
Abstract
Gut microbiota are intrinsic to an herbivorous lifestyle, but very little is known about how plant secondary compounds (PSCs), which are often toxic, influence these symbiotic partners. Here we interrogated the possibility of unique functional core microbiomes in populations of two species of woodrat (Neotoma lepida and bryanti) that have independently converged to feed on the same toxic diet (creosote bush; Larrea tridentata) and compared them to populations that do not feed on creosote bush. Leveraging this natural experiment, we collected samples across a large geographic region in the U.S. desert southwest from 20 populations (~ 150 individuals) with differential ingestion of creosote bush and analyzed three gut regions (foregut, cecum, hindgut) using16S sequencing and shotgun metagenomics. In each gut region sampled, we found a distinctive set of microbes in individuals feeding on creosote bush that were more abundant than other ASVs, enriched in creosote feeding woodrats, and occurred more frequently than would be predicted by chance. Creosote core members were from microbial families e.g., Eggerthellaceae, known to metabolize plant secondary compounds and three of the identified core KEGG orthologs (4-hydroxybenzoate decarboxylase, benzoyl-CoA reductase subunit B, and 2-pyrone-4, 6-dicarboxylate lactonase) coded for enzymes that play important roles in metabolism of plant secondary compounds. The results support the hypothesis that the ingestion of creosote bush sculpts the microbiome across all major gut regions to select for functional characteristics associated with the degradation of the PSCs in this unique diet.
Similar content being viewed by others


Introduction
The microbial communities of the mammalian gut are well known for their roles in defending against pathogens, training the immune system, and synthesizing nutrients [86]. These microbial communities can be highly variable among hosts due to differences in genetics, physiological status, and diet [113, 114]. Despite this variation, it is hypothesized that within a species, population, or dietary strategy, a subset of gut microbes or microbial functions persists, known as the “core microbiome”. These core microbes are proposed to be key ecological and functional members of the community, and recent studies reveal numerous insights into the microbial ecology found in an assortment of host species in a variety of environments [1, 58, 83]. For example, among domestic ruminants, a distinctive core microbiome occurs in the rumens of 32 host species [46]. Microbes belonging to the rumen core may represent taxa that are fundamental to this dietary strategy. However, microbial community structure does not always link with function as some functions are restricted to certain taxa while others are more widespread [103]. Thus, a host population may not share a similar taxonomic core of microbes, but may host microbes with shared functional capability, or a functional core microbiome. While many studies have investigated the presence of a shared, taxonomic core, few have extended the work to examine the functionality of the core.
In herbivores consuming a similar diet, core microbes may be involved in critical functions. Herbivores rely completely on their gut microbiota for fermentation of indigestible, complex plant carbohydrates, such as fiber and cellulose, into simple sugars usable by the host [63]. In addition, to deter herbivores, plants produce plant secondary compounds (PSCs), defensive toxins that can cause a wide range of negative physiological effects on the consumer [36]. It has long been hypothesized that herbivores house gut microbiota to aid in the degradation of PSCs [36] and recently, studies provide evidence for this microbial function in a multiplicity of herbivorous hosts [8, 10, 44, 53, 70, 71]. Plant secondary compounds also significantly influence the taxonomic structure of gut microbial communities [66, 105, 106]. Such a complex diet may select for a taxonomic core microbiome of organisms capable of fiber breakdown or interacting with PSCs. Alternatively, a lack of a discernable taxonomic core microbiome may mean that the ability to break down PSCs is more spread among various taxa, i.e., a functional core. While the presence of a core has been investigated across domestic, ruminant herbivores [46], there has been little investigation across different gut regions within a host, across different hosts consuming the same diet, or hosts in a natural setting.
The herbivore gut is highly specialized for ingestion of their complex diet, including adaptations for housing these communities of symbiotic microbes. Fiber degrading microbes reside in specialized, non-gastric stomach chambers in foregut fermenting animals such as ruminants [63] and in a more distal fermentation chamber, the cecum, for hindgut fermenting animals such as equids, rabbits, and rodents [63]. Some rodents, in addition to the cecum, have a sacculated foregut chamber proximal to the gastric stomach [20]. This chamber does not extensively ferment fiber [69] and houses microbes capable of metabolizing PSCs [69]. Little is known about the extent to which other species of herbivore host PSC degrading microbes and whether these microbes are similar across different host populations consuming the same diet [30].
To advance our understanding of how PSCs affect the gut microbiota of mammalian herbivores, we investigated the presence of a unique core microbiome in woodrat populations (Neotoma spp.) that have converged to feed on the same toxic diet, creosote bush (Larrea tridentata). These herbivorous rodents consume a variety of diets across their range [67, 114]. Some populations of both the desert woodrat (Neotoma lepida) and the Bryant’s woodrat (Neotoma bryanti), species that diverged about 1.6 mya [92], independently converged to feed on creosote bush [80]. Creosote bush leaves are coated in a phenolic-rich resin composed of hundreds of PSCs such as phenolics, flavones, and saponins [3, 101]. Many of these compounds, such as nordihydroguaiaretic acid (NDGA) are toxic to mammals [31, 81, 98] and also antimicrobial [42, 74]. These PSCs not only strongly affect the diversity of the gut microbial community but may also select for microbes that use these compounds as substrates [105, 106]. Additionally, previous dietary intervention studies reveal that the gut microbiota play a critical role in facilitating ingestion of creosote bush in N. lepida [71]. Therefore, the PSCs in creosote bush may have selected for the same taxonomic or functional core microbiome in populations of N. lepida and bryanti that feed on creosote. To investigate the presence of a creosote-related core microbiome, we surveyed the microbial communities in 20 populations of both N. lepida and bryanti that consume creosote (“creosote feeders”) and compared them to microbial communities from populations outside the natural range of creosote bush (“non-creosote feeders”) to identify core microbes specific to a host diet rather than host species. Since microbial communities differ along the alimentary tract and previous work in this system was restricted to primarily microbes in the feces, we evaluated the three major communities in the gut, i.e., foregut, cecum, hindgut. We expected core microbes related to creosote feeding in the foregut as this structure has been documented to house microbes capable of degrading PSCs [67, 85]. Furthermore, we predicted that this chamber would harbor more gut microbiota capable of breaking down PSCs compared to the cecum, which should house primarily fiber degrading bacteria. Finally, because the most abundant PSC produced by creosote bush (NDGA) is a phenolic, we anticipated we would see microbial functions associated with the metabolism of xenobiotics and aromatic compounds in the functional core.
Experimental procedures
Sample collection
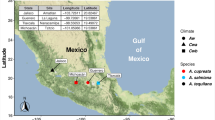
To examine the core microbiota of woodrats consuming a creosote diet, we collected 3–10 individuals from 20 populations across two woodrat species in 2017–2018 (n = 65 creosote feeders, n = 85 non-creosote feeders; Table S1; Figs. S1 and S2). Populations spanned the southwestern United States (California, Utah, and Nevada). We live-trapped animals using Sherman traps baited with oats; previous work has shown that this trapping method does not significantly affect the microbiome of woodrats [68]. For all individuals, a fecal sample was collected at the time of capture and animals were not released after sampling. Fecal samples were used as a representation of the hindgut microbial community because the microbiota in feces often resemble the hindgut of mammalian hosts [32, 33, 69]. Foregut and cecum contents were sampled from a subset of populations. These animals were dispatched after capture and immediately dissected (Table S1). Feces, foregut content, and cecum content were stored in liquid nitrogen in the field and then held at − 80 °C until DNA extraction.
DNA extraction and amplicon sequencing
We isolated DNA from woodrat feces, foregut content, and cecum content using QIAamp PowerFecal DNA kits (Qiagen), following manufacturer protocols. Two negative controls were sequenced for each extraction kit. All DNA amplification, library preparation, and sequencing was conducted at the DNA Service Facility at the University of Illinois-Chicago [87]. To determine the gut microbial community of our sampled woodrats, we amplified the V4 hypervariable region of the 16S rRNA locus using the 515F and 806R primers following the Earth Microbiome Project suggested protocol [19]. To determine dietary content of each population, we amplified the P6 loop of the chloroplast trnL (UAA) intron using the g and h primers as previously validated and described [105, 106, 110]. Analysis was conducted at the population level as previous work has shown that using multiple samples can reduce bias from individual outliers and give a more accurate analysis of the diet [28, 105, 106]. In brief, starting PCR amplifications were performed in 10-μl reactions under the following conditions: 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s. A second PCR amplification was performed wherein each sample well received a unique barcode from an Access Array Barcode Library for Illumina. Conditions for the second PCR were as follows: 95 °C for 5 min, followed by eight cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s. Final libraries were pooled and size selected using AMPure XP cleanup (0.8×, v/v; Agencourt, Beckman-Coulter). All amplicon sequencing was conducted on an Illumina MiniSeq platform (2 × 150 bp paired-end reads).
