
Abstract
Background
Alzheimer’s disease (AD) is a progressive neurodegenerative age-related dementia that results in memory loss of elderly people. Many hypotheses have been formally articulated till now to decipher the pathogenesis of this disease. According to the compelling amyloidogenic hypothesis, β-secretase is a key regulatory enzyme in AD development and is therefore considered as one of the major targets for the development of drugs to treat AD. In this study, 40 plant-derived phytocompounds, proven to have β-secretase inhibitory activity in different laboratory experiments, were evaluated using computational approaches in order to identify the best possible β-secretase inhibitor(s).
Results
Amentoflavone (IFD score: − 7.842 Kcal/mol), Bilobetin (IFD score: − 7.417 Kcal/mol), and Ellagic acid (IFD score: − 6.923 Kcal/mol) showed highest β-secretase inhibitory activities with high binding affinity among all the selected phytocompounds and interacted with key amino acids, i.e., Asp32, Tyr71, and Asp228 in the catalytic site of β-secretase. Moreover, these three molecules exhibited promising results in different drug potential assessment experiments and displayed signs of correlation with significant pharmacological and biological activities.
Conclusion
Amentoflavone, Biolbetin, and Ellagic acid could be investigated further in developing β-secretase-dependent drug for the effective treatment of AD. However, additional in vivo and in vitro experiments might be required to strengthen the findings of this experiment.
Similar content being viewed by others
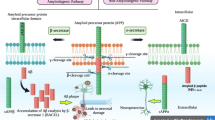


Background
Alzheimer’s disease (AD) is a perilous and progressive neurodegenerative disorder that worsens over time and ultimately culminates in dementia [1, 2]. AD manifests different types of characteristic symptoms, such as cognitive dysfunction (memory loss and language difficulties), behavioral disturbances (depression and hallucinations, also known as non-cognitive symptoms), and impairment in daily activities in affected individuals [3,4,5]. AD predominantly results from genetic heritability, age, and abnormal deposition of proteins inside as well as outside of brain cells. These proteins include amyloid β (Aβ, extracellularly) which is derived from the proteolysis of amyloid precursor protein (APP). APP is not only an integral but also a type 1 transmembrane protein that is found in various types of tissues but predominantly in neurons. It has been suggested that APP might be responsible for the formation of synapses and repairment of neurons [6]. Extracellular deposition of amyloid β proteins called amyloid plaques constitute the fundamental pathogenic driver of AD. Generated by the proteolytic activity of the β-secretase enzyme, it is the prime drug target for the inhibition of Aβ production in AD [7]. Presently, it is a matter of great concern as AD is the sixth prevalent cause of death in the USA, killing more people than other diseases such as breast and prostate cancers. Astonishingly, deaths from AD have increased by 89% since 2000 such that it is now estimated that someone develops AD in every 33 s in the USA. Much to our dismay, the number will continue to rise as high as 16 million by 2050 [8].
Amyloidogenic and non-amyloidogenic pathways and role of β-secretase in AD
β-secretase, also known as beta-site amyloid precursor protein cleavage enzyme 1 (BACE 1), is an aspartyl protease enzyme. Both APP and β-secretase mature in the endoplasmic reticulum (ER) and are later transported to the plasma membrane through the participation of the Golgi body (Fig. 1) [9, 10]. As endosomes internalize more than half of the APP, only a small amount is cleaved by α-secretase enzyme (located on the cell surface). Some of the APP are transported back to the plasma membrane, while some are degraded by lysosomes. The enzyme α-secretase competes with the β-secretase in the trans-Golgi network (TGN) for cleaving and processing APP. β-secretase is located in the TGN and endosomes [11]. It causes impairment in the trafficking of APP in the cell surface upon finding an endosomal dysfunction. The internalization of APP increases with the mediation of the β-secretase cleavage of APP [12, 13]. The cleaved portion of APP by α or β-secretase is further cleaved by γ-secretase (found in the ER), TGN, and endosomes [11].

Role of β-secretase in Alzheimer’s disease and amyloidogenic and non-amyloidogenic pathways. In non-amyloidogenic pathway, cleavage of APP sequentially by α- and γ-secretase produces sAPPα and soluble P3, respectively. On the other hand, in amyloidogenic pathway, β-and γ-secretase produces sAPPβ and insoluble Aβ protein, respectively, leading to the formation of β amyloid plaques. Matured APP and β-secretase are transported to the plasma membrane by Golgi body. A portion of APP is degraded by lysosome
Several hypotheses propose that β-secretase enzyme generates Aβ protein in neurons. The active site of the enzyme β-secretase is located in the extracellular domain and contains two key aspartate residues: Asp332 and Asp28 [11, 12].
In normal health condition (in the absence of β-secretase) and non-amyloidogenic pathway, amyloid precursor protein (APP) is cleaved sequentially. Firstly, the amino-terminal portion of the protein is cleaved by α-secretase. This secretes APP and produces carboxyterminal fragment (CTF83), which precludes the generation of Aβ protein. CTF83 is further cleaved by γ-secretase and produces P3, a soluble and amino-terminal APP intracellular domain (AICD). On the contrary, the β-secretase enzyme processes APP through another pathway named the amyloid pathway (for AD). In this pathway, β-secretase enzyme trims the APP generating secreted APP (sAPPβ) and carboxyterminal fragment (CTF99). They are further cleaved by γ-secretase and generates highly insoluble Aβ protein and AICD. These insoluble Aβ proteins lead to the formation of amyloid plaques and impair the activity of neurons. These plaques destroy synapses, thus contributing to the development of AD [13,14,15,16].
