
Abstract
In the present study, two mitogenomes from the Filobasidium genus were assembled and compared with other Tremellomycetes mitogenomes. The mitogenomes of F. wieringae and F. globisporum both comprised circular DNA molecules, with sizes of 27,861 bp and 71,783 bp, respectively. Comparative mitogenomic analysis revealed that the genetic contents, tRNAs, and codon usages of the two Filobasidium species differed greatly. The sizes of the two Filobasidium mitogenomes varied greatly with the introns being the main factor contributing to mitogenome expansion in F. globisporum. Positive selection was observed in several protein-coding genes (PCGs) in the Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina species, including cob, cox2, nad2, and rps3 genes. Frequent intron loss/gain events were detected to have occurred during the evolution of the Tremellomycetes mitogenomes, and the mitogenomes of 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina have undergone large-scale gene rearrangements. Phylogenetic analyses based on Bayesian inference and the maximum likelihood methods using a combined mitochondrial gene set generated identical and well-supported phylogenetic trees, wherein Filobasidium species had close relationships with Trichosporonales species. This study, which is the first report on mitogenomes from the order Filobasidiales, provides a basis for understanding the genomics, evolution, and taxonomy of this important fungal group.
Similar content being viewed by others

INTRODUCTION
Mitochondria are important organelles of fungi that play important roles in fungal growth and environmental adaptation (Ernster and Schatz 1981; McBride et al. 2006; Murphy 2009). Fungal mitochondria contain their own genomes, which are considered the “second genomes” of fungi (mitogenome). Fungal mitogenomes have several characteristics that differ from nuclear genomes, including a high copy number in one fungal cell, a smaller size relative to nuclear genomes, and uniparental inheritance in most species (Basse 2010). Most fungi contain a set of core protein coding genes, including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3 (Li et al. 2021a). Mitochondrial genes play important roles in maintenance of cell homeostasis and cell energy supplies as well as regulation of fungal growth (Chatre and Ricchetti 2014; Osiewacz 2002). In addition, variations and evolution of the mitogenome have become effective molecular markers reflecting the phylogenetic status and relationships among fungal species (Cheng et al. 2021; Li et al. 2018b, b). However, the number of basidiomycetes mitogenomes available in the National Center for Biotechnology Information (NCBI) database is currently far lower than that of animals (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/). Indeed, although there are about 1 million species of basidiomycetes, there are less than 120 basidiomycetes mitogenomes available in the database. Basidiomycetes are widely distributed worldwide and play important roles in industry, medicine, agriculture, food supply, and maintenance of forest ecosystems (Alves et al. 2013; Elisashvili 2012; Voriskova and Baldrian 2013). Accordingly, analysis of basidiomycete mitogenomes will help us understand their origin and evolution and lay a foundation for better utilization of basidiomycetes.
Tremellomycetes, belonging to Agaricomycotina, is a class of fungi with multiple morphological characteristics (Hibbett 2006; Liu et al. 2015a). To date, hundreds of Tremellomycetes species have been described, including yeasts, dimorphic taxa, and species that form hyphae and/or complex basidiocarps (Millanes et al. 2011; Yurkov and Kurtzman 2019). Classification of Tremellomycetes based on phenotype usually leads to confusion or inaccuracy; however, the introduction of molecular markers has improved our understanding of the phylogenetic relationships among Tremellomycetes (Liu et al. 2015b; Wang and Wang 2015). The genus Filobasidium is an important group of Tremellomycetes found in plant leaves, soil and water (Li et al. 2020a; Nemcova et al. 2015). Some species of Filobasidium have been found to be opportunistic pathogens, some have excellent environmental tolerance, and some are antimicrobial (Luo et al. 2019; Pan et al. 2012; Singh et al. 2013). Because of their diverse physiological characteristics and lifestyles, members of the genus Filobasidium comprise one of the most important groups for analysis of the evolution of Tremellomycetes species. Nevertheless, no mitogenomes from the genus Filobasidium or even the order Filobasidiales have been published to date.
In the present study, the mitogenomes of two Filobasidium species, Filobasidium wieringae, and Filobasidium globisporum, were assembled and compared. The aim of this study was to (1) reveal the genetic characteristics of Filobasidium mitogenomes; (2) characterize the evolutionary dynamics of introns in Tremellomycetes mitogenomes; and (3) determine the phylogenetic placement of Filobasidium in the phylum Basidiomycota based on the combined mitochondrial gene dataset (15 core PCGs). This is the first report of Filobasidium mitogenomes. The availability of these mitogenomes will improve our understanding of the genetics, evolution, and taxonomy of Filobasidium species and other closely related fungal species.
MATERIALS AND METHODS
Mitogenome assembly and annotation
The raw sequencing data used for F. wieringae and F. globisporum mitogenomes assembly were downloaded from the Sequence Read Archive database (accession numbers SRR4171232 and DRR032577, respectively). Quality control steps conducted to remove unqualified sequences from the raw sequencing data consisted of filtering low-quality sequences using ngsShoRT 2.2 (Chen et al. 2014) and removing adapter reads using AdapterRemoval v2 (Schubert et al. 2016). The clean reads were then applied to assemble the mitogenomes of Filobasidium species using NOVOPlasty v4.2.1 with a K-mer size of 29 (Dierckxsens et al. 2017). The circularized assembled mitogenomes of the two Filobasidium species were annotated as previously described (Li et al. 2018a). Briefly, the open reading frames (ORFs), protein-coding genes (PCGs), rRNAs, tRNAs, and introns in the two Filobasidium mitogenomes were initially annotated using M Fannot (Valach et al. 2014) and MITOS (Bernt et al. 2013), which are both based on the mitochondrial genetic code 4. ORFs (> 100 aa) were further modified based on the NCBI Open Reading Frame Finder (Coordinators 2017) and annotated by BLASTP searches against the NCBI non-redundant protein sequence database (Bleasby and Wootton 1990). We determined the intron and exon boundaries in the ORFs using exonerate v2.2 (Slater and Birney 2005). tRNA genes in the two mitogenomes were predicted using the tRNA scan-SE v1.3.1 software (Lowe and Chan 2016). Physical maps of the two Filobasidium mitogenomes were generated using OGDraw v1.2 (Lohse et al. 2013). In the maps, the inner grayscale bar graph shows the GC content of the mitochondrial sequences and the circles inside the GC content graph mark the 50% threshold.