16S rRNA sequence processing
All 16S sequences were processed in QIIME2 version 2021.8 [13]. Primers were removed using Cutadapt [82] and resulting sequences were filtered for quality control in QIIME2. Sequences were grouped into amplicon sequence variants (ASVs) using DADA2 [18], resulting in 5,508 unique ASVs that were assigned to taxonomy using the Silva database release 138.1 [94]. We removed identified chimeras, sequences that appeared in fewer than four samples, sequences that appeared less than ten times total across all samples, and sequences that were identified as chloroplast or mitochondria. After filtering, samples contained 6,884,401 total reads with an average of 27,985 reads per sample. To control for differential sequencing depth, samples were rarefied to a sequencing depth of 4499 reads per sample (the lowest coverage in any one sample that contained sufficient reads). Rarefaction resulted in the removal of four samples. Identification of core microbes was done using unrarefied data, as rarefaction can skew the estimation of core microbes [88, 95]. All other analyses were performed on rarefied data.
trnL sequence processing
To determine the diets of animals, we used trnL plant metabarcoding on fecal samples. Plant sequences were processed using QIIME2 version 2021.8 as previously described and validated [105, 106]. In brief, we retained high-quality sequences by setting minimum sequence length after trimming to 20 bp, increasing minimum acceptable PHRED score to 20, and reducing minimum overlap to 10 bp, all other parameters were left at default settings. Sequences were assigned to operational taxonomic units (OTUs) at the 100% identity level using denovo clustering. Chimeras were removed and, based on the contents of sequencing blanks, we removed OTUs represented by fewer than twenty reads per sample and any OTUs that appeared in less than five total samples. Samples with > 1000 total reads before filtering were removed from this analysis (3 samples). Taxonomy was assigned to OTUs using the Scikit-learn classifier in qiime2 version 2021.8, trained on a custom reference database. To create our database, we used a custom python script to download chloroplast sequences and taxonomies from the NCBI nucleotide database, generate a FASTA file containing each sequence and its reverse complement, and trim the FASTA file using Cutadapt 2.10 with the 5′ and 3′ adapters set to the trnL g and h primers, as previously described [114]. Sequences were not included in the reference database if they contained ambiguous nucleotides, mismatches in either primer, more than three mismatches overall, an amplicon length outside the range of 8–175 bp, contained a taxonomic classification lacking “Viridiplantae”, or if the sequence was flagged as “environmental_samples”. Sequences with taxonomy that did not resolve to at least the family level were considered unclassified (< 4% of sequences). After examining the content of our negative controls and based on previous work [105, 106], families that represented less than 1% of each population’s total relative abundance were removed. Populations were considered ‘creosote feeders’ if creosote reads were present in diet samples after this filtering step and if creosote occurs in the region the population was sampled from Tables S3 and S4.
Identification of core members
To determine the creosote-feeding core microbiome, we used a two-step process. First, we identified the core ASVs across all woodrat samples as those that were present in ≥ 50% of N. lepida and N. bryanti samples [78, 99, 113]. This threshold was chosen based on precedence from other studies that suggest using a more stringent threshold generates diversity scores that correlate poorly with unfiltered data and reduces the ability to compare core microbiomes across studies. Also, because we sampled across many geographically distinct populations, which is known to significantly influence the structure of the microbiome [1, 39, 40, 45, 55, 99, 108, 114]. Then, we removed all core woodrat microbes that were not unique to creosote feeding woodrats to yield the creosote core. Using this two-step process, we defined core ASVs for the cecum, foregut, and hindgut of N. lepida and N. bryanti in creosote feeding populations and in non-creosote feeding populations. We considered ASVs part of the ‘creosote-feeding core microbiome’ for each sampled gut region if they met the ≥ 50% threshold for the general core in both N. lepida and N. bryanti samples and were not considered core in non-creosote feeding populations. Microbes meeting these thresholds are designated as ‘core’ or ‘creosote-feeding core’ hereafter. In addition, because we had more hindgut samples than foregut and cecum, we evaluated the effect of sample size on estimates of core microbes, by restricting the hindgut dataset to only samples that had a matching foregut and cecum sample, and comparing the results from this subset to the results from all hindgut samples.
Statistical analysis
Alpha-diversity of the microbiome was measured using Shannon’s index and Observed ASVs, these values were compared across the foregut, cecum, and hindgut using Kruskal–Wallis tests and between creosote feeders and non-creosote feeders using Mann–Whitney U tests implemented in R. We measured beta-diversity using Bray–Curtis distances (community structure) and Jaccard distances (community membership). Differences in beta-diversity were compared using permutational multivariate analysis of variance (PERMANOVAs) implemented using the vegan package in R with diet type, species, and population as factors. To determine whether core ASVs were under host selection, we fit the prokaryotic neutral model to all ASVs found in each gut region following methods described in [17]. Using this model, all ASVs were classified as either over-represented, neutral, or under-represented. In addition, we applied the neutral model to each population to determine whether there was greater selection on creosote populations than non-creosote populations. Finally, we estimated differentially abundant ASVs using DESeq2 [79]. We used the package ashr to shrink log2fold changes generated by DESeq2, changes in relative abundance were considered significant if the FDR corrected p-value was < 0.01 and the log fold change was ≥ 1.5.
Metagenomic analysis
We performed metagenomic sequencing on 45 fecal samples from 9 populations (n = 3 per species, per population; Table S3). We extracted DNA as previously described, library preparation, amplification, and final sequencing were completed at the DNA Service Facility at the University of Illinois-Chicago. Library prep was performed using the Swift 2S Turbo DNA Library Kit with enzymatic fragmentation (catalog 44,024 Swift Biosciences Ann Arbor, MI) followed by PCR performed according to the manufacturer protocol. Final libraries were size-selected, pooled, and sequenced on an Illumina NovaSeq 6000 with 2 × 150 bp sequencing and a 1% phiX spike-in. Metagenomic sequencing resulted in a total of 452,316,506 reads with an average of 10,051,478 reads per sample (S.D. 1,719,434).
Previous research has shown that read-based and assembly-based methods can produce different results [111], therefore, we characterized the functional profile of the gut microbiota using both unassembled and assembled reads. For the unassembled reads, we used MEGAN6 to conduct an analysis of gene function on forward reads [50]. Adaptors were trimmed from all sequences using FastP [22]. We removed host reads from sequences by mapping the reads to all of the following host genomes: Peromyscus leucopus, P. maniculatus, P. nasutus, and Neotoma lepida. We ran DIAMOND (v. 2.0.9) [16] to blast the remaining reads against the UniRef100 database [109] with an e-value cut-off of 0.001. These host-filtered, annotated forward reads were uploaded into MEGAN6, community edition and classified to KEGG Orthologs (KOs) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [62]. Genes were classified as belonging to the creosote core microbiome as previously described for the taxonomic core. We used DESeq2 to investigate genes that were differentially abundant between creosote and non-creosote feeders as previously described.
In addition, to identify bacteria associated with creosote feeding, we assembled metagenomic sequences into metagenome assembled genomes (MAGs). We used Metaspades (SPAdes v. 3.15.3) on a large memory node to co-assemble all 45 samples resulting in 16,336,611 total contigs. Then, we used MetaBat2 to bin contigs with default parameters, resulting in 649 total bins. We measured MAG completeness and contamination using checkM (v. 1.1.3) and dereplicated the MAGs to 99% ANI using dRep [90]. All unique MAGs identified as > 50% complete and with < 10% contamination were kept for further analysis, resulting in 271 total MAGs identified across all samples. We classified MAGs as part of the ‘core creosote-feeding microbiome’ using the methods described above for ASVs (i.e., creosote-feeding core). We assigned taxonomy to the MAGs using the Genome Taxonomy Database (GTDB) and GTDB-Tk [21]. The functional capability of core MAGs was determined by identifying KOs in each sample. First, coding regions were identified using Prodigal and then extracted using GffRead [93]. To reduce gene redundancy, we clustered the resulting genes at 95% sequence similarity using CD-HIT [37]. Non-redundant genes were kept and blasted against the UniRef100 database using DIAMOND for functional annotation. We then generated gene abundance profiles by mapping reads from each sample to non-redundant genes using BWA [77]. We investigated the presence of differentially abundant genes in creosote and non-creosote feeders using DESeq2, which has been validated for use with metagenomes [57].
