
Abstract
Background
Mammalian lungs comprise a complex microbial ecosystem that interacts with host physiology. Previous research demonstrates that the environment significantly contributes to bacterial community structure in the upper and lower respiratory tract. However, the influence of host genetics on the makeup of lung microbiota remains ambiguous, largely due to technical difficulties related to sampling, as well as challenges inherent to investigating low biomass communities. Thus, innovative approaches are warranted to clarify host-microbe interactions in the mammalian lung.
Results
Here, we aimed to characterize host genomic regions associated with lung bacterial traits in an advanced intercross mouse line (AIL). By performing quantitative microbial profiling (QMP) using the highly precise method of droplet digital PCR (ddPCR), we refined 16S rRNA gene amplicon-based traits to identify and map candidate lung-resident taxa using a QTL mapping approach. In addition, the two abundant core taxa Lactobacillus and Pelomonas were chosen for independent microbial phenotyping using genus-specific primers. In total, this revealed seven significant loci involving eight bacterial traits. The narrow confidence intervals afforded by the AIL population allowed us to identify several promising candidate genes related to immune and inflammatory responses, cell apoptosis, DNA repair, and lung functioning and disease susceptibility. Interestingly, one genomic region associated with Lactobacillus abundance contains the well-known anti-inflammatory cytokine Il10, which we confirmed through the analysis of Il10 knockout mice.
Conclusions
Our study provides the first evidence for a role of host genetic variation contributing to variation in the lung microbiota. This was in large part made possible through the careful curation of 16S rRNA gene amplicon data and the incorporation of a QMP-based methods. This approach to evaluating the low biomass lung environment opens new avenues for advancing lung microbiome research using animal models.
Graphical Abstract

Similar content being viewed by others

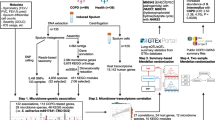

Background
Healthy lungs house a diverse and complex microbial ecosystem that contributes to critical aspects of host biology. Previous surveys of lung microbiota reveal microbial alterations in the context of disease, including cystic fibrosis [1], asthma [2], chronic obstructive pulmonary disease [3, 4], lung cancer [5, 6], as well as COVID-19 [7,8,9]. Thus, revealing the fundamental forces that govern the assembly and stability of bacterial communities in the lung is of critical importance for understanding its role in health and disease. Environmental factors, including smoking status [3, 10], infant feeding mode [11], early life stress [12], household [13, 14], and antibiotic use [15, 16] are identified as significant contributors to upper and lower respiratory tract bacterial community structure. However, there is a paucity of research exploring the influence of host genetics on the makeup of lung microbiota.
The role of host genetics on bacterial community structure at other body sites has been demonstrated using twin designs [17], comparison of mouse inbred strains [18, 19], genome-wide association studies [20,21,22,23,24], and quantitative trait locus (QTL) analyses [25, 26]. These studies are largely limited to the gut, although researchers are beginning to successfully apply these methods to low-biomass communities. For example, we previously used a novel QTL mapping approach in a murine skin microbiota study whereby we extended bacterial trait mapping to both 16S rRNA gene copy (DNA) as well as 16S rRNA gene transcripts (RNA) [27]. Microbial profiling based on RNA as template preferentially reflects living/active cells, and accordingly increased the number of significant associations detected between the host and resident skin microbes [27].
Microbiome research on the human lung environment still lags, largely due to technical sampling challenges unique to this site [28,29,30,31,32]. Bronchoalveolar lavage (BAL), the established best practice for sampling human lung microbiota, is invasive, costly, requires sedation, and poses unnecessary risks to healthy subjects [31]. These barriers impede sampling large numbers of individuals, which is required to detect biological signals with methods such as GWAS [31]. Non-invasive sampling strategies, such as sputum or tracheal aspirate collection, are more accessible, but complicated by poorly defined methods for sputa processing, ambiguous origins of collected microbiota, and a substantial risk of contamination from the oropharynx [31, 33]. Moreover, the low biomass of bacterial samples collected from the lower airways poses further challenges for handling contamination. The so-called “kitome” of nucleic acid extraction kits and reagents, as well as laboratory environments, are well-documented sources of contamination that can radically affect data interpretation, as contaminants tend to be preferentially amplified and sequenced over true microbial signal in low biomass samples [34,35,36,37,38,39,40]. Thus, novel approaches that can be readily translated to human research are needed to advance our understanding of dynamic host-microbe interactions in the mammalian lung.
In this study, we aimed to improve the experimental profiling of resident lung microbes for QTL mapping of the lung microbiota, using a mouse advanced intercross line (AIL) that was previously successful for genetic mapping in both the gut [41] and low biomass environment of the skin [27, 42]. For this, we employed a strategy to first screen for taxa that are likely to be true lung residents using 16S rRNA gene amplicon profiles at the transcript (RNA) level, followed by measurements of overall bacterial load and selected individual taxa using the highly precise method of droplet digital PCR (ddPCR) (Graphical abstract). QTL linkage mapping of lung microbiota using ddPCR-based estimates revealed significant associations with host loci, whose confidence intervals contain genes related to immune and inflammatory responses, cell apoptosis, and DNA repair. Further, a significant association between Lactobacillus abundance and a region of the mouse genome containing Il10, a well-known anti-inflammatory cytokine, was confirmed through the analysis of Il10 knockout mice. These data suggest that incorporating quantitative profiling from ddPCR bacterial load measurements for use in linkage mapping may improve study reliability, and thus open new avenues for advancing lung microbiome research.
Results
AIL mouse population and overall study design
We analyzed 242 lung tissue samples derived from the 15th generation of a previously established AIL population, as described by Belheouane et al. [27]. In brief, the AIL consisted of MRL/MpJ, NZM2410/J, BXD2/TyJ, and CAST/EiJ mice (Jackson Lab, Maine, USA). To create a heterogenous intercross line, mice were intercrossed in equal strain and sex distributions [27, 42].