Treatment of Alzheimer’s disease
Acetylcholinesterase inhibitor drugs are the most prescribed drugs for the treatment of Alzheimer’s disease owing to their beneficial effects in cognitive, non-cognitive, and behavioral symptoms. These drugs include donepezil (reversible and selective inhibitor), rivastigmine (selective and pseudoreversible inhibitor), galantamine, tacrine (irreversible and nonselective inhibitor), metrifonate (pseudoreversible inhibitor), eptastigmine (reversible and selective inhibitor) as well as a newly approved drug, memantine, an NMDA (N-methyl-D-aspartate receptor) channel blocker [11, 17, 18]. Although these drugs are widely used to treat AD, several reports have shown that some of these drugs are not useful in mild cognitive impairment. Some of them are not effective in severe cognitive impairment either and have various side effects including nausea, vomiting, decreased appetite, and gastric acid production. In some cases, patients even show symptoms of hallucination, fatigue, dizziness, etc. Therefore, there are a few detrimental effects that result from the prolonged use of these drugs [19, 20]. Complementary medicines, such as extracts from medicinal plants, also have beneficial effects on cognitive and non-cognitive symptoms of AD. These medicines are thought to have less toxicity or side effects [19].
In this study, we evaluated the potentiality of 40 plant-derived phytocompounds to inhibit β-secretase which have already shown β-secretase inhibition activity in different laboratory experiments. We selected 3 best inhibitors from those 40 compounds and subsequently analyzed them for their drug-like potentials (Fig. 2).

Strategies employed in this study to select best β-secretase inhibitor(s)
Methods
Selection of ligand molecules
An extensive literature survey was carried out to identify phytochemicals from plants that showed β-secretase inhibition in laboratory experiments. Forty ligands were selected from literature review that displayed a β-secretase inhibitory effect to varying extents in different in vitro assays. Table 1 enlists the selected compounds from various plant sources that were used in the next phases of this study.
Molecular docking study
Protein preparation
The three-dimensional crystallographic structure of human β-secretase (PDB ID:2OHM) was downloaded in PDB format from Protein Data Bank (www.rcsb.org) [50]. The structure was then prepared and processed using the Protein Preparation Wizard in Maestro Schrödinger Suite (v11.4). Bond orders were assigned to the structures and hydrogens were added to heavy atoms. All of the water molecules were erased from the atoms, missing side chains were added to the protein backbone using Prime, and the states were generated with Epik at pH 7 ± 2 [51]. At last, the structures were refined and then minimized utilizing the Optimized Potentials for Liquid Simulations force field (OPLS_2005). Minimization was performed by setting the greatest substantial particle RMSD (root-mean-square deviation) to 30 Å, and any extraordinary water under 3H bonds to non-water was again eradicated during the minimization step.
Ligand preparation
A total of 40 selected ligand molecules were downloaded in SDF format from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). These structures were then processed and prepared using the LigPrep wizard of Maestro Schrödinger suite [52]. Minimized 3D structures of ligands were generated using Epik2.2 within pH 7.0 ± 2.0 in the suite. Minimization was again carried out using the OPLS_2005 force field which generated a maximum of 32 possible stereoisomers depending on available chiral centers on each molecule.
Receptor grid generation
Grid usually restricts the active site to a particular area of the receptor protein for the ligand to dock specifically within that area. A grid was generated using default Van der Waals radius scaling factor 1.0 and charge cutoff 0.25, which was then subjected to OPLS_2005 force field for the minimized structure in Glide [53]. A cubic box was generated around the active site (co-crystallized reference ligand) of the target molecule. Then the grid box dimension was adjusted to 14 Å × 14 Å ×14 Å for docking to be carried out.
Glide standard precision (SP) and extra precision (XP) ligand docking
Usually, extra precision (XP) ligand docking is more accurate in predicting the binding affinity of small ligand libraries than standard precision (SP) ligand docking; the latter is generally recommended for large ligand collections and sometimes may come with imperfection [54]. However, both of the docking methods were applied for the selected ligand molecules to make a comparison between two different docking scores. The Van der Waals radius scaling factor and charge cutoff was set to 0.80 and 0.15 respectively for all the ligand molecules under study. The final score was assigned according to the pose of docked ligand within the active site of the receptor molecules. The best possible poses and types of ligand–receptor interactions were then analyzed using the Discovery Studio Visualizer (v4.5) [55].
Prime MM-GBSA rescoring
After SP and XP ligand docking, the ligands were then again subjected to MM-GBSA (Molecular mechanics–generalized born and surface area) rescoring using the Prime module of Maestro Schrödinger suite for further evaluation. This technique utilizes the docked complex and uses an implicit solvent, which then assigns a more accurate scoring function and improves the overall free binding affinity score upon the reprocessing of the complex [54, 56]. It combines OPLS molecular mechanics energies (EMM), a surface generalized born solvation model for polar solvation (GSGB), and a nonpolar salvation term (GNP) for total free energy (ΔGbind) calculation. The total free energy of binding was calculated by the following equation:
Induced fit docking
Ten compounds were selected based on the MM-GBSA score for further evaluation as it is a more rigorous scoring method. After that, these ligands were subjected to induced fit docking (IFD), which is an even more accurate docking method to generate the native poses of the ligands from different sources [57]. Again, OPLS_2005 force field was applied after generating a grid around the co-crystallized ligand of the receptor. This time the ten best ligands docked rigidly. Receptor and Ligand Van Der Waals screening was set at 0.70 and 0.50, respectively. Residues within 2 Å were refined to generate the 2 best possible poses with extra precision. Three best ligands were selected based on the IFD score.
ADME/toxicity profiling
In silico testing of absorption, distribution, metabolism, excretion, and toxicity of candidate drug molecule have become an essential tool in assessing the preclinical safety that contributes to reducing the later stage failure of the investigated drug molecule [58]. The three best-selected ligand molecules were analyzed using admetSAR (http://lmmd.ecust.edu.cn/admetsar2) and pKCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) server to predict different ADME/T parameters [59, 60].
Pharmacological and biological activity prediction
Pharmacological activities of the three best ligand molecules were predicted using PASS online server (http://www.pharmaexpert.ru/passonline/) and the biological activities with GPCR (G protein-coupled receptor) ligand, ion channel, enzymes, etc., were predicted again using Molinspiration Cheminformatics server (https://www.molinspiration.com/cgi-bin/properties) [61, 62]. Prediction of Activity Spectra for Substances (PASS) estimates the pharmacological activities based on the compound’s native chemical structure. PASS predicts the function of a compound based on Structure–Activity Relationship Base (SAR Base), which assumes that the activity of a compound is related to its structure. It functions by comparing the 2D structure of an unknown compound to other well-known compounds possessing specific biological activities existing in the database with almost 95% accuracy [63].