Results
Community composition across and within gut regions
For all woodrat populations, gut microbial diversity varied across the foregut, cecum, and hindgut. Alpha-diversity metrics were significantly different across the three gut communities (Kruskal–Wallis, Shannon index H(2) = 84, p < 0.001; Observed ASVs H(2) = 68, p < 0.001; Fig. S3). The cecum proved highly diverse, with the highest alpha-diversity for both the Shannon index and observed ASVs while the foregut was the least diverse (Table 1). Differences in gut microbial diversity between creosote feeders and non-creosote feeders varied by gut region (Table S4). In the cecum and the hindgut, non-creosote feeders showed increased alpha-diversity compared to creosote feeders when measuring the Shannon index and observed ASVs, respectively (Cecum, Mann–Whitney U, Shannon index U = 160, p < 0.01; Hindgut, Mann–Whitney U, Observed ASVs U = 1878, p < 0.02). There was no difference in alpha-diversity between the gut microbial communities of creosote and non-creosote feeders in the foregut.
The microbial community of the three gut regions significantly differed in both community membership (PERMANOVA, Jaccard, pseudo-F = 4.6, R2 = 0.04, p < 0.001) and community structure (PERMANOVA, Bray–Curtis, pseudo-F = 7.7, R2 = 0.06 p < 0.001). The variable with the most explanatory power was the most abundant plant family in host diet rather than gut region (PERMANOVA, Jaccard, diet R2 = 0.14, p < 0.001; Bray–Curtis, diet R2 = 0.20, p < 0.001). Within each gut region, the microbial communities of creosote-feeding populations significantly differed from populations that do not feed on creosote (PERMANOVA, Jaccard and Bray–Curtis, p < 0.001 for all gut regions; Fig. 1) with levels of dietary creosote explaining 7% of the variation (PERMANOVA, Jaccard and Bray–Curtis, R2 < 0.07, p < 0.001 for all gut regions; Table S5).

Gut microbial community structure and membership between creosote and non-creosote feeding woodrat populations. A Diagram of the gastrointestinal tract of woodrats, highlighted segments represent sampled gut regions: the foregut (blue), cecum (green), and hindgut (purple). Principle coordinate analysis of Bray–Curtis distances between creosote and non-creosote feeders for the foregut (B), cecum (C), and hindgut (D); there were significant differences between creosote and non-creosote feeders across all gut regions (statistics reported in the text)
Creosote-feeding core microbiome membership
The cecum, foregut, and hindgut, predominantly harbored distinct creosote-feeding core microbiomes. With respect to the core microbiome unique to creosote feeders, hereafter ‘core’, 25 ASVs were identified as being core in more than one gut region, while two ASVs, both in the family Lactobacillaceae were core microbes across all three regions (Fig. 2A). The majority of core ASVs shared between two gut regions belonged to Muribaculaceae (Table S6). The cecum harbored the largest overall core microbiome of the gut regions. Of the 2973 total identified ASVs in the cecum, 164 were classified as core (Fig. 2C) represented by 18 microbial families. The families containing the most core ASVs in the cecum were Lachnospiraceae (64 ASVs), Oscillospiraceae (26 ASVs), and Muribaculaceae (29 ASVs).

Gut regions harbored distinct core microbiotas. A UpSet diagrams of core microbes shared between all gut regions. Few core microbes were shared across gut regions. Euler diagrams of core microbes (number of ASVs) that were unique to creosote feeders (left), unique to non-creosote feeders (right), and shared by all populations (center) in the foregut (B), cecum (C), and hindgut (D)
The foregut had the smallest core of the three gut regions (Fig. 2B). The foregut harbored a total of 2207 identified ASVs, 36 of which were identified as core. Foregut core ASVs were represented by 9 families with Muribaculaceae (12 ASVs), Lachnospiraceae (6 ASVs), Desulfovibrionaceae (5 ASVs), and Eggerthellaceae (3 ASVs) containing the most. Finally, hindgut samples contained a total 3781 identified ASVs with 40 ASVs classified as core, belonging to 10 families (Fig. 2D). Muribaculaceae and Oscillospiraceae also contained the most core hindgut ASVs, followed by the family Lachnospiraceae (15, 6, and 5 ASVs respectively).
The sample size of the hindgut dataset affected the size of the core microbiota. When we restricted our hindgut samples to match the animals included in the cecum and foregut samples, the number of microbes identified as core increased. For the smaller hindgut dataset, we saw 2,627 ASVs, 66 of which were classified as core. Core microbes were represented by 13 different families, with the majority of ASVs belonging to the Muribaculaceae, Lachnospiraceae, and Oscillospiraceae (25, 12, and 9 ASVs, respectively). None of the identified core members belonged to the Butyricoccaceae, despite that family being present for core members in the full hindgut core dataset. Notably, though the core microbiota increased compared to the larger hindgut dataset, the cecum still harbored a larger core microbiome.
Core ASVs did not comprise the majority of the microbiome, but were more abundant than other ASVs. The relative abundance of core ASVs was significantly higher than non-core ASVs within each gut region (Kruskal–Wallis, p < 0.001, cecum, foregut, and hindgut). Within each gut region, the average relative abundance of any one core ASVs never exceeded 1.5%. In the cecum, the most abundant core ASV belonged to the family Lachnospiraceae, and had a total relative abundance across all creosote-feeding samples of 0.07%. Collectively, cecum core microbes comprised a total relative abundance of 19.1% while core foregut ASVs comprised 15.7% of the total. The most abundant core ASV in the foregut had the highest relative abundance of any core ASV across the gut regions with a total relative abundance of 6.1% (family Lactobacillaceae). The most abundant core ASV in the hindgut also belonged to the family Lactobacillaceae, and had a total relative abundance in all creosote-feeding samples of 1.1%. The cumulative relative abundance of all core ASVs in the hindgut was 10.4% of the microbiome. Though no core ASVs comprised the majority of the microbiome in any gut region, no single ASV outside the core had a higher relative abundance than 7.6% (foregut, non-core ASV).
Taxonomic differences in creosote feeding populations
Core ASVs were significantly enriched in the cecum, foregut, and hindgut of creosote-feeding populations compared to non-creosote feeding populations. Using DESeq2, within the cecum, we found 111 ASVs belonging to 13 microbial families significantly enriched in creosote feeding animals compared to non-creosote feeding animals; 36 of these ASVs were core (Fig. 3A). Enriched, core ASVs were represented by 9 microbial families (Table S7). Amplicon sequence variants with the largest logfold changes compared to non-creosote feeders belonged to the families Muribaculaceae and Lachnospiraceae (Table S7). Forty-eight ASVs were significantly enriched in the foregut of creosote feeding animals, 16 of which were core (DESeq2; Fig. 3A). Significantly enriched core ASVs in the foregut belonged to the families Eggerthellaceae, Lactobacillaceae, Lachnospiraceae, and Pasteurellaceae (Table S8). The hindgut of creosote feeders contained the highest number of significantly enriched microbes compared to non-creosote feeders with 151 enriched ASVs, 18 of which were classified as core (DESeq2; Fig. 3A). These enriched core microbes were represented by 7 microbial families; the largest logfold changes were observed in ASVs belonging to the Muribaculaceae and Lactobacillaceae (Table S9).

Core microbes enriched and selected for in the foregut, cecum, and hindgut of creosote feeders. A Log2 fold enrichment of core microbes in creosote feeders compared to non-creosote feeders. B Deviance of core ASVs from neutral model fit. Overrepresented ASVs appear above the line and underrepresented ASVs beneath the line. Points are colored by gut region. C Fit of the prokaryotic neutral model to all ASVs in the foregut (blue outline), cecum (green outline), and hindgut (purple outline). Closed circles represent members of the core microbiome, open circles represent non-core ASVs. ASVs are classified as overrepresented (yellow), underrepresented (red), or neutral (gray). The solid line represents the predicted frequency of occurrence and the dashed line is 95% confidence intervals
Neutral selection of ASVs
To determine whether core ASVs were under selection, we assessed the fit of the prokaryotic neutral model to occupancy and abundance distributions of all ASVs in each gut region. Deviations from this model (i.e., reduced model fit) are indicative of non-neutral processes, such as selection from the host or host diet [17]. Model fit was greatest for all ASVs in the hindgut (R2 = 0.46); however, when we restricted the dataset to only include hindgut samples from hosts that had matching cecum and foregut samples, the model fit decreased (R2 = 0.27) and was more similar to the other gut regions (cecum R2 = 0.30; foregut R2 = 0.26). Additionally, there was no significant difference in prokaryotic neutral model fit on all ASVs between creosote and non-creosote populations (student’s t-test, t(13) = − 0.31, p = 0.78) nor was there a relationship between the amount of creosote in each population’s diet and model fit (linear regression, R2 = 0.24, p = 0.12). Amongst the ASVs identified as under selection, many were core microbes from each of the different gut regions. Fewer core microbes selected against, or underrepresented than under selection (Table 2). Across all gut regions, there was underrepresentation of core ASVs belonging to the Lactobacillaceae, Muribaculaceae, Clostridia_UCG-014, Pasteurellaceae, Lachnospiraceae, and Oscillospiraceae and overrepresentation of core ASVs belonging to many families found in the core (Fig. 3B, C).