Sequence analysis
Base compositions of the two Filobasidium mitogenomes and other mitogenomes were analyzed using DNASTAR Lasergene v7.1 (http://www.dnastar.com/). Strand asymmetries of Tremellomycetes mitogenomes were calculated based on the following formulas: AT skew = [A − T]/[A + T], and GC skew = [G − C]/[G + C]. Codon usages of the two Filobasidium mitogenomes were analyzed using Sequence Manipulation Suite (Stothard 2000). The nonsynonymous (Ka) and synonymous (Ks) substitution rates for core PCGs in the 17 mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina were calculated using DnaSP v6.10.01 (Rozas et al. 2017). The genetic distances between each pair of the 15 core PCGs (atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3) in the 17 mitogenomes were analyzed using MEGA v6.06 based on the Kimura-2-parameter (K2P) substitution model (Caspermeyer 2016). To determine if there were intra-genomic duplications of large fragments or interspersed repeats in the two Filobasidium mitogenomes, we conducted BlastN searches (e-value < 10−10) of the two Filobasidium mitogenomes against themselves (Chen et al. 2015). Tandem repeats (> 10 bp in length) in the two Filobasidium mitogenomes were detected using the Tandem Repeats Finder (Benson 1999) with the default parameters.
Comparative mitogenomic analysis and intron analysis
Genome sizes, GC contents, base compositions, and gene and intron numbers of the 17 mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina were compared to assess variations and conservations among different mitogenomes. We further calculated the contribution rate of different regions to the size variations of the two Filobasidium mitogenomes using the following formula: size difference of region/size difference of the entire mitogenome *100%. We classified introns of cox1 genes in the 17 mitogenomes into different position classes (Pcls) according to the method described by Férandon et al. (Ferandon et al. 2010). We first aligned the cox1 genes of the 17 mitogenomes with the cox1 gene of Ganoderma calidophilum (Li et al. 2019d), which was used as a reference in previous studies (Ye et al. 2020) using Clustal W (Thompson et al. 1994). Introns inserted at the same position of the cox1 reference gene belonging to the same Pcl, which were named according to the insert sites (nt) in the corresponding reference gene. When the same Pcls are present, they are considered orthologous introns and usually have high sequence similarity (Ferandon et al. 2010).
Phylogenetic analysis
A phylogenetic tree of 79 Basidiomycota species was constructed using the combined mitochondrial gene set (15 core PCGs) to investigate the phylogenetic status of Filobasidium species in the phylum Basidiomycota. The phylogenetic tree was constructed as previously described (Cheng et al. 2021; Li et al. 2020b). Annulohypoxylon stygium from the phylum Ascomycota was used as the outgroup (Deng et al. 2018). We first aligned individual mitochondrial genes using MAFFT v7.037 (Katoh et al. 2019), after which we concatenated these aligned mitochondrial sequences into a combined mitochondrial gene set using the Sequence Matrix v1.7.8 (Vaidya et al. 2011). A preliminary partition homogeneity test was conducted to detect potential phylogenetic conflicts between different mitochondrial genes. Partition Finder 2.1.1 (Lanfear et al. 2017) was used to detect best-fit models of partitioning schemes and evolution for the combined mitochondrial gene set. Phylogenetic trees were constructed using both the Bayesian inference (BI) and maximum likelihood (ML) methods. We conducted BI analysis using MrBayes v3.2.6 (Ronquist et al. 2012). Two independent runs with four chains (three heated and one cold) each were conducted simultaneously for 2 × 106 generations. Each run was sampled every 100 generations. We assumed that stationarity had been reached when the estimated sample size was greater than 100 and the potential scale reduction factor approached 1.0. The first 25% of samples were discarded as burn-in, and the remaining trees were used to calculate Bayesian posterior probabilities (BPP) in a 50% majority-rule consensus tree (Wu et al. 2021). RAxML v 8.0.0 (Stamatakis 2014) was used to conduct the ML analysis. Bootstrap values (BS) were assessed through an ultrafast bootstrap approach with 1000 replicates.
Data availability
The complete mitogenomes of F. wieringae and F. globisporum were deposited in the GenBank database (Benson et al. 2018) under accession numbers MW039344 and MW039345, respectively.
RESULTS
Characterization and PCGs of Filobasidium mitogenomes
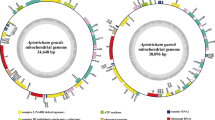
The structures of the two Filobasidium mitogenomes were circular, with total lengths of 27,861 bp and 71,783 bp for F. wieringae and F. globisporum, respectively (Fig. 1). The complete mitogenomes of F. wieringae and F. globisporum had GC contents of 38.51% and 40.32%, respectively (Additional file 1: Table S1). The mitogenome of F. wieringae had a negative AT skew and positive GC skew while the mitogenome of F. globisporum contained positive AT and negative GC skews. A total of 15 and 48 PCGs were detected in the mitogenomes of F. wieringae and F. globisporum, respectively. Both the mitogenomes contained a set of core PCGs shared by basidiomycete mitogenomes, including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and rps3 (Tables S2 and S3). In addition, the mitogenome of F. globisporum contained four non-conserved PCGs encoding proteins with unknown functions and three non-conserved PCGs encoding GIY endonucleases. The F. globisporum mitogenome contained 37 introns distributed in the atp9, cob, cox1, cox2, cox3, nad1, nad4, nad5, rns, and rnl genes, of which 27 belonged to group I, two belonged to group II, and eight were of unknown types. A total of 26 intronic ORFs were detected in these introns, encoding LAGLIDADG homing endonucleases, GIY-YIG homing endonucleases, and proteins with unknown functions. Seven introns were detected in the mitogenome of F. wieringae, six of which belonged to group I. No intronic ORFs were detected in introns of F. wieringae.

Circular maps of the mitochondrial genomes of two Filobasidium species. Genes are represented by different colored blocks. Colored blocks outside each ring indicate that the genes are on the direct strand, while colored blocks within the ring indicates that the genes are located on the reverse strand. The inner grayscale bar graph shows the GC content of the mitochondrial sequences. The circle inside the GC content graph marks the 50% threshold
rRNA and tRNA genes in the Filobasidium mitogenomes
Both Filobasidium mitogenomes contained two rRNA genes, namely the small subunit ribosomal RNA (rns) and the large subunit ribosomal RNA (rnl) (Additional file 1: Table S2). The rnl gene of F. globisporum was 36 bp longer than that of F. wieringae. The two Filobasidium mitogenomes contained identical length rns genes. The mitogenomes of F. wieringae and F. globisporum contained 23 and 22 tRNA genes, respectively, which were all folded into classical cloverleaf structures (Additional file 2: Fig. S1). The two mitogenomes contained two tRNAs with different anticodons coding for serine and leucine. The mitogenome of F. wieringae also contained 2 tRNAs with different anticodons encoding Arginine. The size of individual tRNAs ranged from 71 to 86 bp, mainly due to size variations of the extra arms. Of the 22 tRNA genes shared by the two Filobasidium mitogenomes, 17 contained sites that varied between the two mitogenomes. A total of 159 variable sites were detected in the 22 tRNA genes between the two Filobasidium mitogenomes. The most common variable site was located on the extra arm (36 sites varied between the two mitogenomes), followed by the D arm.