Density functional theory (DFT) calculation
Minimized ligand structures obtained from LigPrep were used for DFT calculation using the Jaguar panel of Maestro Schrödinger Suite. Becke’s three-parameter exchange potential and Lee-Yang-Parr correlation functional (B3LYP) theory with 6-31G* basis set were used for DFT calculation [64,65,66,67]. Quantum chemical properties such as surface properties (MO, density, potential) and multipole moments were calculated along with HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) energy. Afterwards, the global frontier orbital was analyzed, and hardness (η) and softness (S) of selected molecules were calculated using the following equation as per Parr and Pearson interpretation and Koopmans theorem [68, 69].
Results
Molecular docking study
In this experiment, 40 selected ligand molecules that have been shown to have in vitro β-secretase inhibitory effect were docked against an intended target protein. Initially, 10 ligand molecules were selected based on free binding energy (Table 2). A significant correlation among the IC50 value, SP docking score, XP docking score, and free binding energy was observed for the molecules having molecular weight between 330 and 580 Da with minimal exception. However, a slight variation between SP and XP docking scores was observed among the different ligand molecules. A total of 10 ligand molecules were selected based on MM-GBSA docking scores (Table 2). Afterwards, the 10 best-selected ligands were subjected to IFD which is a powerful and accurate docking method for generating poses and assigning binding scores [70]. Finally, 3 best-performed ligands were selected from IFD study which were then analyzed further in the subsequent phases of different experiments.
Binding mode of amentoflavone, bilobetin, and ellagic acid with β-secretase
Amentoflavone, bilobetin, and ellagic acid showed superior IFD score and XP Gscore among the 10 selected ligand molecules (Table 2). Both the hydrogen bonds and hydrophobic interactions play vital roles in drug–receptor interaction by strengthening and specifying the interaction between ligand and target molecules [71]. All of the ligand molecules formed a significant number of hydrogen and hydrophobic interactions with respective amino acids inside the binding pocket of the target (Table 3 and Fig. 3).

a Three-dimensional representation of best possible poses of ligand molecules (green, stick) inside the binding pocket of β-secretase (black, ribbon). b Two-dimensional representation of ligand–receptor interaction. Interacting amino acids are represented in a three-letter code and their respective number in specific chain inside disc. Dotted lines represent type of interactions; green: conventional hydrogen bond; light green: carbon hydrogen bond. Pink: Pi–Pi stacked and Pi–Pi T-shaped
It has been observed that amentoflavone docked with β-secretase with an IFD score of − 823.501 kcal/mol and XP Gscore of − 7.842 kcal/mol interacted with 5 amino acids within the binding pocket and formed a total of 8 interactions (Table 5). It formed 4 conventional hydrogen bonds with Asp228, Phe108, Lys107, and Gly34 amino acid residues at 3.04-, 1.88-, 1.97-, and 2.34-Å distance apart respectively within the binding site of β-secretase. Moreover, amentoflavone also formed additional hydrophobic interactions, i.e., Pi–Pi stacked and Pi–Pi T shaped with Phe108 and Tyr71 amino acid residues (Fig. 3).
The bilobetin docked with β-secretase with an IFD score of − 821.327 kcal/mol and XP Gscore of − 7.417 kcal/mol and interacted with 6 amino acids within the binding pocket and formed a total of 9 interactions within the binding pocket (Table 5). It formed 3 conventional hydrogen bonds with Phe108, Lys107, and Tyr198 amino acid residues at 1.86-, 1.94-, and 2.03-Å distance apart, respectively. Moreover, amentoflavone also formed 2 non-conventional hydrogen bonds with Asp228, and Gly34 amino acid residues at 2.81- and 2.69-Å distance apart respectively, within the binding cleft. It also formed few hydrophobic interactions (i.e., Pi–Pi stacked and Pi–Pi T-shaped interactions) with Tyr71 and Phe108 amino acid residues (Fig. 3).
Ellagic acid docked with β-secretase with an IFD score of − 819.500 kcal/mol and XP Gscore of − 6.923 kcal/mol, interacted with 3 amino acids within the binding pocket and formed a total of 7 interactions within the binding pocket (Table 5). It formed 2 conventional hydrogen bonds with Thr72 amino acid residue at 1.99- and 2.01-Å distance apart respectively. Moreover, amentoflavone also formed another conventional hydrogen bond with Asp32 amino acid residue at 1.97-Å distance apart respectively within the binding cleft. It was also reported to form other hydrophobic interactions (i.e., Pi–Pi stacked and Pi–Pi T-shaped interactions with only Tyr71 amino acid residue (Fig. 3).
ADME/toxicity profiling
The three best-ligand molecules (Table 4) were subjected to analyze their absorption, distribution, metabolism, excretion, and toxicity profiles. The result of ADME/toxicity analysis is represented in Table 5. All of the ligand molecules were reported to be highly absorbed in the intestine and have low Caco-2 permeability. Only ellagic acid exhibited high oral bioavailability.
None of them reported having the ability to cross the blood–brain barrier, and no ligand showed the sign to be P-glycoprotein substrate. Only bilobetin was reported to be a P-glycoprotein inhibitor among the three selected ligand molecules. Ellagic acid was shown to be neither a substrate nor an inhibitor of any of the selected enzymes of the cytochrome P450 family. Both amentoflavone and bilobetin were reported to be the substrate of only CYP3A4. None of the selected ligands was predicted to be OCT2 (organic cation transporter 2) substrate. All of the selected ligands were predicted to induce hepatotoxicity, and only bilobetin was reported to inhibit the hERG (human Ether-a-go-go-related gene) channel. Moreover, ellagic acid was reported to cause eye irritation. Bilobetin showed type III acute oral toxicity, whereas the other two ligand molecules were reported to have type II oral toxicity.