Characterizing the functional core microbiome
To characterize the potential functional core microbiome of creosote feeding woodrats, we compared the gene content of the gut microbial communities of creosote and non-creosote feeding woodrats using KOs. Using the unassembled reads, we investigated the abundance of high-level functional pathways and found that most functions were unclassified (46%) with the next most abundant pathways being functions associated with metabolism (Fig. 4A; Table S10). We also investigated whether there existed a functional core in creosote feeders using KOs associated with degradation of xenobiotics at the protein level. Of the 208 KOs within the database, 4 were identified as part of the creosote feeding functional core. The core KOs unique to creosote feeders coded for the enzymes: enoyl-CoA hydratase (K01692), 4-hydroxybenzoate decarboxylase (K01612), benzoyl-CoA reductase subunit B (K04113), and 2-pyrone-4, 6-dicarboxylate lactonase (K10221). The three most abundant KOs in creosote feeders were associated with biosynthesis or metabolism of pyrimidines and purines (Fig. 4B). The next most abundant KO coded for 4-carboxymuconolactone decarboxylase, an enzyme involved in the degradation of benzoate (Fig. 4B). More than half of the KOs (106) were shared across the functional core of creosote and non-creosote feeders with no hierarchical clustering of functional profiles by diet type (Fig. 4A, B; Table S11). Principal component analysis of the KOs at the xenobiotic degradation protein level did not show separation of the functional profiles of woodrat gut microbial communities by diet (Fig. 5A). In addition, there were no xenobiotic degradation KOs identified as significantly enriched in creosote feeders compared to non-creosote feeders (DESeq2).

There was no difference in the functional profiles of gut microbial communities in creosote and non-creosote feeding woodrats using unassembled reads. Heatmaps of relative abundances of the most abundant KEGG pathways (A) and the most abundant KEGG-assigned xenobiotic degradation proteins (B) in both creosote and non-creosote feeding animals. The dendrogram left of each heatmap represents hierarchical clustering of creosote (dark purple) and non-creosote (light purple) individuals based on Bray–Curtis dissimilarity of KEGG pathway or xenobiotic degradation protein abundance counts. See Tables S11 and S12 for full relative abundance information

There was large overlap of gut microbiome functional profiles between creosote and non-creosote feeders. Principal component analysis of Bray–Curtis distances generated from KO counts at the protein level for A unassembled reads and B assembled MAGs
Co-assembly of the short-read data resulted in 271 total MAGs. These MAGs primarily belonged to the families Muribaculaceae (124), Lachnospiraceae (48), and Ruminococcaceae (15; Tables S12 and S13). Only one MAG belonged to the creosote-feeding core microbiome. This MAG was classified in the family Treponemataceae and has an estimated 447 KOs identified, some of which, such as the multi-drug resistance transporters, could enable microbial survival and growth in a high toxin environment (Table S14). No MAGs were significantly enriched in creosote feeders compared to non-creosote feeders (DESeq2). In addition, there was large overlap of the identified functional profiles of MAGs within creosote and non-creosote feeders (Fig. 5B). However, 367 microbial genes were significantly more abundant in creosote feeders (Table S15). When blasted against the UniRef100 database, we found that these differentially abundant genes with an existing KO classification were associated with a wide range of functions with the majority coding for enzymes or cellular transporters, specifically ABC transporters (Table S15). None were identified as being involved in xenobiotic degradation.
Discussion
Diet heavily influences the mammalian gut microbiome [27, 64, 76, 118]. For mammalian herbivores especially, diet should exert strong selection on the gut microbial community for microbes that provide ecologically relevant functions across hosts, or core microbiota. Little investigation has been done on whether diet exerts this selection for shared microbial taxa and/or functions. Here, we addressed this gap in our knowledge by investigating the taxonomic and functional core microbiome in wild woodrats consuming the same toxic diet. We found core microbes unique to woodrats that consume creosote, and also unique core communities across different gut regions that may aid the host in subsisting on an herbivorous diet. We also identified a functional core unique to creosote feeding woodrats consisting of several KOs that may be involved in the degradation of PSCs.
Based on taxonomy, creosote feeding woodrats harbored distinct gut microbial communities and core microbes in the foregut, cecum, and hindgut. This is consistent with previous studies that also found that microbial communities differ between the midgut and hindgut in various herbivorous hosts [56, 65, 69, 91]. These gut regions carry out different functions within the host and these physiological differences present unique environments for microbes which likely shape the gut microbial community. The cecum, for instance, is a large fermentation chamber that houses high densities of microbes that ferment dietary fiber [63, 69]. Indeed, we found that the cecum had the highest diversity of microbes and the largest creosote-feeding core microbiome. In addition, most core microbes belonged to the families Lachnospiraceae and Oscillospiraceae, families known for fiber fermentation in the rumen [2] and previously identified as part of the ruminant core microbiome [46]. The large cecum core microbiome of woodrats resembling that of ruminant herbivores may indicate that these taxa are widespread across, and functionally important for, fiber degradation in many mammalian herbivores. Future work could investigate this pattern across even more diverse hosts that consume diets high in fiber, such as reptiles and birds, to determine if the same microbial taxa have been selected for, as previously hypothesized [25].
In contrast to the cecum, the foregut is a small, sacculated region thought to house microbes capable of using PSCs as substrates [67, 85]. The foregut had the largest number of core members belonging to the family Eggerthellaceae. Notably, several microbes that belong to Eggerthellaceae are capable of degrading PSCs [7, 9, 11, 43, 72]. These microbes may be metabolizing PSCs within the foregut prior to absorption in the small intestine. More core members were identified as Eggerthellaceae in the foregut than the cecum; however, this family was ubiquitously identified as part of the core microbiome across gut regions. Other core families in the foregut were either families commonly found in the woodrat gut microbiome (Lactobacillaceae, Muribaculaceae, Desulfovibrionaceae) or known fiber degraders (Lachnospiraceae, Oscillospiraceae). Retention time of food in the foregut is not long enough for extensive fermentation [69], but, as it precedes the cecum in the gastrointestinal tract, it is possible some fermentation of simple sugars and volatile fatty begins in this region [69].
The foregut microbiome was less diverse than the cecum, with the core microbiome consisting of ~ 16% as many ASVs as that of the cecum. This lower diversity may stem from higher concentrations of PSCs in undigested diet, which can reduce microbial growth [26], as well as a low pH (~ 4.5) which may make the foregut inhospitable to some gut microbiota [61]. Alternatively, the cecum may harbor a large, and taxonomically diverse core microbiome due to the diversity of fibers, waxes, and pectins found in a plant-based diet. Creosote contains 2–3 times more fiber than resin by dry mass, and different plant species produce various kinds of fibers which may require different microbes to break down [84]. The smaller, more taxonomically varied core in the foregut may indicate that the ability to degrade PSCs is not unique to a few microbial taxa, but is possibly a conserved function across microbial lineages.
The hindgut also harbored a smaller size core than the cecum i.e., about 24% that found in the cecum core. Many of these core microbes belong to families also identified as core in the cecum, such as Muribaculaceae, Oscillospiraceae, and Lachnospiraceae. It is possible that the hindgut core retained fewer members than the cecum because there is little selection from creosote resin once digesta has reached the colon as most of the resin components would be absorbed in the small intestine or fermented by cecal microbes. In addition, we used fecal samples to represent the hindgut and though the feces are often used as a proxy for the hindgut, these microbial communities can be similar or discordant, depending on the host and sampling methods [96, 112, 116]. Some of this variation may stem from the fact that fecal microbial communities may shift after defecation or become contaminated by the environment. In some mammalian species, using feces as a proxy for the hindgut obscured ecological and phylogenetic signals [52]. Though a previous study found that greater than 80% of microbes from woodrat feces collected aseptically were retained in feces collected from a trap [68], even this loss of native microbes may impact the number of shared core microbes found using fecal samples.
Sample size affected the number of core members identified. We collected far more fecal samples than cecum and foregut samples because fecal sample collection is non-invasive. This difference in sample size altered estimates of the core microbiome. When hindgut samples were reduced to the same sample sizes as the other regions, the number of core microbiota increased, though the hindgut microbial community still harbored a smaller core than the cecum. In addition, different families were identified as belonging to the core in the smaller dataset than in the full data set. This has important implications for core microbiome studies as the number of samples used could artificially inflate or shrink the number of microbes identified as core, and even miss taxa of ecological importance. Differential sequencing depth has a similar effect where analyses at different depths identify different core microbiota within different taxonomic groups [95]. While sample size can be difficult to keep consistent in studies of the microbial communities of wild organisms, we caution researchers to take this into consideration when designing core microbiome studies or comparing across studies.