Overlapping nucleotides and composition of mitogenomes
Two overlapping nucleotides were detected in the mitogenome of F. wieringae across the neighboring genes nad2 and nad3 (− 1 bp), as well as between nad4L and nad5 (− 1 bp) (Additional file 1: Table S2). We detected three sets of overlapping nucleotides in the mitogenome of F. globisporum, with the largest set located between cox3 and orf201 (− 19 bp). A total of 6870 bp and 10,849 bp of intergenic sequences were detected in the mitogenomes of F. wieringae and F. globisporum, respectively. The length of these intergenic sequences ranged from 16 to 1291 bp. The longest intergenic sequence was located between nad5 and trns in the F. globisporum mitogenome.
The protein coding regions accounted for the largest proportion of the F. wieringae mitogenome (48.40%) (Fig. 2), while the intronic regions accounted for the largest proportion of the F. globisporum mitogenome, occupying 51.96%. Intergenic regions occupied 15.11–24.66% of the two mitogenomes, while ribonuclease P RNA coding regions accounted for the smallest proportion of the two mitogenomes (0.26%–0.69%). The F. wieringae mitogenome was 43,922 bp smaller than that of F. globisporum. Intronic regions made the greatest contribution to the size expansion of the F. globisporum mitogenome, with a contributing rate of 81.48%. Protein coding regions accounted for 9.56% of the size variation, while intergenic regions contributed 9.06% of the F. globisporum mitogenome expansion.

Mitogenome composition of the entire mitochondrial genomes of the two Filobasidium species. The bottom panel shows the contribution of different gene regions to the expansion of the F. globisporum mitogenome. The y-axis represents the contribution rate of different regions to the size variation of the whole mitogenome, which is calculated by the following formula: size difference of region / size different of the entire mitogenome *100%
Codon usage analysis
Most of the core PCGs in Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina mitogenomes used ATG as the start codon, except for the cox1 gene of Ustilago bromivora and the cox1 and nad2 genes of Ustilago maydis, which used GTG, and the cox2 and rps3 genes of Jaminaea angkorensis, which used TTG (Additional file 1: Table S4). TAA was the most widely used stop codon in the core PCGs of the 17 mitogenomes tested, followed by TAG. We found that the start and stop codons varied greatly between Tremellomycetes species, even those that were closely related. The atp9 gene of F. wieringae used TAG as the stop codon, while that of F. globisporum used TAA as the stop codon. The cob, cox2, nad4, and nad5 genes of F. globisporum used TAG as stop codons, while those of F. wieringae used TAA as stop codons. Within Ustilaginomycotina, the nad2 gene of Ustilago maydis used GTG as the start codon, while Ustilago bromivora used ATG as the start codon.
Codon usage analysis indicated that the most frequently used codons in the two Filobasidium mitogenomes were CAA (for glutamine; Gln), GAA (for glutamic acid; Glu), TGT (for cysteine; Cys), AAA (for lysine; Lys), GAT (for aspartic acid; Asp), and CAT (for histidine; His) (Fig. 3). The frequent use of A and T in codons contributed to a relatively high AT content in the two Filobasidium mitogenomes (average: 60.59%).

Codon usage in the mitochondrial genomes of two Filobasidium species. Frequency of codon usage is plotted on the y-axis. a, F. wieringae; b, F. globisporum
Repetitive sequences analysis
We conducted BLASTn searches of the two Filobasidium mitogenomes against themselves and identified 0 and 16 repeat sequences in the mitogenomes of F. wieringae and F. globisporum, respectively (Additional file 1: Table S5). The length of repeat sequences in the F. globisporum mitogenome ranged from 44 to 695 bp, with pair-wise nucleotide similarities ranging from 71.66 to 95.46%. The largest repeats were located in the fourth intron and fifth exons of the nad5 gene in the F. globisporum mitogenome. The second largest repeats were located in the intergenic region between trnR and orf111, as well as in the exon and intron regions of the nad4 gene in the F. globisporum mitogenome, with a repeating sequence 165 bp long. Repeat sequences accounted for 3.74% of the F. globisporum mitogenome. Both Filobasidium mitogenomes contained two tandem repeats (Additional file 1: Table S6). The longest tandem repeat sequence (66 bp) was detected in the intergenic region between rnl and trnP in the mitogenome of F. globisporum. Tandem repeat sequences accounted for 0.35% and 0.14% of the F. wieringae and F. globisporum mitogenomes, respectively.
Genetic distance and evolutionary rates of core genes
Among the 15 detected core PCGs, the rps3 gene had the largest average Kimura-2-parameter (K2P) genetic distance between the 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, followed by the nad3 and nad6 genes, demonstrating that these genes had differentiated greatly during evolution (Fig. 4). The nad4L gene exhibited the smallest K2P genetic distance between the 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, indicating that this gene was highly conserved. The rps3 gene exhibited the largest non-synonymous substitutions rate (Ka) among the 15 detected core PCGs, while nad4L had the smallest Ka value. The synonymous substitution rate (Ks) of the nad1 gene was largest, while that of the cox2 gene was smallest among the 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina. The Ka/Ks values for most of the 15 core PCGs were < 1, indicating that these genes were subjected to purifying selection pressure. However, the average Ka/Ks values of the cob, cox2, nad2, and rps3 genes were > 1, indicating these genes might have been subjected to positive selection.