Pharmacological and biological activity prediction
The pharmacological connection of the ligand molecules with other neurological diseases, proteins, and enzymes involved in AD was analyzed using PASS online (Table 6). PASS predicts the result in the context of probability of activity (Pa) and inactivity (Pi) of a compound. This result varies between 0.000 and 1.000. The activity is considered possible for a compound only when Pa > Pi [72]. When Pa > 0.7, the compound is very likely to exhibit the activity, but its possibility of being an analogue to a known pharmaceutical is also high. When 0.5 < Pa < 0.7, the compound is likely to exhibit the activity, but the probability along with the chance of being a known pharmaceutical agent is also lower. When Pa < 0.5, the compound is less likely to exhibit the activity [73]. Ellagic acid was reported to have better pharmacological activity with the highest probability scores. Amentoflavone and bilobetin were predicted to have similar pharmacological activity.
The three best ligands were analyzed to predict their involvement in biological activities with GPCR ligand, ion channels, enzyme, etc. The result of this experiment is summarized in Table 7. GPCRs (G protein-coupled receptors), ion channels, enzymes, nuclear receptors, etc., are the most potent drug targets in the human body. Among them, only GPCRs are the targets of almost 50% of currently available drugs [74, 75]. Amentoflavone showed better biological activities followed by bilobetin and ellagic acid. Ellagic acid had the highest positive score. Amentoflavone and bilobetin also reported showing positive scores as enzyme inhibitors. The findings of biological activity prediction indicate the important therapeutic significance of these molecules, but as useless may raise the concern of adverse effects.
Analysis of frontier orbitals
Density functional theory calculation aids to understand the energetics, structure, and properties of molecules at a lower cost. Analysis of the frontier orbitals of molecules allows us to understand the pharmacological properties of the molecule under investigation [76, 77]. The detailed HOMO (highest occupied molecular orbital) energy, LUMO (lowest unoccupied molecular orbital) energy, energy gap (HOMO–LUMO gap), hardness, and softness of the selected four compounds are summarized in Table 8. The occupation of HOMO and LUMO is illustrated in Fig. 4 for each compound. HOMO and LUMO orbitals help to understand the kinetic stability and chemical reactivity of a compound. HOMO usually occupies a small region in a molecule with a higher ability to donate one or more electrons during bond formation. LUMO represents the region capable of accepting electron(s) from another macromolecule (Fig. 4) [78]. The compound with higher HOMO–LUMO gap is considered energetically unfavorable to undergo a chemical reaction [79]. The highest gap was observed for Amentoflavone, whereas Ellagic acid was reported to have the lowest energy gap. According to the energy gap, the order of the compounds is amentoflavone > bilobetin > ellagic acid. The dipole moment of each compound was also calculated along with HOMO and LUMO energy, the scores of which are also reported in Table 8. According to the dipole moment score, the stability order of compounds is ellagic acid > amentoflavone > bilobetin.

The HOMO and LUMO occupation for the selected compounds. Blue and red are positive and negative in its wave function
Discussion
Molecular docking is one of the most commonly used computer-aided techniques in structure-based drug designing. It defines the best possible orientation of a small ligand molecule when bound to the binding site of a second target macromolecular target [80, 81]. This technique utilizes a specific scoring algorithm and assigns binding energy to the ligand molecules based on the poses that fit with the target. It reflects the binding affinity of a ligand molecule for the target. The low binding energy of a ligand–receptor complex indicates high stability of the ligand–receptor complex. It confers that the complex remains more time in contact [82]. Besides being a more rigorous scoring that guides the post-processing of docking experiment, MM-GBSA scoring predicts the most accurate free binding energy of ligand–receptor complex [83,84,85]. In this experiment, the three ligands—amentoflavone, bilobetin, and ellagic acid—were found to be the best inhibitors of β-secretase upon sequential molecular docking experiment (Tables 2 and 3) (Fig. 3).
In silico ADME/toxicity analysis allows rigorous pharmacokinetic property and toxicity testing. This analysis is required to confirm whether a drug should sustain the Phase I clinical trial or not and, in turn, assists in the in vitro assays of the candidate drug [86, 87]. Blood–brain barrier permeability is required to be confirmed for the drugs that primarily target the cells of the central nervous system (CNS). Since the oral delivery system is the most frequently used route of drug delivery and the delivered drug travels through the digestive tract into the intestine, it is expected that the drug is highly absorbed in the human intestinal tissue. P-glycoproteins are the embedded glycoproteins on the cell membrane that are responsible for facilitating the transport of many drugs through the cell membrane. As a result, their inhibition may affect the normal drug transport inside the human body. Caco-2 cell line is usually used for the assessment of drug permeability of a new candidate which, in turn, reflects the human intestinal tissue permeability [88,89,90,91,92]. Cytochrome P450 family of enzymes is responsible to control drug interaction, metabolism, and excretion inside the body. Inhibition of these enzymes may lead to acute drug toxicity, slow clearance, and eventually malfunction of the drug compound inside the human body [93,94,95]. The AMES toxicity examines the toxicity of chemicals [96, 97]. hERG (Human ether-a-go-go-related gene) channels are the voltage-gated potassium ion channels that play key roles in potassium ion transport along the cell membrane. Different structurally and functionally unrelated drugs have been reported to block the hERG potassium channel, raising the concern of off-target drug interaction. Therefore, it is imperative to screen compounds for activity on hERG channels early in the lead optimization process of a drug discovery approach to reduce the risk of a drug candidate failing in preclinical safety studies due to the blockade of hERG channels [98]. Renal OCT2 (organic cation transporter 2) is important for drug and xenobiotic excretion through the kidney. The substrates of this transporter protein are thought to be excreted easily with urine [99]. All of the selected ligand molecules were reported to have similar ADME/T properties (Table 5).