Core microbes were more abundant than other ASVs, enriched in creosote feeding woodrats, and occurred more frequently than would be predicted by chance. While the core microbes did not make up a majority of the gut microbiome, they were more abundant than other ASVs and did represent a cumulative relative abundance of > 15% in the foregut and the cecum. This finding is consistent with other studies that classified their core microbiome at the ASV level [1, 104]. In addition, several core microbes were enriched in creosote feeders and identified as overrepresented by neutral models. These overrepresented core members were taxonomically widespread, indicating that these taxa may be beneficial and are potentially selected for within a host [17]. Indeed, several of these enriched or selected for core microbes belonged to families that can provide useful functions to an herbivore such as fiber fermentation (Ruminococcaceae) or degradation of PSCs (Eggerthellaceae). Taken together, these results suggest that the selected for core members are keystone taxa [6]. Our samples were collected from a large number of geographically distant populations and two different species, factors which significantly influence the community composition of the gut microbial community [114]. Despite samples coming from two species and populations occurring over wide geographic distances (as far as ~ 700 km between creosote feeders), we identified a core microbiome only found in animals consuming creosote, further signifying the possible importance of these cecum and foregut microbes to their host. Experimental manipulation or sequencing of the genome of potential keystone taxa may further elucidate the roles these microbes play in the host gut microbiome.
Some KOs identified as belonging to the functional creosote-feeding core microbiome were related to the metabolism of PSCs. Three of the identified core KOs (4-hydroxybenzoate decarboxylase, benzoyl-CoA reductase subunit B, and 2-pyrone-4, 6-dicarboxylate lactonase) coded for enzymes that play important roles in metabolism of lignans and aromatic rings [12, 14, 48]. Creosote resin is composed of many phenolics, primarily NDGA [101], a lignan with aromatic rings. Therefore, these proteins in the core may play a role in degrading creosote PSCs in the woodrat gut. This finding is consistent with previous work, and warrants further investigation into the particular microbial taxa and pathways that could metabolize creosote resin. We also identified several KOs coding for ABC transporters that were significantly more abundant in the gut microbial communities of creosote feeders compared to non-creosote feeders. Microorganisms use ABC transporters to efflux toxins, including PSCs, to protect the cell from detrimental effects [35, 119]. Thus, microbes in the creosote-feeding gut microbial community may be using transporters to efflux creosote toxins out of the cell. Indeed, the only core MAG had several KOs related to multi-drug resistance, which are usually transporter genes.
When characterizing the functional profile of the gut microbiome of woodrats, nearly half of the metagenomic data was not assigned to a functional pathway, severely limiting our ability to detect a functional core. It is possible that there exists a larger functional core microbiome in creosote feeding woodrats than we were able to observe due to these pathways being largely uncharacterized. One of the major advantages of metagenomic sequencing is that, in addition to taxonomic information, it provides information on the genomic content of microbes found in a region of interest. Indeed, the main goal of most studies utilizing metagenomic sequencing, including ours, is to characterize the metabolic capacity of microbial communities. Metagenomics is often touted as providing more in-depth and accurate results in comparison to 16S rRNA sequencing and this has been demonstrated in well studied systems, such as the human microbiome where microbial functions have been established [34, 47, 59, 75]. However, poorly studied hosts living in natural environments, much of the genetic information recovered cannot be classified with currently available databases, as was the case in this study and many others [38, 49, 97, 107, 117]. This highlights the need for the sequencing and incorporation of more diverse, wild systems in order to access the wealth of novel, microbial functions that currently remain unknown.
In both unassembled reads and assembled MAGs, we identified a creosote-feeding functional core microbiome, however, a majority of the xenobiotic degradation KOs were shared between creosote and non-creosote feeding woodrats. This result could indicate that enzymes capable of xenobiotic degradation are more pervasive across the woodrat gut microbiota than predicted. These enzymes could be conserved across gut microbiomes as they often perform other essential services such as nutrient synthesis. In addition, while not all woodrats consume creosote bush, all woodrats subsist on natural diets that contain PSCs [67]. Therefore, although they are consuming different plant species, these animals may be exposed to similar suites of PSCs. The woodrats in the non-creosote populations feed on plants from the families Krameriaceae, Fagaceae, Polygonaceae, Ephedraceae, and Salicaceae, all of which can produce phenolics-the predominant class of PSCs in creosote resin [4, 5, 51, 54, 60, 89, 102]. Eating a diet that contains any phenolics may select for similar microbial functions, regardless of phenolic structure or abundance. Further evidence for this notion is that, using our neutral models, we did not see increased selection of the microbiota of creosote feeders compared to that of non-creosote feeders. This result may indicate that all woodrat diets exert selective pressure on gut microbiota for microbes that utilize the particular resources ingested by the host [114]. Also, the functional pathways involved in the degradation of PSCs are not well understood. It is possible that many of the same microbial proteins or protein families are involved in metabolizing diverse arrays of PSCs, making them more universally prevalent in hosts consuming toxic diets [73]. Much work has been done to investigate the effect of diet on the structure, composition, and functional profile of the microbiome in domestic herbivores [23, 24, 41]. Comparatively little work has been done on the interplay of PSCs and gut microbial communities, despite the fact that nearly all plants defend themselves with a wide array of these toxins [29, 115]. Future work could focus on how these toxic compounds affect the microbiome and which microbial enzymes and functional pathways are involved in this process.
In conclusion, this work presents a detailed characterization of the unique taxonomic and functional core microbiome of herbivorous mammals feeding on the same toxic diet. Our work demonstrates that there are core microbes and microbial functions found only in populations of woodrats consuming this toxic diet that are not present in other woodrat populations of the same species that consume different diets. In addition, our work advances our knowledge of which microbes and microbial functions may be involved in degradation of these naturally occurring PSCs present in all herbivore diets. Most of the metagenomic data was not able to be classified, suggesting that much remains unknown about the functional profile of the gut microbiome of wild herbivores and highlights the need for further studies. Investigating functional pathways and microbes capable of breaking down these PSCs may better our understanding of the importance of the gut microbiota to an herbivorous host.
Availability of data and materials
Raw sequence data associated with this project are accessible through the NCBI Sequence Read Archive under BioProject PRJNA875083. Code for analyses conducted for this study can be found at the following: https://github.com/tess-stapleton/neotoma-creosote-core.
References
Ainsworth TD, Krause L, Bridge T, Torda G, Raina J-B, Zakrzewski M, et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 2015;9:2261–74. https://doi.org/10.1038/ismej.2015.39.
Anderson CL, Fernando SC. Insights into rumen microbial biosynthetic gene cluster diversity through genome-resolved metagenomics. Commun Biol. 2021;4(1):1–12. https://doi.org/10.1038/s42003-021-02331-7.
Arteaga S, Andrade-Cetto A, Cárdenas R. Larrea tridentata (Creosote bush), an abundant plant of Mexican and US-American deserts and its metabolite nordihydroguaiaretic acid. J Ethnopharmacol. 2005;98(3):231–9.
Atsatt PR, Ingram T. Adaptation to oak and other fibrous, phenolic-rich foliage by a small mammal, Neotoma fuscipes. Oecologia. 1983;60(1):135–42. https://doi.org/10.1007/BF00379333.
Ayer W, Browne L, Kasitu G. Metabolites of Eriogonum umbellatum. Planta Med. 1990;56(03):336–336. https://doi.org/10.1055/s-2006-960977.
Banerjee S, Schlaeppi K, van der Heijden MGA. Keystone taxa as drivers of microbiome structure and functioning. Nat Rev Microbiol. 2018;16(9):567–76. https://doi.org/10.1038/s41579-018-0024-1.
Beltrán D, Romo-Vaquero M, Espín JC, Tomás-Barberán FA, Selma MV. Ellagibacter isourolithinifaciens gen. Nov., sp. Nov., a new member of the family Eggerthellaceae, isolated from human gut. Int J Syst Evol Microbiol. 2018;68(5):1707–12. https://doi.org/10.1099/IJSEM.0.002735/CITE/REFWORKS.
Berasategui A, Salem H, Paetz C, Santoro M, Gershenzon J, Kaltenpoth M, Schmidt A. Gut microbiota of the pine weevil degrades conifer diterpenes and increases insect fitness. Mol Ecol. 2017;26(15):4099–110. https://doi.org/10.1111/MEC.14186.
Bess EN, Bisanz JE, Yarza F, Bustion A, Rich BE, Li X, et al. Genetic basis for the cooperative bioactivation of plant lignans by Eggerthella lenta and other human gut bacteria. Nat Microbiol. 2020;5:56–66. https://doi.org/10.1038/s41564-019-0596-1.
Blyton MDJ, Soo RM, Whisson D, Marsh KJ, Pascoe J, Le Pla M, et al. Faecal inoculations alter the gastrointestinal microbiome and allow dietary expansion in a wild specialist herbivore, the koala. Anim Microbiome. 2019;1(1):1–18. https://doi.org/10.1186/S42523-019-0008-0.