To map genomic regions associated with bacterial traits in the murine lung, we carried out a nested strategy to identify and map candidate resident taxa while minimizing the influence of potential contamination (see Methods). First, we screened for putative bacterial lung residents by analyzing 16S rRNA gene amplicon profiles at the transcript (RNA) level, thereby preferentially identifying live/active taxa. These data were further curated through the application of the “decontam” R package [34], which incorporates information from negative controls and absolute quantification of bacterial load to identify possible contaminants in metagenomic sequencing data (see Methods). For this purpose, we applied ddPCR to obtain precise total bacterial load measurements. Next, a core measurable microbiota (CMM) was defined based on the processed sequences derived from RNA template for further analysis. In a second step, we measured the bacterial loads of individual candidate taxa using ddPCR. Finally, linkage mapping was performed on the following panel of traits: (1) CMM based on conventional relative abundance estimates, derived from RNA template (herein: CMM-RA), (2) CMM based on relative abundance corrected by quantitative microbiome profiling (QMP; i.e., ddPCR estimates of bacterial load), derived from RNA template (herein: CMM-QMP), (3) ddPCR estimates of candidate taxa derived from DNA template (herein: ddPCR-DNA), and (4) ddPCR estimates of candidate taxa derived from RNA template (herein: ddPCR-RNA).
16S rRNA gene sequencing and ddPCR to define bacterial traits
To identify resident candidate taxa from the lung, we first performed 16S rRNA gene amplicon sequencing on both DNA and RNA reverse transcribed into complementary DNA (cDNA) as template (see Methods). After sequence processing, we determined the DNA-based data to be of insufficient quality/quantity, similar to another report that attempted to sequence DNA from murine lung samples using a comparable PCR protocol ([43]; note other studies using a two-step PCR protocol/higher number cycles yielded different results, e.g. [44,45,46,47]). However, rather than adopting a two-step PCR protocol as in Barfod et al. [43], which may be more prone to amplifying contaminants in low biomass samples [48], we instead narrowed the 16S sequencing data analysis to the transcript (RNA) level. This reflects metabolically active cells [49, 50], and may better reveal true resident lung bacteria interacting with the host, as demonstrated in our QTL mapping study exploring gene-microbe interactions in the skin of this same AIL [27]. To assess for potential contamination, we employed the “frequency” method from the “decontam” R package (v.1.4.0) using a threshold of 0.1 (see Methods). This analysis resulted in the removal of potential contaminant amplicon sequence variants (ASVs), including the common contaminants Halomonas and Shewanella. In total, we analyzed 8,414,939 sequences, and after normalizing sequencing coverage to 4,000 sequences per sample, a total of 20,772 ASVs remained in the data set. We first analyzed murine lung bacterial community composition at the genus and ASV levels based on 16S rRNA gene profiles (Fig. 1, Additional file 1: Figure S1). The most abundant genera include Lactobacillus, uncl. Lachnospiraceae, and Pelomonas, with a mean relative abundance of 9.85, 8.79, and 7.50%, respectively (Additional file 2: Table S1). Lactobacillus and Pelomonas were additionally selected as bacterial candidate traits for targeted linkage mapping using ddPCR (see below).

Lung bacterial community composition of AIL mice. Samples are ordered according to bacterial load in both panels, with the taxonomy displayed for the ten most abundant genera, determined separately, for (A) QMP and (B) relative abundance data. The lowest 10% of the samples based on bacterial load is shown in the zoomed window of panel A, for which the three negative extraction controls (NEC) are the lowest
Next, we defined a CMM [25, 26] (see Methods) consisting of 58 taxa ranging from the genus- to phylum-level, as well as 13 ASVs. The number of CMM traits represents a small fraction of the total number of lung microbiota (e.g., 1.74% of genera and 0.06% of ASVs), but their abundances represent 55.16% and 22.38% of the respective taxa at these levels.
Because 16S rRNA gene amplicon profiling is prone to biases and errors related to relative abundance compositional analysis [51], we performed QMP using ddPCR-based bacterial load estimates at the RNA level to account for these concerns. 16S rRNA sequence-derived relative abundances of the CMM traits were then subsequently transformed into corrected, absolute quantitative abundances [51] (see Methods), i.e., CMM-QMP traits. Thereafter, the quantitative profiles of the 58 taxa and 13 ASVs were included as an independent set of bacterial traits.
Finally, as indicated above, we selected two candidate bacterial traits, Lactobacillus and Pelomonas, for further independent analysis, as they were the two most abundant classifiable genera and are frequently identified as lung residents [43, 52, 53]. Droplet digital PCR is ideal for quantifying low biomass samples, as the process of fractionating a sample into thousands of individual droplets, in which independent PCR reactions occur, allows for the amplification of even very low levels of target strains [4, 54,55,56]. This was demonstrated by e.g., Gobert et al., who effectively measured low levels of Lactobacillus in fecal samples using ddPCR [54]. We thus generated load estimates for Lactobacillus and Pelomonas using genus-specific primers adapted for ddPCR (see Methods). This was performed at both the DNA and RNA level, as (i) ddPCR is sensitive enough to allow for absolute quantification and (ii) taxon-specific primers are expected to be less prone to contaminating taxa than universal PCR primers. Importantly, these specific estimates are significantly correlated to those based on 16S rRNA gene sequencing for both Lactobacillus (ddPCR-RNA vs. CMM-RA: Spearman’s r = 0.5848, p < 2.2 × 10− 16; ddPCR-RNA vs. CMM-QMP: r = 0.7131, p < 2.2 × 10− 16) and Pelomonas (ddPCR-RNA vs. CMM-RA: r = 0.2519, p = 7.411 × 10− 05; ddPCR-RNA vs. CMM-QMP: r = 0.2916, p = 3.796 × 10− 06), whereby the correlation is stronger for the CMM-QMP traits.
Summary statistics and evaluation of sources of variation for lung bacterial traits
Prior to genetic mapping, it is important to determine whether (i) a given phenotype displays sufficient variation between individuals, and (ii) any residual variation remains after accounting for potential covariates such as cage, sex, or age. Summary statistics for each of the four categories of bacterial traits are provided in Additional file 2: Tables S1-3. This reveals considerable interindividual variation among traits. In particular, traits with large mean abundances, such as Lactobacillus and Pelomonas, display a large range, from 0 to 88.15% and 0-82.85% across the dataset for the CMM-RA measurements (Additional file 2: Tables S1-3).