The best-selected molecules were also analyzed for respective pharmacological activity and biological activity. Ellagic acid and amentoflavone reported having better pharmacological and biological activity, respectively (Tables 6 and 7). The analysis of frontier orbitals revealed that the best-selected molecules were also stable to undergo chemical reactions (Table 8) (Fig. 4).
Finally, medicinal plants are potential sources of numerous phytocompounds of great therapeutic values, with many being considered in alleviating Alzheimer’s disease conditions [100]. In this experiment, a total of 40 plant-derived phytochemicals were analyzed in stepwise computational molecular docking approaches to identify potential inhibitors of β-secretase. Eventually, 3 ligands (i.e., amentoflavone, bilobetin, and ellagic acid) were selected as the best inhibitors. A significant correlation among the IC50 values, SP docking scores, XP docking scores, and free binding energies for the molecules having a molecular weight in a specific range was observed with minimal exception (Table 2).
Asp32 and Asp228 amino acid residues from the catalytic dyad inside the active site of β-secretase. Portray crucial contributions for the cleavage of the amyloid precursor protein. Again, Tyr71 residue within the active site responds to inhibitor binding by changing its conformation [101, 102]. In this experiment, amentoflavone formed 1 conventional hydrogen bond with Asp228 and 3 hydrophobic interactions with Tyr71 amino acid residues (Table 3 and Fig. 4). Again, bilobetin formed 1 non-conventional hydrogen bond with Asp228 and 3 hydrophobic interactions with Tyr71 amino acid residues. Moreover, ellagic acid also formed 1 conventional hydrogen bond with Asp32 and 4 hydrophobic interactions with Tyr71 amino acid residues. Hence, these compounds are expected to interfere with the normal function of β-secretase. Later, they were predicted to perform almost similar when analyzed in different post-screening studies. Impermeability of the selected compounds to the blood–brain barrier might require further modification since this characteristic is a major concern for AD drug development (Table 5) [103].
Finally, the de novo drug discovery process involves prolonged time, multiple steps and elevated cost. Computer-aided drug designing nowadays offers great fidelity of prediction and has become increasingly popular in the last few decades. It is used in conjunction with in vitro drug discovery and greatly helps in reducing time and cost of any de novo drug discovery initiatives. In this study, we screened 40 known inhibitors (phytocompounds) of β-secretase and eventually found amentoflavone, bilobetin, and ellagic acid as the best inhibitors. The compounds were found to interact with the key amino acids in the active site of β-secretase. Moreover, the best molecules proved to be adequately harmless when they were analyzed in the different steps of drug-likeness property analysis experiment. Therefore, these molecules can be investigated further to develop potent anti-AD drugs. The other molecules of this study could also be investigated since they also performed very similarly in docking experiments. However, computational exploration is largely based on the modeling of the molecules and sometimes may generate faulty outcomes [104, 105]. Therefore, further in vivo and in vitro studies might be required to strengthen the findings of this study.
Conclusion
The underlying mechanism of AD development still remains unclear. On top of that, the presence of the blood–brain barrier makes it difficult to design a drug to treat AD. As a result, not a single drug has yet been proven to treat this progressive neurological disease to the extent of any satisfactory margin. In this study, we analyzed 40 phytocompounds that showed a β-secretase inhibitory effect in laboratory studies via different computational experiments. Upon continuous exploration, we found amentoflavone, bilobetin, and ellagic acid as the most potent inhibitors of β-secretase, which could be the best possible drugs for β-secretase dependent AD treatment. These compounds also performed well in different post-screening studies unveiling the potential druggable properties. Nonetheless, amentoflavone, bilobetin, and ellagic acid could be investigated further for potential AD drug discovery. Additionally, the other selected compounds could also be analyzed further since they also performed well in different experiments of this study. This study should contribute to the development of an effective drug for AD treatment in the very near future. However, further supportive laboratory experiments and interventions might be required to support the findings of the study.
Availability of data and materials
Authors made all the data generated during experiment available in this manuscript.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADME/T:
-
Absorption, distribution, metabolism, excretion and toxicity
- AICD:
-
APP intracellular domain
- APP:
-
Amyloid precursor protein
- BACE 1:
-
Beta-site amyloid precursor protein cleavage enzyme 1
- BL3YP:
-
Becke’s three-parameter exchange potential and Lee-Yang-Parr correlation
- CTF83:
-
Carboxyterminal fragment
- DFT:
-
Density functional theory
- ER:
-
Endoplasmic reticulum
- HOMO:
-
Highest occupied molecular orbital
- IFD:
-
Induced fit docking
- LUMO:
-
Lowest unoccupied molecular orbital
- MM-GBSA:
-
Molecular mechanics–generalized born and surface area
- NMDA:
-
N-methyl-D-aspartate receptor
- OPLS:
-
Optimized potentials for liquid simulations
- PASS:
-
Prediction of activity spectra for substances
- PDB:
-