Bode LM, Bunzel D, Huch M, Cho GS, Ruhland D, Bunzel M, et al. In vivo and in vitro metabolism of trans-resveratrol by human gut microbiota. Am J Clin Nutr. 2013;97(2):295–309. https://doi.org/10.3945/AJCN.112.049379.
Boll M, Fuchs G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur J Biochem. 1995;234:921–33.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7. https://doi.org/10.1038/s41587-019-0209-9.
Breinig S, Schiltz E, Fuchs G. Genes involved in anaerobic metabolism of phenol in the bacterium Thauera aromatica. J Bacteriol. 2000;182(20):5849. https://doi.org/10.1128/JB.182.20.5849-5863.2000.
Brucker R, Kolodkin-Gal I, Andersen SB, Kohl KD, Dearing MD. The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. (2016) https://doi.org/10.3389/fmicb.2016.01165
Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18(4):366–8. https://doi.org/10.1038/s41592-021-01101-x.
Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, Bohannan BJM. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2015;10(3):655–64. https://doi.org/10.1038/ismej.2015.142.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3. https://doi.org/10.1038/nmeth.3869.
Caporaso JG, Ackermann G, Apprill A, Bauer M, Berg-Lyons D, Betley J et al. Earth microbiome project: EMP 16S Illumina amplicon protocol. Protoc Io 2018; 1–7. https://doi.org/10.17504/protocols.io.nuudeww
Carleton MD. A survey of gross stomach morphology in New World Cricetinae (Rodentia, Muroidea), with comments on functional interpretations. Misc Publ Museum Zool Univ Mich. 1973;146:1–43.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk v2: memory friendly classification with the Genome Taxonomy Database. BioRxiv. 2022;39:105. https://doi.org/10.1101/2022.07.11.499641.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/BIOINFORMATICS/BTY560.
Cholewińska P, Czyż K, Nowakowski P, Wyrostek A. The microbiome of the digestive system of ruminants-a review. Anim Health Res Rev. 2020;21:3–14. https://doi.org/10.1017/S1466252319000069.
Cholewińska P, Górniak W, Wojnarowski K. Impact of selected environmental factors on microbiome of the digestive tract of ruminants. BMC Vet Res. 2021;17(1):1–10. https://doi.org/10.1186/S12917-021-02742-Y/FIGURES/1.
Colston TJ, Jackson CR. Microbiome evolution along divergent branches of the vertebrate tree of life: what is known and unknown. Mol Ecol. 2016;25(16):3776–800. https://doi.org/10.1111/MEC.13730.
Compean KL, Ynalvez RA. Antimicrobial activity of plant secondary metabolites: a review. Res J Med Plant. 2014;8(5):204–13. https://doi.org/10.3923/RJMP.2014.204.213.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–63. https://doi.org/10.1038/nature12820.
Deagle BE, Thomas AC, McInnes JC, et al. Counting with DNA in metabarcoding studies: How should we convert sequence reads to dietary data? Mol Ecol. 2019;28:391–406. https://doi.org/10.1111/mec.14734.
Dearing MD, Foley WJ, McLean S. The influence of plant secondary metabolites on the nutritional ecology of herbivorous terrestrial vertebrates. Annu Rev Ecol Evol Syst. 2005;36:169–89. https://doi.org/10.1146/ANNUREV.ECOLSYS.36.102003.152617.
Dearing MD, Weinstein SB. Metabolic enabling and detoxification by mammalian gut microbes. Annu Rev Microbiol. 2022;76:579–96. https://doi.org/10.1146/ANNUREV-MICRO-111121-085333.
Dearing MD, Orr TJ, Klure DM, Greenhalgh R, Weinstein SB, Stapleton TE, Yamada KYH, Nelson MD, Doolin ML, Nielsen DP, Matocq MD, Shapiro MD. Toxin tolerance across landscapes: ecological exposure not a prerequisite. Funct Ecol. 2022;36:2119–31.
Dill-Mcfarland KA, Weimer PJ, Pauli JN, Peery MZ, Suen G. Diet specialization selects for an unusual and simplified gut microbiota in two- and three-toed sloths. Environ Microbiol. 2016;18(5):1391–402. https://doi.org/10.1111/1462-2920.13022.
Dougal K, Harris PA, Edwards A, Pachebat JA, Blackmore TM, Worgan HJ, Newbold CJ. A comparison of the microbiome and the metabolome of different regions of the equine hindgut. FEMS Microbiol Ecol. 2012;82(3):642–52. https://doi.org/10.1111/J.1574-6941.2012.01441.X.
Durazzi F, Sala C, Castellani G, Manfreda G, Remondini D, De Cesare A. Comparison between 16S rRNA and shotgun sequencing data for the taxonomic characterization of the gut microbiota. Sci Rep. 2021;11(1):1–10. https://doi.org/10.1038/s41598-021-82726-y.
Fleißner A, Sopalla C, Weltring KM. An ATP-binding cassette multidrug-resistance transporter is necessary for tolerance of Gibberella pulicaris to phytoalexins and virulence on potato tubers. Mol Plant Microbe Interact. 2002;15(2):102–8. https://doi.org/10.1094/mpmi.2002.15.2.102.
Freeland WJ, Janzen DH. Strategies in herbivory by mammals: the role of plant secondary compounds. Am Nat. 1974;108(961):269–89. https://doi.org/10.1086/282907.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–2. https://doi.org/10.1093/BIOINFORMATICS/BTS565.
Gibson KM, Nguyen BN, Neumann LM, Miller M, Buss P, Daniels S, et al. Gut microbiome differences between wild and captive black rhinoceros—implications for rhino health. Sci Rep. 2019;9(1):1–11. https://doi.org/10.1038/s41598-019-43875-3.
Gomez-Arango LF, Barrett HL, McIntyre HD, Callaway LK, Morrison M, Nitert MD. Contributions of the maternal oral and gut microbiome to placental microbial colonization in overweight and obese pregnant women. Sci Rep. 2017;7(1):1–10. https://doi.org/10.1038/s41598-017-03066-4.
Graystock P, Rehan SM, McFrederick QS. Hunting for healthy microbiomes: determining the core microbiomes of Ceratina, Megalopta, and Apis bees and how they associate with microbes in bee collected pollen. Conserv Genet. 2017;18(3):701–11. https://doi.org/10.1007/s10592-017-0937-7.
Gruninger RJ, Ribeiro GO, Cameron A, McAllister TA. Invited review: application of meta-omics to understand the dynamic nature of the rumen microbiome and how it responds to diet in ruminants. Animal. 2019;13(9):1843–54. https://doi.org/10.1017/S1751731119000752.
Guzmán-Beltrán S, Rubio-Badillo MÁ, Juárez E, Hernández-Sánchez F, Torres M. Nordihydroguaiaretic acid (NDGA) and α-mangostin inhibit the growth of Mycobacterium tuberculosis by inducing autophagy. Int Immunopharmacol. 2016;31:149–57. https://doi.org/10.1016/j.intimp.2015.12.027.
Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341(6143):295–8. https://doi.org/10.1126/SCIENCE.1235872/SUPPL_FILE/1235872.HAISER.SM.PDF.
Hammer TJ, Bowers MD. Gut microbes may facilitate insect herbivory of chemically defended plants. Oecologia. 2015;179(1):1–14. https://doi.org/10.1007/S00442-015-3327-1.
Hammer TJ, Janzen DH, Hallwachs W, Jaffe SP, Fierer N. Caterpillars lack a resident gut microbiome. Proc Natl Acad Sci USA. 2017;114(36):9641–6. https://doi.org/10.1073/PNAS.1707186114/SUPPL_FILE/PNAS.1707186114.SAPP.PDF.
Henderson G, Cox F, Ganesh S, Jonker A, Young W, Janssen PH, et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015;5:14567. https://doi.org/10.1038/srep14567.
Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, et al. Evaluating the information content of shallow shotgun metagenomics. MSystems. 2018. https://doi.org/10.1128/msystems.00069-18.
Hobbs ME, Malashkevich V, Williams HJ, Xu C, Sauder JM, Burley SK, et al. Structure and catalytic mechanism of Lig I: insight into the amidohydrolase enzymes of cog3618 and lignin degradation. Biochemistry. 2012;51(16):3497. https://doi.org/10.1021/BI300307B.
Huang P, Zhang Y, Xiao K, Jiang F, Wang H, Tang D, et al. The chicken gut metagenome and the modulatory effects of plant-derived benzylisoquinoline alkaloids. Microbiome. 2018;6(1):1–17. https://doi.org/10.1186/S40168-018-0590-5/FIGURES/5.
Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, et al. MEGAN community edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLOS Comput Biol. 2016;12(6):e1004957. https://doi.org/10.1371/JOURNAL.PCBI.1004957.
Ibragic S, Sofić E. Chemical composition of various ephedra species. Bosn J Basic Med Sci. 2015;15(3):21–7. https://doi.org/10.17305/bjbms.2015.539.