Next, we evaluated the influence of host and environmental factors on bacterial trait variation in each of the four categories by constructing a mixed effects model that includes sex and age as fixed explanatory variables, cage as a random effect, and the bacterial trait values as responses (see Methods; Additional file 2: Table S4-6). These variables are associated with variation in trait values to varying degrees. For example, cage explains 0% of the total variance for Lactobacillus, but explains 28.83% of the total variance for Pelomonas among CMM-RA traits. However, importantly, the remaining residual variation in all four categories of traits accounts for the greatest proportion of total variance after accounting for sex, age, and cage effects. This suggests that other variables differing between individual mouse hosts, such as genotype, may contribute to variation in bacterial trait abundances.
QTL mapping of lung microbiota traits
To test for associations between resident lung bacteria and the host genome, we performed QTL mapping for the CMM-RA and CMM-QMP traits, as well as for the ddPCR-DNA and -RNA for Lactobacillus and Pelomonas. In total, this yielded seven significant (p ≤ 0.05) and six suggestive (p ≤ 0.1) host loci involving eight bacterial traits (CMM-RA: one significant, five suggestive; CMM-QMP: four significant, one suggestive; ddPCR-DNA/RNA: two significant), with narrow confidence intervals ranging from 0.08 to 3.59 Mb, with an average of 1.80 Mb (Table 1, Additional file 3: Table S7). The number of protein-coding genes within these confidence intervals ranged from one to 36. For example, two significant associations were detected among the CMM-QMP traits at the genus-level, including Pelomonas and Streptococcus at chromosomes 11 and 16, for which the confidence intervals contained one and eight genes, respectively. For ddPCR-DNA and -RNA, Lactobacillus and Pelomonas were each significantly (p < 0.05) associated with a single genomic locus (Fig. 2; Table 1).

Manhattan plots from linkage mapping of bacterial traits. From inner to outer circles: Enterobacteriales CMM-RA, Enterobacteriaceae CMM-RA, Deltaproteobacteria CMM-QMP, Pelomonas CMM-QMP, Streptococcus CMM-QMP, ASV8_Propionibacterium CMM-QMP, Pelomonas ddPCR-RNA, and Lactobacillus ddPCR-DNA. CMM-RA traits are shown in yellow, CMM-QMP in blue, and ddPCR-DNA/RNA in green. Peak SNPs are highlighted with red dots. LOD score on the y-axis indicates the -log p value of the association between a locus and a phenotypic trait

Overview and comparison of the Manhattan plots of Lactobacillus and Pelomonas traits from CMM-RA, CMM-QMP, and ddPCR-DNA and ddPCR-RNA, with each column dedicated to the chromosome where the QTL was detected; Lactobacillus ddPCR-DNA at chromosome 1, Pelomonas ddPCR-RNA at chromosome 4, and Pelomonas CMM-QMP at chromosome 11, from left to right. Solid black lines indicate significant thresholds and dashed lines indicate suggestive thresholds of each trait. Red vertical lines indicate confidence intervals of the QTL.
Overall, the phenotyping performed for the two abundant core taxa Lactobacillus and Pelomonas was based on four different methods, which differ according to (i) primers (universal vs. taxon-specific), (ii) relative abundance vs. quantitative estimates, and (iii) DNA vs. RNA. To compare these methods, we generated four respective QTL profile plots for each of the three significant associations involving these two taxa (Fig. 3). For the Lactobacillus association on chromosome 1, only the ddPCR-DNA measurements revealed a significant association, which suggests that this region of the genome associates only to their cell number rather than their activity. In contrast, the Pelomonas QTL on chromosome 4 involves only the ddPCR-RNA estimates. Lastly, the Pelomonas QTL on chromosome 11 involves only the CMM-QMP estimates. A similar peak structure is apparent for the CMM-RA estimate, but does not reach the significant or suggestive threshold. Thus, with regard to the three categories mentioned above, the involvement of quantitative- over relative abundance information appears to be most important for the detection of gene-microbe interactions, while differences between primers and/or cell number vs. activity may lead to discrepancies between phenotyping methods.
Analysis of candidate genomic regions
The narrow confidence intervals afforded by the AIL population allowed us to identify several promising candidate genes. In Table 2, we report a list of candidate genes from significant QTLs and their functions, summarized from experimental evidence. Most genes within the candidate regions are related to immune response, inflammatory response, cell apoptosis, and/or DNA repair. A number of genes are notable due to their role in lung functioning and disease susceptibility. For the QTL on chromosome 1 associated with variation in Lactobacillus ddPCR-DNA, the Mk2 (mitogen-activated protein kinase-activated protein kinase 2) and the Il10 (Interleukin 10) genes are the two closest genes in proximity to peak SNP UNC1677482. In humans, MK2 is a downstream product of the p38MAPK pathway acting as a pro-inflammatory kinase, and induces various signals such as cytokines in response to lipopolysaccharide (LPS)- and virus-induced infections [58,59,60,61]. MK2 is involved in transcriptional and post-transcriptional regulation of cytokine expression and was previously shown to affect the stability of Il10 transcript [59]. IL-10, a well-known anti-inflammatory cytokine, is e.g., also involved in mitigating disease severity in Mycobacterium tuberculosis infection [62]. For the Pelomonas (ddPCR-RNA) QTL on chromosome 4, the peak SNP lies within an intergenic region. However, several genes are found within the confidence interval, including Pou3f2 and Mms22l. POU family transcription factors were previously shown to be highly expressed in small-cell lung carcinoma (SCLC) cell lines and to contribute to neuroendorcrine differentiation in non-small cell lung carcinoma (NSCLC) cell lines[63]. MMS22L was shown to accelerate cancer cell growth in lung cancer cell lines [64]. In contrast to the other candidate genes, Klhl32 is poorly characterized. However, recent work by de Vries et al. [65] identified KLHL32 as a protein-coding gene that strongly associates with DNA methylation levels of a specific CpG-site (a cytosine base located adjacent to a guanine base) in patients with chronic obstructive pulmonary disease (COPD).