Genetic analysis of 15 protein coding genes conserved in 17mitogenomesfrom Agaricomycotina, Pucciniomycotina and Ustilaginomycotina. K2P, the Kimura-2-parameter distance; Ka, the number of nonsynonymous substitutions per nonsynonymous site; Ks, the number of synonymous substitutions per synonymous site
Intron dynamics of cox1 genes
A total of 197 introns were detected in the in the rns, rnl, atp6, atp9, cob, cox1, cox2, cox3, nad1, nad3, and nad5 genes of the 17 mitogenomes from Agaricomycotina, Pucciniomycotina and Ustilaginomycotina (Additional file 1: Table S1). The cox1 gene was the largest host gene of mitochondrial introns, harboring 78 introns accounting for 39.59% of the total introns in mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina. Therefore, the intron dynamics in the cox1 gene could significantly influence mitogenome size and organization. Introns in the cox1 genes of the 17 mitogenomes were classified into different position classes (Pcls) using the cox1 gene of the medical fungus Ganoderma calidophilum (Li et al. 2019d) as a reference. The same Pcl genes from different species were considered to be orthologous introns. The 78 introns in cox1 genes of the 17 mitogenomes were classified into 25 Pcls (Fig. 5). The class and number of introns in different species varied greatly, indicating potential intron loss/gain events. Pcls present in more than one-fifth of species were considered to be common introns, while others were considered rare introns. In the present study, nine common Pcls and 16 rare Pcls were detected in the 17 mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina. The most widely distributed intron was P706, which was detected in eight of the 17 species. Intron P383 was the second most common intron, being distributed in seven of the 17 mitogenomes. Several rare Pcls (P166, P170, P237, P311, P900, P1030, P1057, P1117, P1281, and P1287) were only detected in one of the 17 species. However, some rare introns in Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, including P237, P900, P1030, and P1057, were detected in distantly related species, such as Hygrophorus russula (Li et al. 2019c), Pleurotus citrinopileatus (Li et al. 2018a), Armillaria solidipes (Kolesnikova et al. 2019), and Rhizopogon salebrosus (Li et al. 2019a) from Agaricomycetes, indicating possible gene transfer events. P166, P170, P311, P1117, P1281, and P1287 were only detected in Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina species, and no homologous introns were found from other Basidiomycota species. The cox1 gene of F. globisporum contained 10 Pcls, while no introns were identified in the cox1 gene of F. wieringae. These results indicated that the ancestors of Filobasidium species lost or gained introns on a large-scale during evolution.

Position class (Pcl) information of cox1 genes. Introns in cox1 genes of 17 mitogenomes from Agaricomycotina, Pucciniomycotina and Ustilaginomycotina were classified into different position classes (Pcls) using the cox1 gene of Ganoderma calidophilum as the reference. Each Pcl was constituted by introns inserted at the same position of corresponding cox1 gene and named according to its insertion site in the aligned corresponding reference sequence (nt). The Pcls present in more than 1/5 of the 17 mitogenomes were considered as common Pcls, while introns detected in less than 1/5 of species were considered to be rare introns. Phylogenetic positions of the 17 species were established using the Bayesian inference (BI) method and Maximum Likelihood (ML) method based on concatenated mitochondrial genes. Species information is shown in Additional file 1: Table S7
Gene arrangement and comparative mitogenomic analysis
In the present study, we analyzed mitochondrial gene arrangements, including 15 core PCGs and two rRNA genes, of the 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina (Fig. 6). The results showed that the mitochondrial gene arrangements varied greatly between species. Large-scale gene rearrangements were detected between species from different genera, including gene relocations, and position exchanges. We also observed several gene rearrangements between species from the same genera, such as Cryptococcus neoformans and Cryptococcus amylolentus, Microbotryum cf. violaceum and Microbotryum lychnidisdioicae, and Ustilago bromivora and Ustilago maydis. The same gene arrangements were only observed between Cryptococcus gattii and Cryptococcus neoformans, as well as between Tilletia indica and Tilletia walker, which had a close phylogenetic relationship. Large-scale gene rearrangements have also occurred in the two Filobasidium mitogenomes, and 13 of the 17 mitochondrial genes have undergone positional changes.

Mitochondrial gene arrangement analyses of 17 mitochondrial genomes from Agaricomycotina, Pucciniomycotina and Ustilaginomycotina. The same gene were represented by same color blocks. Phylogenetic positions of the 17 species were established using the Bayesian inference (BI) method and Maximum Likelihood (ML) method based on concatenated mitochondrial genes. Species information is shown in Additional file 1: Table S7
The sizes of 17 mitogenomes tested varied greatly, ranging from 24,874 to 177,540 bp, with an average size of 46,800 bp (Additional file 1: Table S1). The mitogenome of F. globisporum (71,783 bp) was the fourth largest among the 17 mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, which was only smaller than that of Ustilagobromivora (177,540 bp, acc. LT558140 in the NCBI database) from the order Ustilaginales, Microbotryum cf. violaceum (92,107 bp), and Microbotryum lychnidisdioicae (107,808 bp) from the order Microbotryales. The GC content of the 17 mitogenomes ranged from 28.79 to 40.43%, with an average of 34.15%. The GC content of the two Filobasidium species was much higher than the average value. Eight and five of the 17 mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina had positive AT skews and GC skews, respectively. Each mitogenome contained 15–51 PCGs, and the mitogenome of Microbotryum lychnidisdioicae contained the most PCGs. The mitogenome of F. globisporum contained the greatest number of introns (37) and intronic ORFs (26) among the 17 mitogenomes detected. All 17 mitogenomes contained two rRNA genes. In addition, 20–31 tRNA genes were detected in the17 species from Agaricomycotina, Pucciniomycotina and Ustilaginomycotina.
Phylogenetic analysis
We obtained an identical and well-supported phylogenetic tree based on mitochondrial gene sets using both BI and ML methods (15 core PCGs) (Fig. 7). All major clades within the phylogenetic tree were well supported (BPP ≥ 0.96; BS ≥ 98). According to the phylogenetic tree, the 79 Basidiomycota species could be divided into 16 major clades corresponding to the orders Agaricales, Boletales, Cantharellales, Filobasidiales, Gomphales, Hymenochaetales, Microbotryales, Microstromatales, Polyporales, Pucciniales, Russulales, Sporidiobolales, Tilletiales, Tremellales, Trichosporonales, and Ustilaginales (Additional file 1: Table S7). Phylogenetic analysis indicated that the two Filobasidium species branched basally to the other two Tremellomycete orders (Trichosporonales and Tremellales) (Liu et al. 2015a, b). Phylogenetic analysis based on the mitochondrial gene set also showed that the mitogenome was an effective molecular marker for analysis of the phylogenetic relationship of basidiomycetes.

