHS1 (dachsous cadherin-related 1) gene. Patients with MVP2 have non-syndromic MVP with an autosomal-dominant pattern and severe variable expression [25]. Neubauer et al. have studied five young sudden unexplained death cases by focusing on 184 genes associated with heart disease by the WES. They found that there were different variants of the DCHS1 gene in one of these individuals, which are involved in mitral valve prolapse [26]. Besides, in agreement with our result, Freed et al. have examined five generations of a large family and found that at least 12 members had mitral valve prolapse without any manifestation of connective tissue abnormalities [27].
Conclusion
Secondary findings are very important and must be considered differently from sequencing results in a diagnostic setting. Secondary findings can be especially important in the autosomal-dominant state because autosomal-dominant pattern can be affected by unique features such as incomplete penetrance or variable expression, which may explain the absence of clinical manifestations during the experiment and this is controversial. Finally, in addition to the benefits of identifying secondary findings, there are some important points to consider: (1) they need approval in families, (2) some of them may be related to the primary disease, and (3) they may carry a psychological burden.
Availability of data and materials
Data are available from the corresponding author upon reasonable request.
Abbreviations
- CVD:
-
Cardiovascular disease
- FCHL:
-
Familial combined hyperlipidemia
- IR:
-
Insulin resistance
- MS:
-
Metabolic syndrome
- NAFLD:
-
Non-alcoholic fatty liver disease
- T2D:
-
Type 2 diabetes mellitus
- VLDL:
-
Very low-density lipoprotein
- WGS/WES:
-
Whole-genome or whole-exome sequencing
References
Saelaert M, Mertes H, De Baere E, Devisch I (2018) Incidental or secondary findings: an integrative and patient-inclusive approach to the current debate. Eur J Hum Genet 26(10):1424–1431
Hart MR, Biesecker BB, Blout CL, Christensen KD, Amendola LM, Bergstrom KL et al (2019) Secondary findings from clinical genomic sequencing: prevalence, patient perspectives, family history assessment, and health-care costs from a multisite study. Genet Med 21(5):1100–1110
Luijten J, van Greevenbroek MMJ, Schaper NC, Meex SJR, van der Steen C, Meijer LJ et al (2019) Incidence of cardiovascular disease in familial combined hyperlipidemia: a 15-year follow-up study. Atherosclerosis 280:1–6
Brouwers M, van Greevenbroek M, Stehouwer C, de Graaf J, Stalenhoef A (2012) The genetics of familial combined hyperlipidaemia. Nat Rev Endocrinol 8(6):352
Shah A, Wilson D (2015) Primary hypertriglyceridemia in children and adolescents. J Clin Lipidol 9(5 Suppl):S20
Skoumas I, Masoura C, Aznaouridis K, Metaxa V, Tsokanis A, Papadimitriou L et al (2013) Impact of cardiometabolic risk factors on major cardiovascular events in patients with familial combined hyperlipidemia. Circ J 77(1):163
Sentinelli F, Minicocci I, Montali A, Nanni L, Romeo S, Incani M et al (2013) Association of RXR-gamma gene variants with familial combined hyperlipidemia: genotype and haplotype analysis. J Lipids 2013:517943
Taghizadeh E, Esfehani R, Sahebkar A, Parizadeh S, Rostami D, Mirinezhad M et al (2019) Familial combined hyperlipidemia: an overview of the underlying molecular mechanisms and therapeutic strategies. IUBMB Life 71:1221–1229
Bello-Chavolla O, Kuri-García A, Ríos-Ríos M, Vargas-Vázquez A, Cortés-Arroyo J, Tapia-González G et al (2018) Familial combined hyperlipidemia: current knowledge, perspectives, and controversies. Revista de investigacion clinica organo del Hospital de Enfermedades de la Nutricion 70(5):224
Skoumas I, Ioakeimidis N, Vlachopoulos C, Chrysohoou C, Michalakeas C, Georgakopoulos C et al (2018) Statin therapy and risk of diabetes mellitus in aging patients with heterozygous familial hypercholesterolemia or familial combined hyperlipidemia: a 10-year follow-up. Angiology 69(3):242–248
Taghizadeh E, Mardani R, Rostami D, Taghizadeh H, Bazireh H, Hayat SMG (2019) Molecular mechanisms, prevalence, and molecular methods for familial combined hyperlipidemia disease: a review. J Cell Biochem 120(6):8891–8898