Evaluation of Lactobacillus in a Il10 knockout model
Given the promising association detected between Lactobacillus load (DNA) and a locus containing the well-known anti-inflammatory cytokine IL-10, we aimed to confirm this potential gene-microbe association in an Il10 knockout model using the same phenotyping method as for our mapping population. We accordingly performed ddPCR to quantify the Lactobacillus load at the DNA level using genus-specific primers in Il10+/+, Il10+/− and Il10−/− mice. Interestingly, we observe significant differences in Lactobacillus according to genotype, with Il10+/+ mice displaying higher abundances than Il10+/− and Il10−/− mice (Fig. 4). We confirmed that the loads were associated only with genotype and not with any other variables including sex, cage, age, and cross (Kruskal Wallis; p > 0.05).

Lactobacillus load according to Il10 genotype. Bacterial loads determined by ddPCR (DNA) were log10-transformed and compared using Kruskal-Wallis tests with Wilcoxon signed-rank post-hoc tests. P values were corrected for multiple testing according to Benjamini and Hochberg [108]. Error bars depict standard error. p < 0.01 **, p < 0.05 *, not significant “ns”
Discussion
In this study, we applied a rigorous combination of experimental- and data curation procedures that enabled us to carry out the first systematic assessment of host genetic effects on the mammalian lung microbiota. From the panel of traits defined for mapping, we identified seven significant and six suggestive host gene-microbial associations. Importantly, we found that incorporating quantitative bacterial load estimates in defining microbial phenotypes to be more effective in identifying gene-microbe associations than 16S rRNA gene amplicon profiles alone. While we identified only two significant associations among CMM-RA traits, five further significant associations were revealed after incorporating bacterial load information (i.e., CMM-QMP and ddPCR). Previous studies of the lung microbiota have identified two of the bacterial taxa involved, Lactobacillus [32, 53, 109] and Pelomonas [43], and these may represent key inhabitants of this host habitat. Moreover, we confirmed an association between Lactobacillus and a genomic region containing the Il10 gene in an independent Il10 knockout mouse model. This confirmation is of particular significance given the expected impact of environmental differences on the lung microbiota, as the Il10 mice were housed and analyzed in a completely separate facility with differences in food, bedding, caretakers, etc.
Lactobacillus are known to inhabit the mammalian lung and are generally regarded as probiotic bacteria [52]. Additionally, previous research showed Lactobacillus to modulate host immune responses and to reduce injury during lung infection [110,111,112]. Our targeted mapping approach revealed Lactobacillus load to be associated with a region containing the important candidate genes Il10 and Mk2. The functional relationship between Lactobacillus and these genes remains unclear, although a number of studies suggest a clinically relevant link. A previous study administered the Lactobacillus casei strain Shirota (LcS) to 2-week-old mice, which were then challenged with ovalbumin to induce allergic symptoms in the lungs [113]. This revealed modified immune response, including increased IL-10 levels, in mice administered LcS compared to controls. Although it seems that the effect is dependent on species or strain [114, 115], research continues to demonstrate a beneficial role of Lactobacillus in immunity. For example, nasal administration of L. casei aided lung recovery from S. pneumoniae infection [112], and oral administration of L. rhamnosus caused changes in cytokine levels that aided recovery from lung injury and inflammation induced by a synthetic analog of viral double stranded RNA, poly(I:C) [111]. Given this evidence, it is possible that Lactobacillus and/or its metabolites modulate the phosphorylation of p38MAPK, perhaps through bacterial byproducts or components of the bacterial cell wall [116, 117], which affect downstream signals that support lung function and recovery. Alternatively, Mk2 gene expression and subsequent IL-10 production may affect the growth or modify the abundance of Lactobacillus in the lung.
Likewise, Pelomonas was previously identified as a member of the murine lung bacterial community [43]. In humans, recent studies identified Pelomonas in the oropharynx of patients infected with SARS-CoV-2 virus [118] and also in breast cancer patients [119], with associations with multiple cytokines and immune genes. Yet, the role of Pelomonas in the mammalian lung remains largely unknown. A potential mechanism might be via \(\gamma\)-aminobutyric acid (GABA) found in the pulmonary neuroendocrine cells (PNEC) distributed along the alveolar airway epithelium [120]. GABA acts as a mediator in mucus production and airway smooth muscle toning and contraction [121,122,123]. Interestingly, Pelomonas was found to contribute to contraction frequency in hydra [124]. Although the potential interplay between Pelomonas and GABA in the lungs awaits experimental investigation, we speculate that this may contribute to crosstalk between the host and bacteria. Moreover, given its association with Pou3f2 and Mms22l, Pelomonas might play a role in susceptibility to lung cancer and other related lung diseases. Additionally, POU transcription factors are specifically expressed in small cell lung cancer (SCLC), contributing to accelerating cell growth, and POU3F2 was revealed to maintain the proneuronal/neuroendocrine phenotype of SCLC [125]. MMS22L was found to be over-expressed in clinical and esophageal cancers, playing a role in growth and survival of cancer cells [64]. This gene might impact the efficacy of DNA-damaging agents, as the knockout of the gene enhances cancer cell apoptosis [64]. Based on these observations, both Pou3f2 and Mms22l might serve as cancer therapy targets, which could be aided by the mechanistic understanding of a possible interaction with Pelomonas.
Other QTL intervals also include interesting genes associated with lung or respiratory tract development, functioning, and/or diseases that are potentially supported by the host-microbe interactions. Enterobacteriaceae and Enterobacteriales were found to be associated with QTL on chromosome 1 with a narrow confidence interval of 1.79 Mb. Within this interval, Chrnd and Chrng were previously shown to contribute to nicotine dependence [66], and Kcnj13 was shown to take part in smooth muscle morphogenesis and trachea development in the mouse respiratory tract [71]. Yin et al. [71] found that mice deficient in Kcnj13 developed shorter trachea due to loss of function of the potassium channel KCNJ13, which is critical during trachea tubulogenesis. Another candidate region on chromosome 16 is associated with variation in Deltaproteobacteria abundance. Within this interval, the core circadian clock gene Per2 was found near the peak SNP. Interestingly, members of the Per subfamily act as tumor suppressor genes in mice, and the downregulation and loss of PER2 is associated with tumor proliferation and metastasis, including NSCLC [72,73,74]. In contrast, increased PER2 expression inhibits cell growth and NSCLC growth [72, 73].