Molecular phylogeny of 79Basidiomycota species based on Bayesian inference (BI) and Maximum likelihood (ML) analysis of 15 protein coding genes. Support values are Bayesian posterior probabilities (before slash) and bootstrap (BS) values (after slash).The asterisk on the evolutionary clades indicates that the BPP value is 1 and the BS value is 100.Species and NCBI accession numbers for mitogenomes used in the phylogenetic analysis are provided in Additional file 1: Table S7
DISCUSSION
The mitogenome was obtained from endosymbiotic bacteria by the ancestors of eukaryotes (Gray et al. 2001). During the long-term evolution and differentiation of eukaryotes, the mitogenomes of most eukaryotes contracted. Many ancient mitochondrial genes have been transferred into the nuclear genome, which is considered to have several advantages (Adams and Palmer 2003; Adams et al. 2002). However, a small number of mitochondrial genes have been retained, including a set of core PCGs for energy metabolism, two rRNA genes, and 5–35 tRNA genes (Allen 2015; Wang et al. 2020a). These retained genes play important roles in regulation of cell homeostasis and mitochondrial function (Allen 2015; Bjorkholm et al. 2015). In the present study, we found that the core PCGs of the two Filobasidium species varied in length and base composition. In addition, possible positive selections on cob, cox2, nad2, and rps3 genes were detected between some species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina. Core PCGs, including atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, and nad6, are used for energy metabolism, and the rps3 gene is likely involved in assembly of the mitochondrial small (37S) ribosomal subunit (Seif et al. 2005). The effects of size and length variations of these genes on fungal phenotypes need to be further verified. Tremellomycetes species have diverse lifestyles and morphological characteristics. Some species are parasitic, some are saprophytic, some are symbiotic, some can form basidiocarps, and some are yeast like (Millanes et al. 2011; Yurkov and Kurtzman 2019). This diversity may result in positive selection pressure on core PCGs of Tremellomycetes mitogenomes. In addition, the lengths, and base compositions of rRNA genes and tRNA genes in two Filobasidium species also varied in this study. Previous studies have shown that the base mutation of mitochondrial tRNA can affect protein synthesis (Ding et al. 2019; Lin et al. 2019); however, the effects of rRNA and tRNA variations on the growth, development and physiological activities of Filobasidium species need to be further analyzed.
In the present study, large mitogenome size variations were detected between two Filobasidium species. Specifically, the mitogenome of F. globisporum was 2.58 times greater than that of F. wieringae. The intronic region was considered to be the most important factor leading to size expansion of the F. globisporum mitogenome, with a contribution rate of 81.48%. These results were consistent with those of previous studies, suggesting that introns played an important role in the size variations of fungal mitogenomes (Li et al. 2020c; Ye et al. 2020). Introns are considered mobile genetic elements in the fungal mitogenome, and changes in their dynamics have a significant impact on the size and organization of the fungal mitogenome (Hamari et al. 2002; Repar and Warnecke 2017; Sandor et al. 2018). In the present study, intron classes and numbers varied greatly between the 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, indicating that frequent intron loss/gain events occurred during evolution of the species. Some introns were observed to be widely distributed in species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, including P209, P273, P383, P612, P706, P867, P1107, and P1125. Interestingly, P273 was considered a rare intron in Basidiomycete species, indicating that the different intron classes were unevenly distributed in basidiomycetes (Ye et al. 2020). Some introns were detected in only one of the 17 species, while homologous introns were detected in distant species from Agaricomycetes, indicating potential gene transfer events. In addition, some rare introns were detected only in Tremellomycetes, and no homologous introns were detected in other basidiomycete species. Further studies are needed to reveal the origin and evolution of these rare introns in Tremellomycetes species to clarify the functions of mobile genetic elements in mitochondria.
The arrangement of mitochondrial genes can be used as an important reference to reflect the phylogenetic status and genetic relationship of species (Li et al. 2018a, b; Wang et al. 2020b). In the present study, we found that the mitochondrial gene arrangement varied greatly in Tremellomycetes, and that species from different families had different gene arrangements. In addition, large-scale gene rearrangements were observed between species from the same genera in Tremellomycetes, indicating that the orders of mitochondrial genes in Tremellomycetes species were highly variable in evolution. The rearrangement of fungal mitochondrial genes has been less studied than that of animal mitochondrial genes. Several models have been proposed to reveal mitochondrial gene rearrangement in animals (Lavrov et al. 2002; Xia et al. 2016); however, the mechanism of mitochondrial gene rearrangement in fungi has not been revealed. Higher repeat sequences than animal mitogenomes may be one of the reasons for frequent rearrangements of fungal mitogenomes (Aguileta et al. 2014).
Basidiomycetes are a diverse group that are distributed worldwide. Basidiomycetes play an important role in industry, medicine, agriculture, and ecological maintenance (Alves et al. 2013; Elisashvili 2012; Voriskova and Baldrian 2013). Accurate classification and identification of basidiomycetes will contribute to their efficient utilization (Hibbett et al. 2007; James et al. 2006). However, some basidiomycetes have limited and overlapping morphological characteristics, which makes them difficult to classify and identify based only on morphology. To date, the nuclear genome and molecular markers have been used for classification of basidiomycetes (Hibbett 2006; James et al. 2006; Spatafora et al. 2016). However, the mitogenome of basidiomycetes is easier to obtain than the nuclear genome and contains more genetic information than individual molecular markers. These advantages make the mitogenome a potential tool for the phylogeny and classification of basidiomycetes (Li et al. 2019b, 2021b, 2020d). However, basidiomycete mitogenomes have been less studied than animal and Ascomycetes mitogenomes, with less than 120 complete mitogenomes of basidiomycetes published in the NCBI database. Moreover, the mitogenome of the order Filobasidiales has not previously been reported. In the present study, we obtained a phylogenetic tree with a good support rate by using the combined mitochondrial gene set (15 core PCGs), indicating that the mitochondrial gene is an effective tool for analysis of the phylogenetic relationship of basidiomycetes. More mitogenomes are needed to promote the classification or identification of basidiomycetes and reconstruct the phylogeny of fungi.