Protein Data Bank
- RMSD:
-
Root-mean-square deviation
- SAR:
-
Structure–activity relationship
- SP :
-
Standard precision
- TGN:
-
Trans-Golgi network
- XP:
-
Extra precision
References
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E (2011) Alzheimer’s disease. Lancet 377(9770):1019–1031
Alzheimer’s Association (2018) 2018 Alzheimer’s disease facts and figures. Alzheimers Dement 14(3):367–429
Mendez MF (2012) Early-onset Alzheimer’s disease: nonamnestic subtypes and type 2 AD. Arch Med Res 43(8):677–685
Wilson RS, Segawa E, Boyle PA, Anagnos SE, Hizel LP, Bennett DA (2012) The natural history of cognitive decline in Alzheimer’s disease. Psychol Aging 27(4):1008–1017
Burns A, Jacoby R, Levy R (1990) Psychiatric phenomena in Alzheimer’s disease. I: disorders of thought content. Br J Psychiatry 157(1):72–76
Du X, Wang X, Geng M (2018) Alzheimer’s disease hypothesis and related therapies. Transl Neurodegeneration 7(1):1–7
Humpel C (2011) Chronic mild cerebrovascular dysfunction as a cause for Alzheimer’s disease? Exp Gerontol 46(4):225–232
De Gage SB, Moride Y, Ducruet T, Kurth T, Verdoux H, Tournier M, Pariente A, Bégaud B (2014) Benzodiazepine use and risk of Alzheimer’s disease: case-control study. BMJ 349:g5205
Haass C, Kaether C, Thinakaran G, Sisodia S (2012) Trafficking and proteolytic processing of APP. Cold Spring Harbor Perspect Med 2(5):a006270
Vassar R et al (1999) β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286(5440):735–741
Sanabria-Castro A, Alvarado-Echeverría I, Monge-Bonilla C (2017) Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci 24(1):46–54
Nixon RA (2017) Amyloid precursor protein and endosomal–lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB J 31(7):2729–2743
Alzheimer’s Association (2017) 2017 Alzheimer’s disease facts and figures. Alzheimers Dement 13(4):325–373
Chow VW, Mattson MP, Wong PC, Gleichmann M (2010) An overview of APP processing enzymes and products. NeuroMolecular Med 12(1):1–12
Allinson TM, Parkin ET, Turner AJ, Hooper NM (2003) ADAMs family members as amyloid precursor protein α-secretases. J Neurosci Res 74(3):342–352
Games D, Adams D, Alessandrini R, Barbour R, Borthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373(6514):523
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Cummings J, Lee G, Ritter A, Zhong K (2018) Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement 4:195–214
Kısa D, Korkmaz N, Taslimi P, Tuzun B, Tekin Ş, Karadag A, Şen F (2020) Bioactivity and molecular docking studies of some nickel complexes: new analogues for the treatment of Alzheimer, glaucoma and epileptic diseases. Bioorg Chem 101:104066
Raschetti R, Albanese E, Vanacore N, Maggini M (2007) Cholinesterase inhibitors in mild cognitive impairment: a systematic review of randomised trials. PLoS Med 4(11):e338
Sasaki H, Miki K, Kinoshita K, Koyama K, Juliawaty LD, Achmad SA, Hakim EH, Kaneda M, Takahashi K (2010) β-Secretase (BACE-1) inhibitory effect of biflavonoids. Bioorg Med Chem Lett 20(15):4558–4560
Razzaghi-Asl N, Sepehri S, Ebadi A, Miri R, Shahabipour S (2015) Molecular docking and quantum mechanical studies on biflavonoid structures as BACE-1 inhibitors. Struct Chem 26(2):607–621
Zhang YM, Zhan R, Chen YG, Huang ZX (2014) Two new flavones from the twigs and leaves of Cephalotaxus lanceolata. Phytochem Lett 9:82–85
Ghosh AK, Brindisi M, Tang J (2012) Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J Neurochem 120:71–83
Das B, Mahender G, Rao YK, Prabhakar A, Jagadeesh B (2005) Biflavonoids from Cycas beddomei. Chem Pharm Bull 53(1):135–136
Matsumura S, Murata K, Yoshioka Y, Matsuda H (2016) Search for β-secretase inhibitors from natural spices. Nat Prod Commun 11(4):1934578X1601100423
Jung HA, Ali MY, Jung HJ, Jeong HO, Chung HY, Choi JS (2016) Inhibitory activities of major anthraquinones and other constituents from Cassia obtusifolia against β-secretase and cholinesterases. J Ethnopharmacol 191:152–160
Gao B, Yao CS, Zhou JY, Chen RY, Fang WS (2006) Active constituents from Aloe arborescens as BACE inhibitors. Yao Xue Xue Bao 41(10):1000–1003
Thapa A, Chi EY (2015) Biflavonoids as potential small molecule therapeutics for Alzheimer’s disease. In: Natural compounds as therapeutic agents for amyloidogenic diseases. Springer, Cham, pp 55–77
Jung HJ, Sung WS, Yeo SH, Kim HS, Lee IS, Woo ER, Lee DG (2006) Antifungal effect of amentoflavone derived from Selaginella tamariscina. Arch Pharm Res 29(9):746
Choi YH, Yon GH, Hong KS, Yoo DS, Choi CW, Park WK, Kong JY, Kim YS, Ryu SY (2008) In vitro BACE-1 inhibitory phenolic components from the seeds of Psoralea corylifolia. Planta Med 74(11):1405–1408
Park JH, Jun M (2016) Neuroprotective effect of biochanin A via the inhibition of β-secretase (BACE1). 한국식품영양과학회 학술대회발표집:484–484
Saviranta NM, Anttonen MJ, von Wright A, Karjalainen RO (2008) Red clover (Trifolium pratense L.) isoflavones: determination of concentrations by plant stage, flower colour, plant part and cultivar. J Sci Food Agric 88(1):125–132
Wang X, Kim JR, Lee SB, Kim YJ, Jung MY, Kwon HW, Ahn YJ (2014) Effects of curcuminoids identified in rhizomes of Curcuma longa on BACE-1 inhibitory and behavioral activity and lifespan of Alzheimer’s disease Drosophila models. BMC Complement Altern Med 14(1):88
Park SH, Yang EJ, Kim SI, Song KS (2014) β-Secretase (BACE1)-inhibiting C-methylrotenoids from Abronia nana suspension cultures. Bioorg Med Chem Lett 24(13):2945–2948
Marumoto S, Miyazawa M (2012) Structure–activity relationships for naturally occurring coumarins as β-secretase inhibitor. Bioorg Med Chem 20(2):784–788
Jeon SY, Bae K, Seong YH, Song KS (2003) Green tea catechins as a BACE1 (β-secretase) inhibitor. Bioorg Med Chem Lett 13(22):3905–3908