The success of our mapping study relied on our approach to mitigate contamination and the employment of quantitative microbial profiling. Studies comparing ddPCR and qPCR find that while qPCR has a lower detection limit, it yields a worse signal-to-noise ratio when tested with negative controls [56], and yields inconsistent results when tested against intentionally contaminated samples diluted to replicate low-biomass conditions [126]. Further, Taylor et al. [127] note that ddPCR is also particularly advantageous for studies requiring long sample processing times, e.g. where a study cohort might be constructed slowly over time, as the method is robust to batch effects.
In our study, we find that incorporating ddPCR-based quantitative profiling improved the detection of gene-microbe interactions. In particular, it is known that QTL mapping significance thresholds are influenced by the underlying phenotype distribution [128]. Our significance thresholds were assigned based on random and repetitive shuffling of microbial phenotype values across genotypes. The probability of a given genotype/sample to be randomly assigned its original phenotype value is higher when interindividual variation is low, ultimately leading to higher significance thresholds. Notably, our QTL analysis using CMM-QMP phenotypes were assigned lower QTL significance thresholds compared to the QTL analysis using CMM-RA phenotypes (see e.g., Pelomonas CMM-RA vs. CMM-QMP QTL on chromosome 11, Fig. 3), as variation within the CMM-QMP is greater than that within the CMM-RA (Additional file 2: Tables S1-2). Thus, it is possible that the increase in interindividual variability among CMM-QMP traits, made possible through ddPCR measurements, better reflects the underlying biological distribution of the trait, which also contributes to lower significance thresholds.
The effectiveness of our mapping approach is congruent with previous employing absolute abundance profiling to characterize resident microbial communities [129,130,131,132,133,134]. Vandeputte et al. [51] found microbial load to be a key feature of Crohn’s increase disease, with disease being characterized by a reduced bacterial load rather than differences in abundance among disease-associated taxa per se. Similarly, Sibila et al. [135] identified an association between bacterial load and airway inflammation in patients with bronchiectasis. A subsequent clinical trial conducted using the same patient cohort revealed that those with a high bacterial load showed significant improvement after receiving antibiotic therapy in comparison to those with a low bacterial load [135].
Conclusions
In summary, by combining the unique resource of a high-resolution mouse genetic mapping population together with experimental and computational advances in studying low biomass microbial communities, we demonstrate a novel role for host genetic variation in shaping lung microbiota composition. We find several promising associations for the commonly identified lung taxa Lactobacillus and Pelomonas. Given that mouse models of human lung diseases, including asthma, chronic obstructive pulmonary disease, and pulmonary fibrosis, have served as powerful tools for understanding pathophysiology and identifying new drug targets [136], our results suggest that the functional relevance of these taxa that may be exploited for future preventative/therapeutic purposes. These approaches outlined here may find useful application in future experimental models of host-microbe interactions in the lung.
Methods
Animal husbandry
The analysis of G15 intercross mice was approved by the “Ministerium für Energiewende, Landwirtschaft, Umwelt und ländliche Räume des Landes Schleswig- Holstein” in Kiel, Germany (reference number: V 312– 72,241. 122–5 (12 − 2/09)). The G15 AIL mouse population was generated by intercrossing four strains, MRL/MpJ, NZM2410/J, BXD2/TyJ, and CAST/EiJ, with equal sex and strain distributions for 15 generations as previously described [27, 42]. The analysis of Il10 KO and wildtype C57BL/6 mice was performed according to approved animal protocols and institutional guidelines of the Max Planck Institute for Evolutionary Biology in Plön. Mice were maintained and handled in accordance with FELASA guidelines and German animal welfare law (Tierschutzgesetz § 11, permit from Veterinäramt Kreis Plön: 1401–144/PLÖ–004697).
Il10 KO and wildtype C57BL/6 mice (Jackson Laboratories, Maine, USA) were mated at age of 8–10 weeks to produce F1 mice, using both directions of the cross to reduce potential “grandmother,” or legacy effects [137, 138]. Heterozygous F1 mice were mated within each cross at 10 weeks and included 11 pairs from each cross. F2 mice were weaned at 3 weeks; males and females were housed in separate cages according to family, with mixed genotypes. Mice were maintained in individual ventilated cages (IVCs), type II long (Tecniplast®, Greenline) in a specific pathogen-free facility (MPI für Evolutionsbiologie, Plön, Germany) with a 12-h light/dark cycle. Decalcified water and food (1324, fortified, from Altromin) were provided ad libidum. An average of ten mice (range: 9–15) were selected from each sex of each genotype from each cross for tissue extraction at 17 weeks of age.
Sampling and nucleic acid extraction
The entire lower respiratory tract was dissected and preserved in RNALater (Thermo Fisher Scientific) at 4 °C overnight. Samples were stored at -20 °C after removing RNALater. Approximately the bottom half of the left lobe was obtained for nucleic acid extraction, using tools sterilized with 70% ethanol, RNase-Away (Thermo Fisher Scientific), and sterilizing beads (Fine Science Tools) heated to at least 120 °C. Lung tissues were first homogenized using Lysing Matrix E (MP Biomedicals) and nucleic acid extraction was conducted using the AllPrep 96 DNA/RNA kit (QIAGEN) with on-column DNase I treatment (QIAGEN), according to manufacturer’s protocol, with the exception of TCEP instead of β-mercaptoethanol for the lysis step. A total of 40µL of RNA was eluted by adding 20µL of RNase-free water twice; 30µL of EB buffer was added twice for DNA elution. Concentrations were measured using NanoDrop 1000 (Thermo Fisher Scientific), with RNA samples diluted to equal concentrations (200ng/µL). Reverse transcription was performed according to the manufacturer’s instructions using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) using 10µL of template RNA.