Ingala MR, Simmons NB, Wultsch C, Krampis K, Speer KA, Perkins SL. Comparing microbiome sampling methods in a wild mammal: Fecal and intestinal samples record different signals of host ecology, evolution. Front Microbiol. 2018;9(MAY):803. https://doi.org/10.3389/FMICB.2018.00803/BIBTEX.
Itoh H, Tago K, Hayatsu M, Kikuchi Y. Detoxifying symbiosis: microbe-mediated detoxification of phytotoxins and pesticides in insects. Nat Prod Rep. 2018;35(5):434–54. https://doi.org/10.1039/C7NP00051K.
Jiménez-Estrada M, Velázquez-Contreras C, Garibay-Escobar A, Sierras-Canchola D, Lapizco-Vázquez R, Ortiz-Sandoval C, et al. In vitro antioxidant and antiproliferative activities of plants of the ethnopharmacopeia from northwest of Mexico. BMC Complement Altern Med. 2013;13(1):1–8. https://doi.org/10.1186/1472-6882-13-12.
John Wallace R, Sasson G, Garnsworthy PC, Tapio I, Gregson E, Bani P, et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci Adv. 2019;5(7):8391–4. https://doi.org/10.1126/SCIADV.AAV8391/SUPPL_FILE/AAV8391_SM.PDF.
Jones J, DiBattista JD, Stat M, Bunce M, Boyce MC, Fairclough DV, et al. The microbiome of the gastrointestinal tract of a range-shifting marine herbivorous fish. Front Microbiol. 2018;9:2000. https://doi.org/10.3389/FMICB.2018.02000/BIBTEX.
Jonsson V, Österlund T, Nerman O, Kristiansson E. Statistical evaluation of methods for identification of differentially abundant genes in comparative metagenomics. BMC Genomics. 2016;17(1):1–14. https://doi.org/10.1186/S12864-016-2386-Y/FIGURES/5.
Jorge F, Dheilly NM, Poulin R. Persistence of a core microbiome through the ontogeny of a multi-host parasite. Front Microbiol. 2020;11:954. https://doi.org/10.3389/FMICB.2020.00954/BIBTEX.
Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T, et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol. 2016;7(4):459. https://doi.org/10.3389/FMICB.2016.00459/BIBTEX.
Julkunen-Tiitto R. Phenolic constituents of Salix: A chemotaxonomic survey of further finnish species. Phytochemistry. 1989;28(8):2115–25. https://doi.org/10.1016/S0031-9422(00)97930-5.
Kamada N, Chen GY, Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14(7):685–90. https://doi.org/10.1038/ni.2608.
Kanehisa M, Sato Y, Kawashima M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022;31(1):47–53. https://doi.org/10.1002/PRO.4172.
Karasov WH, Martínez-del-Rio C. Physiological ecology: how animals process energy, nutrients, and toxins, vol. 45. Princeton: Princeton University Press; 2008. https://doi.org/10.5860/choice.45-4381.
Knight R. Dietary effects on human gut microbiome diversity. Br J Nutr. 2015;113:S1–5. https://doi.org/10.1017/S0007114514004127.
Kohl KD, Brun A, Magallanes M, Brinkerhoff J, Laspiur A, Acosta JC, et al. Gut microbial ecology of lizards: insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol Ecol. 2017;26(4):1175–89. https://doi.org/10.1111/MEC.13921.
Kohl KD, Dearing MD. Experience matters: prior exposure to plant toxins enhances diversity of gut microbes in herbivores. Ecol Lett. 2012;15(9):1008–15. https://doi.org/10.1111/j.1461-0248.2012.01822.x.
Kohl KD, Dearing MD. The woodrat gut microbiota as an experimental system for understanding microbial metabolism of dietary toxins. Front Microbiol. 2016;7:1165. https://doi.org/10.3389/fmicb.2016.01165.
Kohl KD, Luong K, Dearing MD. Validating the use of trap-collected feces for studying the gut microbiota of a small mammal (Neotoma lepida). J Mammal. 2015;96(1):90–3. https://doi.org/10.1093/jmammal/gyu008.
Kohl KD, Miller AW, Marvin JE, Mackie R, Dearing MD. Herbivorous rodents (Neotoma spp.) harbour abundant and active foregut microbiota. Environ Microbiol. 2014;16(9):2869–78. https://doi.org/10.1111/1462-2920.12376.
Kohl KD, Stengel A, Dearing MD. Inoculation of tannin-degrading bacteria into novel hosts increases performance on tannin-rich diets. Environ Microbiol. 2016;18(6):1720–9. https://doi.org/10.1111/1462-2920.12841.
Kohl KD, Weiss RB, Cox J, Dale C, Dearing MD. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol Lett. 2014;17(10):1238–46. https://doi.org/10.1111/ele.12329.
Koppel N, Bisanz JE, Pandelia ME, Turnbaugh PJ, Balskus EP. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. ELife. 2018;7:33953. https://doi.org/10.7554/eLife.33953.
Koppel N, Rekdal VM, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science. 2017;356(6344):1246–57. https://doi.org/10.1126/SCIENCE.AAG2770/ASSET/7F3BFA3D-4441-425F-A5E7-A8C2BB8E8AEF/ASSETS/GRAPHIC/356_AAG2770_F4.JPEG.
Kyselka J, Rabiej D, Dragoun M, Kreps F, Burčová Z, Němečková I, et al. Antioxidant and antimicrobial activity of linseed lignans and phenolic acids. Eur Food Res Technol. 2017;243(9):1633–44. https://doi.org/10.1007/S00217-017-2871-9.
Laudadio I, Fulci V, Palone F, Stronati L, Cucchiara S, Carissimi C. Quantitative assessment of shotgun metagenomics and 16S rDNA amplicon sequencing in the study of human gut microbiome. OMICS. 2018;22(4):248–54. https://doi.org/10.1089/omi.2018.0013.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, et al. Beltrán, Romo-VaquerBeltrán. Romo-Vaquer Sci. 2008;320(5883):1647–51. https://doi.org/10.1126/SCIENCE.1155725/SUPPL_FILE/LEY.SOM.PDF.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2009;25(14):1754–60. https://doi.org/10.1093/BIOINFORMATICS/BTP324.
Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017;550(7674):61–6. https://doi.org/10.1038/nature23889.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Malenke JR, Skopec MM, Dearing MD. Evidence for functional convergence in genes upregulated by herbivores ingesting plant secondary compounds. BMC Ecol. 2014;14(1):1–16. https://doi.org/10.1186/1472-6785-14-23/TABLES/6.
Mangione AM, Dearing MD, Karasov WH. Creosote bush (Larrea tridentata) resin increases water demands and reduces energy availability in desert woodrats (Neotoma lepida). J Chem Ecol. 2004;30(7):1409–29. https://doi.org/10.1023/B:JOEC.0000037748.19927.a1.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10. https://doi.org/10.14806/ej.17.1.200.
Mendes LW, Kuramae EE, Navarrete AA, Van Veen JA, Tsai SM. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014;8:1577–87. https://doi.org/10.1038/ismej.2014.17.
Meyer MW, Karasov WH. Antiherbivore chemistry of Larrea tridentata: effects on woodrat (Neotoma lepida) feeding and nutrition. Ecology. 1989;70(4):953–61. https://doi.org/10.2307/1941362.
Miller AW, Oakeson KF, Dale C, Dearing MD. Microbial community transplant results in increased and long-term oxalate degradation. Microb Ecol. 2016;72(2):470–8. https://doi.org/10.1007/s00248-016-0800-2.
Moran NA, Ochman H, Hammer TJ. Evolutionary and ecological consequences of gut microbial communities. Annu Rev Ecol Evol Syst. 2019;50(1):451–75. https://doi.org/10.1146/ANNUREV-ECOLSYS-110617-062453.
Naqib A, Poggi S, Wang W, Hyde M, Kunstman K, Green SJ. Making and sequencing heavily multiplexed, high-throughput 16S ribosomal RNA gene amplicon libraries using a flexible, two-stage PCR protocol. Methods Mol Biol. 2018;1783:149–69. https://doi.org/10.1007/978-1-4939-7834-2_7/FIGURES/4.
Neu AT, Allen EE, Roy K. Defining and quantifying the core microbiome: challenges and prospects. Proc Natl Acad Sci. 2021;118(51):e2104429118. https://doi.org/10.1073/PNAS.2104429118/-/DCSUPPLEMENTAL.
Ochmian I, Oszmiański J, Skupień K. Chemical composition, phenolics, and firmness of small black fruits. J Appl Bot Food Qual. 2009;83(1):64–9.
Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11(12):2864–8. https://doi.org/10.1038/ismej.2017.126.