CONCLUSIONS
In the present study, two complete mitogenomes from the Filobasidiales order were reported and compared with other mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, including F. wieringae and F. globisporum. The mitochondrial genome size of the two Filobasidium species varied greatly, ranging from 27,861 to 71,783 bp, and the intronic region was considered to be the main factor contributing to mitogenome size variations in the Filobasidium genus. We further found intron loss/gain events in Tremellomycetes and other mitogenomes occurred during evolution. Comparative mitogenomic analysis revealed that the genetic contents, codon usages, and repetitive sequence differentiated greatly in the two Filobasidium species. In addition, a large number of base and sequence length variations were found in the core coding genes, tRNA genes and rRNA genes of the two Filobasidium mitogenomes. Several core PCGs have experienced strong pressure of positive selection in mitogenomes from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, including cob, cox2, nad2, and rps3 genes. In addition, large-scale gene rearrangements were detected between the 17 species from Agaricomycotina, Pucciniomycotina, and Ustilaginomycotina, which showed that the mitochondrial gene arrangement was highly variable. We further analyzed the phylogenetic status of Filobasidium species based on BI and ML methods using a combined mitochondrial gene set. This study serves as the first investigation of mitogenomes from the order Filobasidiales, and the results presented herein will help improve our understanding of Filobasidiales genomics, evolution, and taxonomy.
Availability of data and materials
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
Abbreviations
- Mitogenome:
-
Mitochondrial genome
- PCG:
-
Protein-coding gene
- Pcls:
-
Position classes
- Ks :
-
Synonymous substitution rates
- Ka :
-
Nonsynonymous substitution rates
- BI:
-
Bayesian inference
- ML:
-
Maximum likelihood
References
Adams KL, Palmer JD (2003) Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol 29(3):380–395
Adams KL, Qiu YL, Stoutemyer M, Palmer JD (2002) Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc Natl Acad Sci U S A 99(15):9905–9912
Aguileta G, de Vienne DM, Ross ON, Hood ME, Giraud T, Petit E, Gabaldon T (2014) High variability of mitochondrial gene order among fungi. Genome Biol Evol 6(2):451–465
Allen JF (2015) Why chloroplasts and mitochondria retain their own genomes and genetic systems: colocation for redox regulation of gene expression. Proc Natl Acad Sci U S A 112(33):10231–10238
Alves MJ, Ferreira ICFR, Dias J, Teixeira V, Martins A, Pintado M (2013) A review on antifungal activity of mushroom (basidiomycetes) extracts and isolated compounds. Curr Top Med Chem 13(21):2648–2659
Basse CW (2010) Mitochondrial inheritance in fungi. Curr Opin Microbiol 13(6):712–719
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucl Acids Res 27(2):573–580
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, Sayers EW (2018) GenBank Nucl Acids Res 46(D1):D41–D47
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69(2):313–319
Bjorkholm P, Harish A, Hagstrom E, Ernst AM, Andersson SG (2015) Mitochondrial genomes are retained by selective constraints on protein targeting. Proc Natl Acad Sci U S A 112(33):10154–10161
Bleasby AJ, Wootton JC (1990) Construction of validated, non-redundant composite protein sequence databases. Protein Eng 3(3):153–159
Caspermeyer J (2016) MEGA evolutionary software re-engineered to handle today’s big data demands. Mol Biol Evol 33(7):1887
Chatre L, Ricchetti M (2014) Are mitochondria the Achilles’ heel of the Kingdom Fungi? Curr Opin Microbiol 20:49–54
Chen C, Khaleel SS, Huang H, Wu CH (2014) Software for pre-processing Illumina next-generation sequencing short read sequences. Source Code Biol Med 9:8
Chen Y, Ye W, Zhang Y, Xu Y (2015) High speed BLASTN: an accelerated MegaBLAST search tool. Nucl Acids Res 43(16):7762–7768
Cheng J, Luo Q, Ren YH, Luo Z, Liao WL, Wang X, Li Q (2021) Panorama of intron dynamics and gene rearrangements in the phylum Basidiomycota as revealed by the complete mitochondrial genome of Turbinellus floccosus. Appl Microbiol Biotechnol 105(5):2017–2032
Coordinators NR (2017) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 36:D13–D21
Deng Y, Hsiang T, Li S, Lin L, Wang Q, Chen Q, Xie B, Ming R (2018) Comparison of the mitochondrial genome sequences of six Annulohypoxylon stygium isolates suggests short fragment insertions as a potential factor leading to larger genomic size. Front Microbiol 9:2079
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucl Acids Res 45(4):e18
Ding Y, Teng YS, Zhuo GC, Xia BH, Leng JH (2019) The mitochondrial tRNAHis G12192A mutation may modulate the clinical expression of deafness-associated tRNAThr G15927A mutation in a Chinese pedigree. Curr Mol Med 19:136–146
Elisashvili V (2012) Submerged cultivation of medicinal mushrooms: bioprocesses and products (review). Int J Med Mushrooms 14(3):211–239
Ernster L, Schatz G (1981) Mitochondria: a historical review. J Cell Biol 91(3 Pt 2):227s–255s
Ferandon C, Moukha S, Callac P, Benedetto JP, Castroviejo M, Barroso G (2010) The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS ONE 5(11):e14048
Gray MW, Burger G, Lang BF (2001) The origin and early evolution of mitochondria. Genome Biol 2(6):1–5
Hamari Z, Juhasz A, Kevei F (2002) Role of mobile introns in mitochondrial genome diversity of fungi (a mini review). Acta Microbiol Immunol Hung 49(2–3):331–335
Hibbett DS (2006) A phylogenetic overview of the Agaricomycotina. Mycologia 98(6):917–925
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lucking R, Thorsten Lumbsch H, Lutzoni F, Matheny PB, McLaughlin DJ, Powell MJ, Redhead S, Schoch CL, Spatafora JW, Stalpers JA, Vilgalys R, Aime MC, Aptroot A, Bauer R, Begerow D, Benny GL, Castlebury LA, Crous PW, Dai YC, Gams W, Geiser DM, Griffith GW, Gueidan C, Hawksworth DL, Hestmark G, Hosaka K, Humber RA, Hyde KD, Ironside JE, Koljalg U, Kurtzman CP, Larsson KH, Lichtwardt R, Longcore J, Miadlikowska J, Miller A, Moncalvo JM, Mozley-Standridge S, Oberwinkler F, Parmasto E, Reeb V, Rogers JD, Roux C, Ryvarden L, Sampaio JP, Schussler A, Sugiyama J, Thorn RG, Tibell L, Untereiner WA, Walker C, Wang Z, Weir A, Weiss M, White MM, Winka K, Yao YJ, Zhang N (2007) A higher-level phylogenetic classification of the fungi. Mycol Res 111(Pt 5):509–547