16S rRNA gene amplicon sequencing
The V1-V2 hypervariable region of the 16S rRNA gene was amplified using primers 27 F and 338R, barcoded with unique eight-based MIDs (multiplex identifiers), using a dual indexing approach on Illumina MiSeq platform for both cDNA and DNA template [27]. We chose the V1-V2 region of the 16S rRNA gene for multiple reasons. In particular, we made previous experience of this primer pair performing well in an earlier low biomass study of the same G15 mice [27], which also enables future cross-body site analyses. Three negative extraction controls (NEC) from each extraction plate and one microbial community DNA standard (20ng/µL) (ZymoBIOMICS) were included. Host reads were removed from DNA extracts using “Kneaddata” (v.0.6.1), which removes host reads based on the provided reference database, Mus musculus. However, the sequence qualities and read counts were not sufficient after this procedure, and we thus did not further proceed with these sequences generated from the extracted DNA (Additional file 4: Table S8). Sequences were processed using the “DADA2” package (v.1.14.1) [139] with the Ribosomal Database Project (RDP) training set 16 [140] for taxonomic classification, resulting in abundance tables of amplicon sequence variants (ASVs). Results were merged with metadata including sex, age, weight, and cage information, using “phyloseq” (v.1.30.0) [141] for further analysis in R (v.3.6.3).
ddPCR
A 20µL ddPCR master mix was prepared with QX200™ ddPCR™ EvaGreen® SuperMix (BioRad) following the manufacturer’s instructions (BioRad), with a final primer concentration of 120nM and with 10ng of nucleic acid template. PCR was performed on Bio-Rad C1000 Touch Thermal Cycler with the following conditions: 95 °C for 5 min, 40 cycles at 95 °C for 15 s and 60 °C for 1 min, 4 °C for 5 min, 90 °C for 5 min, and incubation at 10 °C. Final products were transferred to QX200™ Droplet Reader and quantified as gene copies (per 20µL) using Bio-Rad QuantaSoft (v.1.7.4.0917). Lactobacillus and Pelomonas loads were quantified using genus specific primers on 242 DNA and cDNA samples each (Table 3).
Contamination assessment and defining core measurable microbiota
To assess for potential contamination, total bacterial load estimates were measured by ddPCR, which together with negative extraction controls (n = 3, i.e., one per 96 well extraction plate) were used for the “frequency” method in the R package “decontam” (v.1.6.0) with a threshold of 0.1. This method identifies contaminants using a de novo classification method based on identifying a negative correlation between concentration (bacterial load) and the frequency of the putative contaminants appearing in samples - the lower the bacterial load, the higher the proportion/frequency of contaminant taxa is expected [34, 38]. ASVs identified as likely contaminants, including those belonging to Halomonas and Shewanella, were removed from the dataset prior to further analysis. After the decontamination process, samples were rarefied to even sampling depth of 4,000 reads per sample. CMM thresholds are study-specific and reflect the design, body site, and the aims of the study [142]. Here, using the cDNA template samples (RNA-level), we defined the CMM of resident lung microbiota using a 25% prevalence threshold together with a minimum relative abundance threshold of 1%. We reasoned that a 25% prevalence threshold cut-off was appropriate, as taxa below 20% prevalence can be limited in their statistical testability [143, 144] and because prevalence thresholds above 30% may be unnecessarily stringent for statistical reliability [144]. The final CMM among 242 samples included 13 ASVs and 58 taxa from the genus- to phylum-level (CMM-RA).
Quantitative microbiome profiling
We used ddPCR-based total bacterial load (RNA level) estimates for quantitative microbiome profiling (QMP), whereby 16S rRNA gene relative abundances were corrected using bacterial load measurements and transformed to “absolute” or quantitative abundances. For this, we used R function rarefy_even_sampling_depth (https://github.com/raeslab/QMP) [51], which rarefies samples to even sampling depth, defined as the ratio between sequencing depth and bacterial load (here, based on ddPCR). Among the QMP dataset (CMM-QMP), taxa and ASVs included in the CMM were further selected for mapping.
QTL mapping
Prior to mapping, summary statistics were performed on all traits including CMM-RA, CMM-QMP, and ddPCR-DNA/RNA in R studio (v.1.2.1335) with R (v.3.6.3) (Additional file 2: Tables S1-3). In order to perform a log10 transformation of relative abundance values, a value of 0.5 was added to the absolute abundances of all CMMs prior to converting the absolute abundances into relative abundances. Then, linear mixed effects analysis was performed on these traits using “lme4” (v.1.1–10) [145]. Variance was estimated using “r.squaredGLMM” in “MuMIn” (v.1.43.6) [146] and “VarCorr” in “lme4” for fixed and random effects, respectively.
Linkage mapping was performed using “DOQTL” (1.6.0) [147] and “QTLRel” (0.2.14) [148] in R (v3.2.2), whereby we fit an additive model that regresses the log10-transformed traits on the four founder haplotype contributions. Genotype data were previously collected and described in Belheouane et al. [27]. Briefly, the data was obtained by extracting DNA from liver tissue, which was processed using the MegaMuga (Illumina) array to obtain host genotypes. A kinship matrix was defined using “kinship.probs” in “DOQTL” to adjust for the kinship between animals [27]. A 3D-array of founder haplotype contributions (sample # x 4 founders x marker #) [27] and kinship matrix, along with sex and age as fixed variables and cage information as random variable, were included for linkage mapping.
Permutations were run in R (v.3.2.2) for each trait by shuffling the phenotypic data to define significance thresholds at both the 90th and 95th percentiles of LOD scores [27]. Permutations were run 10,000 times, a ten-fold increase from minimum recommendations by Gatti et al. [147]. QTL confidence intervals were defined at 1.5 LOD score drops on either side of the QTL peak. After QTL mapping, genes located within confidence intervals were examined and then plotted using “get.mgi.features” and “gene.plot” in “DOQTL” to identify potential candidate genes.