Taghizadeh E, Ghayour-Mobarhan M, Ferns G, Pasdar A (2020) A novel variant in LPL gene is associated with familial combined hyperlipidemia. BioFactors (Oxford, England) 46(1):94
Taghizadeh E, Mirzaei F, Jalilian N, Ghayour MM, Ferns G, Pasdar A (2020) A novel mutation in USF1 gene is associated with familial combined hyperlipidemia. IUBMB Life 72(4):616
Directors ABo (2015) ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med 17(1):68
Hart MR, Biesecker BB, Blout CL, Christensen KD, Amendola LM, Bergstrom KL et al (2019) Secondary findings from clinical genomic sequencing: Prevalence, patient perspectives, family history assessment, and healthcare costs from a multi-site study. Genet Med 21(5):1100
Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK et al (2012) Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet 91(1):97–108
Chen W, Li W, Ma Y, Zhang Y, Han B, Liu X et al (2018) Secondary findings in 421 whole exome-sequenced Chinese children. Hum Genomics 12(1):42
Liu Y-P, Ding M, Zhang X-C, Liu Y, Xuan J-F, Xing J-X et al (2019) Association between polymorphisms in the GRIN1 gene 5′ regulatory region and schizophrenia in a northern Han Chinese population and haplotype effects on protein expression in vitro. BMC Med Genet 20:26
Jang M, Lee S, Kim N, Ki C (2015) Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet Med 17(12):1007
Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH et al (2015) Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res 25(3):305
Committee OG, American COMG, Genomics S, Legal IC, Bioethics CO (2013) Ethical and policy issues in genetic testing and screening of children. Pediatrics 131(3):620
Jalkh N, Mehawej C, Chouery E (2020) Actionable exomic secondary findings in 280 Lebanese participants. Front Genet 11:208
Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M et al (2012) Deleterious-and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet 91(6):1022
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ et al (2013) Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet 93(4):631
Delling FN, Vasan RS (2014) Epidemiology and pathophysiology of mitral valve prolapse: new insights into disease progression, genetics, and molecular basis. Circulation 129(21):2158
Neubauer J, Haas C, Bartsch C, Medeiros-Domingo A, Berger W (2016) Post-mortem whole-exome sequencing (WES) with a focus on cardiac disease-associated genes in five young sudden unexplained death (SUD) cases. Int J Legal Med 130(4):1011–1021
Freed LA, Acierno JS Jr, Dai D, Leyne M, Marshall JE, Nesta F et al (2003) A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am J Hum Genet 72(6):1551
Benson DW, Sharkey A, Fatkin D, Lang P, Basson CT, McDonough B et al (1998) Reduced penetrance, variable expressivity, and genetic heterogeneity of familial atrial septal defects. Circulation 97(20):2043–2048
Acknowledgements
The grant of this study was paid by Mashhad University of Medical Sciences, Mashhad, Iran.
Funding
This study was funded by Mashhad University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
GAF and ET presented the idea, MZ, MSM and FT performed the bioinformatics analysis, ET carried out the experiment, MZ, MSM and FT wrote the manuscript, and MGM and AP supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
An informed written consent was obtained for all patients before enrollment, and the study was approved by ethical committee of Mashhad university of medical sciences (IRCT: IR.MUMS.MEDICAL.REC.1397.179). All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent for publication
An informed consent was obtained from all individual participants included in this study.
Competing interests
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zakeri, M., Safaiee, M.S., Taheri, F. et al. Secondary findings from whole-exome sequencing data in families with familial combined hyperlipidemia (FCHL). Egypt J Med Hum Genet 22, 79 (2021). https://doi.org/10.1186/s43042-021-00195-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00195-4