James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, Celio G, Gueidan C, Fraker E, Miadlikowska J, Lumbsch HT, Rauhut A, Reeb V, Arnold AE, Amtoft A, Stajich JE, Hosaka K, Sung GH, Johnson D, O’Rourke B, Crockett M, Binder M, Curtis JM, Slot JC, Wang Z, Wilson AW, Schussler A, Longcore JE, O’Donnell K, Mozley-Standridge S, Porter D, Letcher PM, Powell MJ, Taylor JW, White MM, Griffith GW, Davies DR, Humber RA, Morton JB, Sugiyama J, Rossman AY, Rogers JD, Pfister DH, Hewitt D, Hansen K, Hambleton S, Shoemaker RA, Kohlmeyer J, Volkmann-Kohlmeyer B, Spotts RA, Serdani M, Crous PW, Hughes KW, Matsuura K, Langer E, Langer G, Untereiner WA, Lucking R, Budel B, Geiser DM, Aptroot A, Diederich P, Schmitt I, Schultz M, Yahr R, Hibbett DS, Lutzoni F, McLaughlin DJ, Spatafora JW, Vilgalys R (2006) Reconstructing the early evolution of fungi using a six-gene phylogeny. Nature 443(7113):818–822
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20(4):1160–1166
Kolesnikova AI, Putintseva YA, Simonov EP, Biriukov VV, Oreshkova NV, Pavlov IN, Sharov VV, Kuzmin DA, Anderson JB, Krutovsky KV (2019) Mobile genetic elements explain size variation in the mitochondrial genomes of four closely-related Armillaria species. BMC Genomics 20(1):351
Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B (2017) PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol 34(3):772–773
Lavrov DV, Boore JL, Brown WM (2002) Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol Biol Evol 19(2):163–169
Li Q, Chen C, Xiong C, Jin X, Chen Z, Huang W (2018a) Comparative mitogenomics reveals large-scale gene rearrangements in the mitochondrial genome of two Pleurotus species. Appl Microbiol Biotechnol 102(14):6143–6153
Li Q, Wang Q, Chen C, Jin X, Chen Z, Xiong C, Li P, Zhao J, Huang W (2018b) Characterization and comparative mitogenomic analysis of six newly sequenced mitochondrial genomes from ectomycorrhizal fungi (Russula) and phylogenetic analysis of the Agaricomycetes. Int J Biol Macromol 119:792–802
Li Q, Ren Y, Shi X, Peng L, Zhao J, Song Y, Zhao G (2019a) Comparative mitochondrial genome analysis of two ectomycorrhizal fungi (Rhizopogon) reveals dynamic changes of intron and phylogenetic relationships of the subphylum agaricomycotina. Int J Mol Sci 20(20):5167
Li Q, Wang Q, Jin X, Chen Z, Xiong C, Li P, Liu Q, Huang W (2019b) Characterization and comparative analysis of six complete mitochondrial genomes from ectomycorrhizal fungi of the Lactarius genus and phylogenetic analysis of the Agaricomycetes. Int J Biol Macromol 121:249–260
Li Q, Wang Q, Jin X, Chen Z, Xiong C, Li P, Zhao J, Huang W (2019c) The first complete mitochondrial genome from the family Hygrophoraceae (Hygrophorus russula) by next-generation sequencing and phylogenetic implications. Int J Biol Macromol 122:1313–1320
Li Q, Xiang D, Wan Y, Wu Q, Wu X, Ma C, Song Y, Zhao G, Huang W (2019d) The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. Int J Biol Macromol 139:397–408
Li AH, Yuan FX, Groenewald M, Bensch K, Yurkov AM, Li K, Han PJ, Guo LD, Aime MC, Sampaio JP, Jindamorakot S, Turchetti B, Inacio J, Fungsin B, Wang QM, Bai FY (2020a) Diversity and phylogeny of basidiomycetous yeasts from plant leaves and soil: Proposal of two new orders, three new families, eight new genera and one hundred and seven new species. Stud Mycol 96:17–140
Li Q, He X, Ren Y, Xiong C, Jin X, Peng L, Huang W (2020b) Comparative mitogenome analysis reveals mitochondrial genome differentiation in ectomycorrhizal and asymbiotic amanita species. Front Microbiol 11:1382
Li Q, Ren Y, Xiang D, Shi X, Zhao J, Peng L, Zhao G (2020c) Comparative mitogenome analysis of two ectomycorrhizal fungi (Paxillus) reveals gene rearrangement, intron dynamics, and phylogeny of basidiomycetes. IMA Fungus 11(1):12
Li Q, Yang L, Xiang D, Wan Y, Wu Q, Huang W, Zhao G (2020d) The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): features, intron dynamics and phylogenetic implications. Int J Biol Macromol 145:974–984
Li Q, Li L, Feng H, Tu W, Bao Z, Xiong C, Wang X, Qing Y, Huang W (2021a) Characterization of the complete mitochondrial genome of Basidiomycete yeast hannaella oryzae: intron evolution, gene rearrangement, and its phylogeny. Front Microbiol 12:646567
Li Q, Wu P, Li L, Feng H, Tu W, Bao Z, Xiong C, Gui M, Huang W (2021b) The first eleven mitochondrial genomes from the ectomycorrhizal fungal genus (Boletus) reveal intron loss and gene rearrangement. Int J Biol Macromol 172:560–572
Lin L, Cui P, Qiu Z, Wang M, Yu Y, Wang J, Sun Q, Zhao H (2019) The mitochondrial tRNA(Ala) 5587T>C and tRNA(Leu(CUN)) 12280A>G mutations may be associated with hypertension in a Chinese family. Exp Ther Med 17(3):1855–1862
Liu XZ, Wang QM, Goker M, Groenewald M, Kachalkin AV, Lumbsch HT, Millanes AM, Wedin M, Yurkov AM, Boekhout T, Bai FY (2015a) Towards an integrated phylogenetic classification of the Tremellomycetes. Stud Mycol 81:85–147
Liu XZ, Wang QM, Theelen B, Groenewald M, Bai FY, Boekhout T (2015b) Phylogeny of tremellomycetous yeasts and related dimorphic and filamentous basidiomycetes reconstructed from multiple gene sequence analyses. Stud Mycol 81:1–26
Lohse M, Drechsel O, Kahlau S, Bock R (2013) OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res 41(Web Server issue):W575–W581
Lowe TM, Chan PP (2016) tRNAscan-SE on-line: integrating search and context for analysis of transfer RNA genes. Nucl Acids Res 44(W1):W54-57
Luo BL, Sun HL, Zhang Y, Gu YL, Yan WL, Zhang RR, Ni YQ (2019) Habitat-specificity and diversity of culturable cold-adapted yeasts of a cold-based glacier in the Tianshan Mountains, northwestern China. Appl Microbiol Biotechnol 103(5):2311–2327
McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse. Curr Biol 16(14):R551-560