Data Availability
Sequencing data generated and analyzed is available in the NCBI Sequence Read Archive (SRA) under BioProject PRJNA856849 (https://dataview.ncbi.nlm.nih.gov/object/PRJNA856849?reviewer=3ko45p5hm7jq0b3bkp2vstedtc). Scripts used for the mapping as well as raw genotype and meta data are available at https://github.com/ceciliajchung/G15_lung.
Abbreviations
- AIL:
-
Advanced intercross line
- ASV:
-
Amplicon sequence variant
- BAL:
-
Bronchoalveolar lavage
- CMM:
-
Core measurable microbiota
- ddPCR:
-
Droplet digital PCR
- LOD:
-
Logarithm of odds
- PCR:
-
Polymerase chain reaction
- QMP:
-
Quantitative microbiome profiling
- qPCR:
-
quantitative PCR
- QTL:
-
Quantitative trait loci
- RA:
-
Relative abundance.
References
Coburn B, Wang PW, Caballero JD, Clark ST, Brahma V, Donaldson S, et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci Rep. 2015;5:10241.
Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS ONE. 2010;5(1):e8578.
Erb-Downward JR, Thompson DL, Han MK, Freeman CM, McCloskey L, Schmidt LA, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE. 2011;6(2):e16384.
Sze MA, Dimitriu PA, Hayashi S, Elliott WM, McDonough JE, Gosselink JV, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(10):1073–80.
Tsay J-CJ, Wu BG, Sulaiman I, Gershner K, Schluger R, Li Y, et al. Lower airway dysbiosis affects lung cancer progression. Cancer Discov. 2021;11(2):293–307.
Zheng L, Sun R, Zhu Y, Li Z, She X, Jian X, et al. Lung microbiome alterations in NSCLC patients. Sci Rep. 2021;11(1):1–11.
Fan J, Li X, Gao Y, Zhou J, Wang S, Huang B, et al. The lung tissue microbiota features of 20 deceased patients with COVID-19. J Infect. 2020;81(3):e64–e7.
Gaibani P, Viciani E, Bartoletti M, Lewis RE, Tonetti T, Lombardo D, et al. The lower respiratory tract microbiome of critically ill patients with COVID-19. Sci Rep. 2021;11(1):1–11.
Han Y, Jia Z, Shi J, Wang W, He K. The active lung microbiota landscape of COVID-19 patients. medRxiv. 2020.
Yu G, Gail MH, Consonni D, Carugno M, Humphrys M, Pesatori AC, et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 2016;17(1):1–12.
Biesbroek G, Bosch AA, Wang X, Keijser BJ, Veenhoven RH, Sanders EA, et al. The impact of breastfeeding on nasopharyngeal microbial communities in infants. Am J Respir Crit Care Med. 2014;190(3):298–308.
Schlosser-Brandenburg J, Ebner F, Klopfleisch R, Kühl AA, Zentek J, Pieper R et al. Influence of Nutrition and maternal bonding on postnatal Lung Development in the Newborn Pig. Front Immunol. 2021:3144.
Rylance J, Kankwatira A, Nelson DE, Toh E, Day RB, Lin H, et al. Household air pollution and the lung microbiome of healthy adults in Malawi: a cross-sectional study. BMC Microbiol. 2016;16(1):1–7.
Hosgood HD III, Sapkota AR, Rothman N, Rohan T, Hu W, Xu J, et al. The potential role of lung microbiota in lung cancer attributed to household coal burning exposures. Environ Mol Mutagen. 2014;55(8):643–51.
Barfod KK, Vrankx K, Mirsepasi-Lauridsen HC, Hansen JS, Hougaard KS, Larsen ST, et al. The murine lung microbiome changes during lung inflammation and intranasal vancomycin treatment. open Microbiol J. 2015;9:167.
Heirali AA, Workentine ML, Acosta N, Poonja A, Storey DG, Somayaji R, et al. The effects of inhaled aztreonam on the cystic fibrosis lung microbiome. Microbiome. 2017;5(1):51.
Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19(5):731–43.
Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, Wymore A, et al. Host genetic and environmental effects on mouse intestinal microbiota. ISME J. 2012;6(11):2033–44.
Kovacs A, Ben-Jacob N, Tayem H, Halperin E, Iraqi FA, Gophna U. Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb Ecol. 2011;61(2):423–8.
Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015;16(1):191.
Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48(11):1407–12.
Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, Gilad Y. Genome-wide association studies of the human gut microbiota. PLoS ONE. 2015;10(11):e0140301.
Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, Hov JR, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet. 2016;48(11):1396–406.
Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, Smith MI, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet. 2016;48(11):1413–7.
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proceedings of the National Academy of Sciences. 2010;107(44):18933-8.
Wang J, Kalyan S, Steck N, Turner LM, Harr B, Künzel S et al. Analysis of intestinal microbiota in hybrid house mice reveals evolutionary divergence in a vertebrate hologenome. Nat Commun. 2015;6.
Belheouane M, Gupta Y, Künzel S, Ibrahim S, Baines JF. Improved detection of gene-microbe interactions in the mouse skin microbiota using high-resolution QTL mapping of 16S rRNA transcripts. Microbiome. 2017;5(1):59.
Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184(8):957–63.
Goddard AF, Staudinger BJ, Dowd SE, Joshi-Datar A, Wolcott RD, Aitken ML et al. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proceedings of the National Academy of Sciences. 2012;109(34):13769-74.
Whiteson KL, Bailey B, Bergkessel M, Conrad D, Delhaes L, Felts B, et al. The upper respiratory tract as a microbial source for pulmonary infections in cystic fibrosis. Parallels from island biogeography. Am J Respir Crit Care Med. 2014;189(11):1309–15.
Carney SM, Clemente JC, Cox MJ, Dickson RP, Huang YJ, Kitsios GD, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62(3):283–99.
Baker JM, Hinkle KJ, McDonald RA, Brown CA, Falkowski NR, Huffnagle GB, et al. Whole lung tissue is the preferred sampling method for amplicon-based characterization of murine lung microbiota. Microbiome. 2021;9(1):1–14.
Man WH, de Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat Rev Microbiol. 2017;15(5):259–70.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6(1):226.