Jeon SY, Kwon SH, Seong YH, Bae K, Hur JM, Lee YY, Suh DY, Song KS (2007) β-Secretase (BACE1)-inhibiting stilbenoids from Smilax Rhizoma. Phytomedicine 14(6):403–408
Kwak HM, Jeon SY, Sohng BH, Kim JG, Lee JM, Lee KB, Jeong HH, Hur JM, Kang YH, Song KS (2005) β-Secretase (BACE1) inhibitors from pomegranate (Punica granatum) husk. Arch Pharm Res 28(12):1328–1332
Jung HA, Min BS, Yokozawa T, Lee JH, Kim YS, Choi JS (2009) Anti-Alzheimer and antioxidant activities of Coptidis Rhizoma alkaloids. Biol Pharm Bull 32(8):1433–1438
Dhananjayan K, Arunachalam S, Raj BA (2014) Targeting BACE1 (Beta secretase) through polyphenolic compounds-a computational insilico approach with emphasis on binding site analysis. J Comput Methods Mol Des 4(1):14–24
Ban JY, Jeon SY, Bae K, Song KS, Seong YH (2006) Catechin and epicatechin from Smilacis chinae rhizome protect cultured rat cortical neurons against amyloid β protein (25–35)-induced neurotoxicity through inhibition of cytosolic calcium elevation. Life Sci 79(24):2251–2259
Kwak WJ, Han CK, Son KH, Chang HW, Kang SS, Park BK, Kim HP (2002) Effects of Ginkgetin from Ginkgo biloba leaves on cyclooxygenases and in vivo skin inflammation. Planta Med 68(04):316–321
Wang YH, Du GH (2009) Ginsenoside Rg1 inhibits β-secretase activity in vitro and protects against A β-induced cytotoxicity in PC12 cells. J Asian Nat Prod Res 11(7):604–612
Karpagam V, Sathishkumar N, Sathiyamoorthy S, Rasappan P, Shila S, Kim YJ, Yang DC (2013) Identification of BACE1 inhibitors from Panax ginseng saponins—an in silico approach. Comput Biol Med 43(8):1037–1044
Park IH, Jeon SY, Lee HJ, Kim SI, Song KS (2004) A β-secretase (BACE1) inhibitor hispidin from the mycelial cultures of Phellinus linteus. Planta Med 70(02):143–146
Tian XY, Zhao Y, Yu SS, Fang WS (2010) BACE1 (beta-secretase) inhibitory phenolic acids and a novel sesquiterpenoid from Homalomena occulta. Chem Biodivers 7(4):984–992
Omar SH, Scott CJ, Hamlin AS, Obied HK (2018) Biophenols: Enzymes (β-secretase, cholinesterases, histone deacetylase and tyrosinase) inhibitors from olive (Olea europaea L.). Fitoterapia 128:118–129
Shimmyo Y, Kihara T, Akaike A, Niidome T, Sugimoto H (2008) Flavonols and flavones as BACE-1 inhibitors: structure–activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochim Biophys Acta 1780(5):819–825
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Schrödinger release 2018-4: Protein preparation wizard; Epik, Schrödinger, LLC, New York, NY, 2016; Impact, Schrödinger, LLC, New York, NY, 2016; Prime, Schrödinger, LLC, New York, NY, 2018
Schrödinger release 2018-4: LigPrep, Schrödinger, LLC, New York, NY, 2018
Schrödinger release 2018-4: Glide, Schrödinger, LLC, New York, NY, 2018
Ramírez D, Caballero J (2016) Is it reliable to use common molecular docking methods for comparing the binding affinities of enantiomer pairs for their protein target? Int J Mol Sci 17(4):525
Dassault Systèmes BIOVIA (2019) Discovery studio visualizer, 19.1. Dassault Systèmes, San Diego
Schrödinger release 2018-4: Prime, Schrödinger, LLC, New York, NY, 2018
Zhong H, Tran LM, Stang JL (2009) Induced-fit docking studies of the active and inactive states of protein tyrosine kinases. J Mol Graph Model 28(4):336–346
Tian S, Wang J, Li Y, Li D, Xu L, Hou T (2015) The application of in silico drug-likeness predictions in pharmaceutical research. Adv Drug Deliv Rev 86:2–10
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y (2012) admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties
Cheminformatics M (2014) Bratislava, Slovak Republic
Filimonov DA, Lagunin AA, Gloriozova TA, Rudik AV, Druzhilovskii DS, Pogodin PV, Poroikov VV (2014) Prediction of the biological activity spectra of organic compounds using the PASS online web resource. Chem Heterocycl Compd 50(3):444–457
Parasuraman S (2011) Prediction of activity spectra for substances. J Pharmacol Pharmacother 2(1):52
Schrödinger Release 2018-4: Jaguar, Schrödinger, LLC, New York, NY, 2018
Becke AD (1993) A new mixing of Hartree–Fock and local density-functional theories. J Chem Phys 98(2):1372–1377
Gill PM, Johnson BG, Pople JA, Frisch MJ (1992) The performance of the Becke—Lee—Yang—Parr (B—LYP) density functional theory with various basis sets. Chem Phys Lett 197(4-5):499–505
Stephens PJ, Devlin FJ, Chabalowski CFN, Frisch MJ (1994) Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem 98(45):11623–11627
Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci 83(22):8440–8441
Parr RG (1980) Density functional theory of atoms and molecules. In: Horizons of quantum chemistry. Springer, Dordrecht, pp 5–15
Tripathi A, Bankaitis VA (2017) Molecular docking: from lock and key to combination lock. J Mol Med Clin Appl 2(1)
Davis AM, Teague SJ (1999) Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew Chem Int Ed 38(6):736–749
Stepanchikova AV, Lagunin AA, Filimonov DA, Poroikov VV (2003) Prediction of biological activity spectra for substances: evaluation on the diverse sets of drug-like structures. Curr Med Chem 10(3):225–233
Lagunin A, Stepanchikova A, Filimonov D, Poroikov V (2000) PASS: prediction of activity spectra for biologically active substances. Bioinformatics 16(8):747–748
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5(12):993
Lundstrom K (2009) An overview on GPCRs and drug discovery: structure-based drug design and structural biology on GPCRs. In: G protein-coupled receptors in drug discovery. Humana Press, Totowa, pp 51–66
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103(5):1793–1874