Millanes AM, Diederich P, Ekman S, Wedin M (2011) Phylogeny and character evolution in the jelly fungi (Tremellomycetes, Basidiomycota, Fungi). Mol Phylogenet Evol 61(1):12–28
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Nemcova K, Breierova E, Vadkertiova R, Molnarova J (2015) The diversity of yeasts associated with grapes and musts of the Strekov winegrowing region, Slovakia. Folia Microbiol 60(2):103–109
Osiewacz HD (2002) Aging in fungi: role of mitochondria in Podospora anserina. Mech Ageing Dev 123(7):755–764
Pan WH, Liao WQ, Hagen F, Theelen B, Shi WM, Meis JF, Boekhout T (2012) Meningitis caused by Filobasidium uniguttulatum: case report and overview of the literature. Mycoses 55(2):105–109
Repar J, Warnecke T (2017) Mobile introns shape the genetic diversity of their host genes. Genetics 205(4):1641–1648
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61(3):539–542
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302
Sandor S, Zhang YJ, Xu JP (2018) Fungal mitochondrial genomes and genetic polymorphisms. Appl Microbiol Biotechnol 102(22):9433–9448
Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9:88
Seif E, Leigh J, Liu Y, Roewer I, Forget L, Lang BF (2005) Comparative mitochondrial genomics in zygomycetes: bacteria-like RNase P RNAs, mobile elements and a close source of the group I intron invasion in angiosperms. Nucl Acids Res 33(2):734–744
Singh H, Kim H, Song H, Joe M, Kim D, Bahn YS, Choi JI, Lim S (2013) A novel radiation-resistant strain of Filobasidium sp. isolated from the West Sea of Korea. J Microbiol Biotechnol 23(11):1493–1499
Slater GS, Birney E (2005) Automated generation of heuristics for biological sequence comparison. BMC Bioinform 6:31
Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML, Bonito G, Corradi N, Grigoriev I, Gryganskyi A, James TY, O’Donnell K, Roberson RW, Taylor TN, Uehling J, Vilgalys R, White MM, Stajich JE (2016) A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108(5):1028–1046
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28(6):1102–1104
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res 22(22):4673–4680
Vaidya G, Lohman DL, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27(2):171–180
Valach M, Burger G, Gray MW, Lang BF (2014) Widespread occurrence of organelle genome-encoded 5S rRNAs including permuted molecules. Nucl Acids Res 42(22):13764–13777
Voriskova J, Baldrian P (2013) Fungal community on decomposing leaf litter undergoes rapid successional changes. Isme J 7(3):477–486
Wang L, Wang QM (2015) Molecular phylogenetic analysis of ballistoconidium-forming yeasts in trichosporonales (tremellomycetes): a proposal for Takashimella gen. nov. and Cryptotrichosporon tibetense sp. nov. PLoS ONE 10(7):e0132653
Wang X, Jia LH, Wang MD, Yang H, Chen MY, Li X, Liu HY, Li Q, Liu N (2020a) The complete mitochondrial genome of medicinal fungus Taiwanofungus camphoratus reveals gene rearrangements and intron dynamics of Polyporales. Sci Rep 10(1):1–4
Wang X, Song A, Wang F, Chen M, Li X, Li Q, Liu N (2020b) The 206 kbp mitochondrial genome of Phanerochaete carnosa reveals dynamics of introns, accumulation of repeat sequences and plasmid-derived genes. Int J Biol Macromol 162:209–219
Wu P, Bao Z, Tu W, Li L, Xiong C, Jin X, Li P, Gui M, Huang W, Li Q (2021) The mitogenomes of two saprophytic Boletales species (Coniophora) reveals intron dynamics and accumulation of plasmid-derived and non-conserved genes. Comput Struct Biotechnol J 19:401–414
Xia Y, Zheng Y, Murphy RW, Zeng X (2016) Intraspecific rearrangement of mitochondrial genome suggests the prevalence of the tandem duplication-random loss (TDLR) mechanism in Quasipaa boulengeri. BMC Genomics 17(1):965
Ye J, Cheng J, Ren Y, Liao W, Li Q (2020) The first mitochondrial genome for geastrales (Sphaerobolus stellatus) reveals intron dynamics and large-scale gene rearrangements of basidiomycota. Front Microbiol 11:1970
Yurkov AM, Kurtzman CP (2019) Three new species of Tremellomycetes isolated from maize and northern wild rice. FEMS Yeast Res 19(2):foz004
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31800070 and 42107233) and the Foundation of Chengdu Medical College (CYZYB20-03).
Author information
Authors and Affiliations
Contributions
Conceived and designed experiments: QL, CZ, and MC. Analyzed the data: LL, HF, ZB, KT, YH, and WT. Wrote and reviewed the paper: QL and CZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Adherence to national and international regulations
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Comparison on mitogenomes among 17 species from Agaricomycotina, Pucciniomycotina and Ustilaginomycotina. Table S2. Annotation and characterization of the two Filobasidium mitogenomes. Table S3. Core protein coding gene information of the two Filobasidium species. Table S4. Start and stop codons analysis of 17 species from Agaricomycotina, Pucciniomycotina and Ustilaginomycotina. Table S5. Local BLAST analysis of the Filobasidium mitogenomes against themselves. Table S6. Tandem repeats detected in the mitogenomes of Filobasidium using the online program Tandem Repeats Finder. Table S7. Species, and GenBank accession number used for phylogenetic analysis in this study.
Additional file 2: Figure S1.
Putative secondary structures of tRNA genes identified in the mitochondrial genomes of two Filobasidium species. The 22 tRNAs in green or red fonts represent tRNAs shared by the two Filobasidium species, while the tRNA in blue font represent tRNA only in F. wieringae. Residues conserved across the two mitochondrial genomes are shown in green, while variable sites are shown in red. All genes are shown in order of occurrence in the mitochondrial genome of F. wieringae, starting from trnL.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Bao, Z., Tang, K. et al. First two mitochondrial genomes for the order Filobasidiales reveal novel gene rearrangements and intron dynamics of Tremellomycetes. IMA Fungus 13, 7 (2022). https://doi.org/10.1186/s43008-022-00094-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43008-022-00094-2