Pardesi B, Roberton AM, Lee KC, Angert ER, Rosendale DI, Boycheva S, et al. Distinct microbiota composition and fermentation products indicate functional compartmentalization in the hindgut of a marine herbivorous fish. Mol Ecol. 2022;00:1–16. https://doi.org/10.1111/MEC.16394.
Patton A, Huckaby JL, Álvarez-Castañeda DG, Ticul S. The evolutionary history and a systematic revision of woodrats of the Neotoma lepida group. Berkley: University of California Press; 2014.
Pertea M, Pertea G. GFF utilities: GffRead and GffCompare. F1000 Res. 2020;9:304. https://doi.org/10.12688/F1000RESEARCH.23297.1.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–6. https://doi.org/10.1093/nar/gks1219.
Ramakodi MP. Effect of amplicon sequencing depth in environmental microbiome research. Curr Microbiol. 2021;78(3):1026–33. https://doi.org/10.1007/S00284-021-02345-8.
Reyman M, van Houten MA, Arp K, Sanders EAM, Bogaert D. Rectal swabs are a reliable proxy for faecal samples in infant gut microbiota research based on 16S-rRNA sequencing. Sci Rep. 2019;9(1):1–8. https://doi.org/10.1038/s41598-019-52549-z.
Riiser ES, Haverkamp THA, Varadharajan S, Borgan Ø, Jakobsen KS, Jentoft S, Star B. Switching on the light: using metagenomic shotgun sequencing to characterize the intestinal microbiome of Atlantic cod. Environ Microbiol. 2019;21(7):2576–94. https://doi.org/10.1111/1462-2920.14652.
Ríos JM, Mangione AM, Gianello JC. Effects of natural phenolic compounds from a desert dominant shrub Larrea divaricata Cav. on toxicity and survival in mice. Revis Chilena de Historia Nat. 2008;81(2):292–302. https://doi.org/10.4067/S0716-078X2008000200011.
Risely A. Applying the core microbiome to understand host–microbe systems. J Anim Ecol. 2020;89:1549–58. https://doi.org/10.1111/1365-2656.13229.
Risely A, Gillingham MAF, Béchet A, Brändel S, Heni AC, Heurich M, et al. Phylogeny- and abundance-based metrics allow for the consistent comparison of core gut microbiome diversity indices across host species. Front Microbiol. 2021;12:659918. https://doi.org/10.3389/FMICB.2021.659918/FULL.
Schmutz EM, Mabry TJ, Hunziker JH, Difeo DR. Creosote bush. Biology and chemistry of Larrea in new world deserts. J Range Manag. 1978;31(5):399. https://doi.org/10.2307/3897371.
Schofield JA, Hagerman AE, Harold A. Loss of tannins and other phenolics from willow leaf litter. J Chem Ecol. 1998;24(8):1409–21. https://doi.org/10.1023/A:1021287018787.
Shade A, Handelsman J. Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol. 2012;14(1):4–12. https://doi.org/10.1111/J.1462-2920.2011.02585.X.
Simonin M, Dasilva C, Terzi V, Ngonkeu ELM, Diouf D, Kane A, et al. Influence of plant genotype and soil on the wheat rhizosphere microbiome: evidences for a core microbiome across eight African and European soils. FEMS Microbiol Ecol. 2020;96(6):fiaa067. https://doi.org/10.1093/FEMSEC/FIAA067.
Stapleton TE, Kohl KD, Denise Dearing M. Plant secondary compound- and antibiotic-induced community disturbances improve the establishment of foreign gut microbiota. FEMS Microbiol Ecol. 2022;98(1):1–13. https://doi.org/10.1093/FEMSEC/FIAC005.
Stapleton TE, Weinstein SB, Greenhalgh R, Dearing MD. Successes and limitations of quantitative diet metabarcoding in a small, herbivorous mammal. Mol Ecol Resour. 2022. https://doi.org/10.1111/1755-0998.13643.
Stewart RD, Auffret MD, Warr A, Wiser AH, Press MO, Langford KW, et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat Commun. 2018;9(1):1–11. https://doi.org/10.1038/s41467-018-03317-6.
Sullam KE, Rubin BER, Dalton CM, Kilham SS, Flecker AS, Russell JA. Divergence across diet, time and populations rules out parallel evolution in the gut microbiomes of Trinidadian guppies. ISME J. 2015;9(7):1508–22. https://doi.org/10.1038/ismej.2014.231.
Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31(6):926–32. https://doi.org/10.1093/BIOINFORMATICS/BTU739.
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A, et al. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007;35(3):e14. https://doi.org/10.1093/NAR/GKL938.
Tamames J, Cobo-Simón M, Puente-Sánchez F. Assessing the performance of different approaches for functional and taxonomic annotation of metagenomes. BMC Genomics. 2019;20(1):1–16. https://doi.org/10.1186/S12864-019-6289-6/FIGURES/10.
Tang Q, Jin G, Wang G, Liu T, Liu X, Wang B, Cao H. Current sampling methods for gut microbiota: a call for more precise devices. Front Cell Infect Microbiol. 2020;10:151. https://doi.org/10.3389/FCIMB.2020.00151/BIBTEX.
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature. 2007;449(7164):804. https://doi.org/10.1038/NATURE06244.
Weinstein SB, Martinez-Mota R, Stapleton TE, Klure DM, Greenhalgh R, Orr TJ, et al. Microbiome stability and structure is governed by host phylogeny over diet and geography in woodrats (Neotoma spp.). Proc Natl Acad Sci USA. 2021;118(47):2108787118. https://doi.org/10.1073/pnas.2108787118.
Wink M. Plant secondary metabolism: diversity, function and its evolution. Nat Prod Commun. 2008;3:1205–16. https://doi.org/10.1177/1934578x0800300801.
Yan W, Sun C, Zheng J, Wen C, Ji C, Zhang D, et al. Efficacy of fecal sampling as a gut proxy in the study of chicken gut microbiota. Front Microbiol. 2019;10:2126. https://doi.org/10.3389/FMICB.2019.02126/BIBTEX.
Youngblut ND, de la Cuesta-Zuluaga J, Reischer GH, Dauser S, Schuster N, Walzer C, et al. Large-scale metagenome assembly reveals novel animal-associated microbial genomes, biosynthetic gene clusters, and other genetic diversity. MSystems. 2020;5(6):10–1128. https://doi.org/10.1128/MSYSTEMS.01045-20.
Youngblut ND, Reischer GH, Walters W, Schuster N, Walzer C, Stalder G, et al. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat Commun. 2019;10(1):1–15. https://doi.org/10.1038/s41467-019-10191-3.
Zwiers LH, Stergiopoulos I, Gielkens MMC, Goodall SD, De Waard MA. ABC transporters of the wheat pathogen Mycosphaerella graminicola function as protectants against biotic and xenobiotic toxic compounds. Mol Genet Genomics. 2003;269(4):499–507. https://doi.org/10.1007/s00438-003-0855-x.
Acknowledgements
We thank Dr. Dylan Klure, KayLene Yamada, Lane Mulvey, Madeline Nelson, Dr. Sara Weinstein, Dr. Teri Orr, Dr. Jim Patton, Dr. Rodolfo Martínez-Mota, and Dr. Cecilia Noecker for their assistance in the field, collection of samples, and assistance with data analysis. We also thank Drs. Margaret Doolin, Phyllis Coley, Kevin Kohl, William Brazelton, June Round, and Colin Dale for their comments on the manuscript. We thank the Scientific Computing and Imaging Institute at the University of Utah for computing resources. The computing resources on Frontera were provided through an allocation by the Texas Advanced Computing Center PHY20033. We also thank Mission Creek Preserve and Ronald W. Casper’s Wilderness Park for housing in the field and collection of samples. We thank three anonymous reviewers for their helpful comments that improved this manuscript.
Funding
This project was funded by NSF DEB-1342615 and NSF IOS 1656497 to MDD and NSF Grants 1808652 and 2008772 to HS, and was also supported through the NSF IR/D program to MDD.
Author information
Authors and Affiliations
Contributions
TES conceptualized the project, conducted the fieldwork, conducted the wet bench work, conducted the 16S and trnL sequencing analysis, contributed to the shotgun metagenomic sequencing analysis, wrote the manuscript, and created all figures. LML conducted the shotgun metagenomic sequencing analysis and contributed to the manuscript. HS supplied funding to LML, MDD conceptualized the project, contributed to the writing of the manuscript, and supplied funding to the project.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal use and procedures were approved by the University of Utah IACUC (16-02011). Animal samples were collected under permits from California (SC-8123), Utah (1COLL5194-1, 2), and Nevada (333663) issued to MDD and California Department of Fish and Wildlife permit SC-2105 issued to Jim Patton.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Stapleton, T.E., Lindsey, L.M., Sundar, H. et al. Rodents consuming the same toxic diet harbor a unique functional core microbiome. anim microbiome 6, 43 (2024). https://doi.org/10.1186/s42523-024-00330-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-024-00330-7