Matysiak J (2007) Evaluation of electronic, lipophilic and membrane affinity effects on antiproliferative activity of 5-substituted-2-(2, 4-dihydroxyphenyl)-1, 3, 4-thiadiazoles against various human cancer cells. Eur J Med Chem 42(7):940–947
Zhan CG, Nichols JA, Dixon DA (2003) Ionization potential, electron affinity, electronegativity, hardness, and electron excitation energy: molecular properties from density functional theory orbital energies. J Phys Chem A 107(20):4184–4195
Becke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A 38(6):3098
Gohlke H, Hendlich M, Klebe G (2000) Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol 295(2):337–356
Gschwend DA, Good AC, Kuntz ID (1996) Molecular docking towards drug discovery. J Mol Recognit 9(2):175–186
Shoichet BK, McGovern SL, Wei B, Irwin JJ (2002) Lead discovery using molecular docking. Curr Opin Chem Biol 6(4):439–446
Sun H, Li Y, Tian S, Xu L, Hou T (2014) Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys 16(31):16719–16729
Chen F, Liu H, Sun H, Pan P, Li Y, Li D, Hou T (2016) Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Phys Chem Chem Phys 18(32):22129–22139
Greenidge PA, Kramer C, Mozziconacci JC, Wolf RM (2012) MM/GBSA binding energy prediction on the PDBbind data set: successes, failures, and directions for further improvement. J Chem Inf Model 53(1):201–209
Yu H, Adedoyin A (2003) ADME–Tox in drug discovery: integration of experimental and computational technologies. Drug Discov Today 8(18):852–861
Wang Y, Xing J, Xu Y, Zhou N, Peng J, Xiong Z, Liu X, Luo X, Luo C, Chen K, Zheng M (2015) In silico ADME/T modelling for rational drug design. Q Rev Biophys 48(4):488–515
Paul Gleeson M, Hersey A, Hannongbua S (2011) In-silico ADME models: a general assessment of their utility in drug discovery applications. Curr Top Med Chem 11(4):358–381
Li AP (2001) Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today 6(7):357–366
Geerts T, Vander Heyden Y (2011) In silico predictions of ADME-Tox properties: drug absorption. Comb Chem High Throughput Screen 14(5):339–361
Ullah A, Prottoy NI, Araf Y, Hossain S, Sarkar B, Saha A (2019) Molecular docking and pharmacological property analysis of phytochemicals from Clitoria ternatea as potent inhibitors of cell cycle checkpoint proteins in the cyclin/CDK pathway in cancer cells. Comput Mol Biosci 9(03):81
Sarkar B, Islam SS, Ullah MA, Hossain S, Prottoy MNI, Araf Y, Taniya MA (2019) Computational assessment and pharmacological property breakdown of eight patented and candidate drugs against four intended targets in Alzheimer’s disease. Adv Biosci Biotechnol 10(11):405
Hossain S, Sarkar B, Prottoy MNI, Araf Y, Taniya MA, Ullah MA (2019) Thrombolytic activity, drug likeness property and ADME/T analysis of isolated phytochemicals from ginger (Zingiber officinale) using in silico approaches. Mod Res Inflamm 8(3):29–43
Anzenbacher P, Anzenbacherova E (2001) Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci 58(5-6):737–747
Lamb DC, Waterman MR, Kelly SL, Guengerich FP (2007) Cytochromes P450 and drug discovery. Curr Opin Biotechnol 18(6):504–512
Ames BN, Gurney EG, Miller JA, Bartsch H (1972) Carcinogens as frameshift mutagens: metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. Proc Natl Acad Sci 69(11):3128–3132
Xu C, Cheng F, Chen L, Du Z, Li W, Liu G, Lee PW, Tang Y (2012) In silico prediction of chemical Ames mutagenicity. J Chem Inf Model 52(11):2840–2847
Priest B, Bell IM, Garcia M (2008) Role of hERG potassium channel assays in drug development. Channels 2(2):87–93
Hacker K, Maas R, Kornhuber J, Fromm MF, Zolk O (2015) Substrate-dependent inhibition of the human organic cation transporter OCT2: a comparison of metformin with experimental substrates. PLoS One 10(9):e0136451
Rao RV, Descamps O, John V, Bredesen DE (2012) Ayurvedic medicinal plants for Alzheimer’s disease: a review. Alzheimers Res Ther 4(3):22
Barman A, Prabhakar R (2014) Computational insights into substrate and site specificities, catalytic mechanism, and protonation states of the catalytic Asp Dyad of β-secretase. Scientifica 2014:598728
Sabbah DA, Zhong HA (2016) Modeling the protonation states of β-secretase binding pocket by molecular dynamics simulations and docking studies. J Mol Graph Model 68:206–215
Pardridge WM (2009) Alzheimer’s disease drug development and the problem of the blood-brain barrier. Alzheimers Dement 5(5):427–432
Sacan A, Ekins S, Kortagere S (2012) Applications and limitations of in silico models in drug discovery. In: Bioinformatics and drug discovery. Humana Press, Totowa, pp 87–124
Korkmaz N, Ceylan Y, Taslimi P, Karadağ A, Bülbül AS, Şen F (2020) Biogenic nano silver: synthesis, characterization, antibacterial, antibiofilms, and enzymatic activity. Adv Powder Technol 31(7):2942–2950
Acknowledgements
The authors are thankful to the members of Swift Integrity Computational Lab, Dhaka, Bangladesh for their kind support to carry out the experiments.
Funding
Authors received no funding from any external sources.
Author information
Authors and Affiliations
Contributions
MU, FJ, BS, and YA wrote the draft manuscript. MU, BS, and NA carried out the experiment. MU conceived and designed the study. YA, NA, AN, and TA edited the paper. MU, YA, BS, NA, and AN revised the paper. The authors approved the final version for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare no conflict of interest
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ullah, M., Johora, F.T., Sarkar, B. et al. Computer-assisted evaluation of plant-derived β-secretase inhibitors in Alzheimer’s disease. Egypt J Med Hum Genet 22, 26 (2021). https://doi.org/10.1186/s43042-021-00150-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-021-00150-3