Drengenes C, Wiker HG, Kalananthan T, Nordeide E, Eagan TM, Nielsen R. Laboratory contamination in airway microbiome studies. BMC Microbiol. 2019;19(1):1–13.
Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 2019;27(2):105–17.
Jervis-Bardy J, Leong LE, Marri S, Smith RJ, Choo JM, Smith-Vaughan HC, et al. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome. 2015;3(1):19.
Karstens L, Asquith M, Davin S, Fair D, Gregory WT, Wolfe AJ et al. Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. MSystems. 2019;4(4).
Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12(1):87.
Weyrich LS, Farrer AG, Eisenhofer R, Arriola LA, Young J, Selway CA, et al. Laboratory contamination over time during low-biomass sample analysis. Mol Ecol Resour. 2019;19(4):982–96.
Vorobyev A, Gupta Y, Sezin T, Koga H, Bartsch YC, Belheouane M, et al. Gene-diet interactions associated with complex trait variation in an advanced intercross outbred mouse line. Nat Commun. 2019;10(1):1–15.
Srinivas G, Möller S, Wang J, Künzel S, Zillikens D, Baines JF et al. Genome-wide mapping of gene–microbiota interactions in susceptibility to autoimmune skin blistering. Nat Commun. 2013;4.
Barfod KK, Roggenbuck M, Hansen LH, Schjørring S, Larsen ST, Sørensen SJ, et al. The murine lung microbiome in relation to the intestinal and vaginal bacterial communities. BMC Microbiol. 2013;13(1):303.
Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016;1(10):1–9.
Dickson RP, Erb-Downward JR, Falkowski NR, Hunter EM, Ashley SL, Huffnagle GB. The lung microbiota of healthy mice are highly variable, cluster by environment, and reflect variation in baseline lung innate immunity. Am J Respir Crit Care Med. 2018;198(4):497–508.
Ashley SL, Sjoding MW, Popova AP, Cui TX, Hoostal MJ, Schmidt TM, et al. Lung and gut microbiota are altered by hyperoxia and contribute to oxygen-induced lung injury in mice. Sci Transl Med. 2020;12(556):eaau9959.
Lipinski JH, Falkowski NR, Huffnagle GB, Erb-Downward JR, Dickson RP, Moore BB, et al. Toll-like receptors, environmental caging, and lung dysbiosis. Am J Physiology-Lung Cell Mol Physiol. 2021;321(2):L404–L15.
Drengenes C, Eagan TM, Haaland I, Wiker HG, Nielsen R. Exploring protocol bias in airway microbiome studies: one versus two PCR steps and 16S rRNA gene region V3 V4 versus V4. BMC Genomics. 2021;22(1):1–15.
Männistö MK, Kurhela E, Tiirola M, Häggblom MM. Acidobacteria dominate the active bacterial communities of Arctic tundra with widely divergent winter-time snow accumulation and soil temperatures. FEMS Microbiol Ecol. 2013;84(1):47–59.
Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. Activity of abundant and rare bacteria in a coastal ocean. Proceedings of the National Academy of Sciences. 2011;108(31):12776-81.
Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017;551(7681):507–11.
Yildiz S, Lopes JPPB, Bergé M, González-Ruiz V, Baud D, Kloehn J, et al. Respiratory tissue-associated commensal bacteria offer therapeutic potential against pneumococcal colonization. Elife. 2020;9:e53581.
Singh N, Vats A, Sharma A, Arora A, Kumar A. The development of lower respiratory tract microbiome in mice. Microbiome. 2017;5(1):61.
Gobert G, Cotillard A, Fourmestraux C, Pruvost L, Miguet J, Boyer M. Droplet digital PCR improves absolute quantification of viable lactic acid bacteria in faecal samples. J Microbiol Methods. 2018;148:64–73.
Abellan-Schneyder I, Schusser AJ, Neuhaus K. ddPCR allows 16S rRNA gene amplicon sequencing of very small DNA amounts from low-biomass samples. BMC Microbiol. 2021;21(1):1–14.
Sze MA, Abbasi M, Hogg JC, Sin DD. A comparison between droplet digital and quantitative PCR in the analysis of bacterial 16S load in lung tissue samples from control and COPD GOLD 2. PLoS ONE. 2014;9(10).
Delroisse J-M, Boulvin A-L, Parmentier I, Dauphin RD, Vandenbol M, Portetelle D. Quantification of Bifidobacterium spp. and Lactobacillus spp. in rat fecal samples by real-time PCR. Microbiol Res. 2008;163(6):663–70.
Ehlting C, Ronkina N, Böhmer O, Albrecht U, Bode KA, Lang KS, et al. Distinct functions of the mitogen-activated protein kinase-activated protein (MAPKAP) kinases MK2 and MK3: MK2 mediates lipopolysaccharide-induced signal transducers and activators of transcription 3 (STAT3) activation by preventing negative regulatory effects of MK3. J Biol Chem. 2011;286(27):24113–24.
Ehlting C, Trilling M, Tiedje C, Le-Trilling VTK, Albrecht U, Kluge S, et al. MAPKAP kinase 2 regulates IL-10 expression and prevents formation of intrahepatic myeloid cell aggregates during cytomegalovirus infections. J Hepatol. 2016;64(2):380–9.
Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk H-D, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat Cell Biol. 1999;1(2):94–7.
Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, et al. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27(1):170–81.
Redford PS, Boonstra A, Read S, Pitt J, Graham C, Stavropoulos E, et al. Enhanced protection to Mycobacterium tuberculosis infection in IL-10‐deficient mice is accompanied by early and enhanced Th1 responses in the lung. Eur J Immunol. 2010;40(8):2200–10.
Ishii J, Sato H, Yazawa T, Shishido-Hara Y, Hiramatsu C, Nakatani Y, et al. Class III/IV POU transcription factors expressed in small cell lung cancer cells are involved in proneural/neuroendocrine differentiation. Pathol Int. 2014;64(9):415–22.
Nguyen MH, Ueda K, Nakamura Y, Daigo Y. Identification of a novel oncogene, MMS22L, involved in lung and esophageal carcinogenesis. Int J Oncol. 2012;41(4):1285–96.
de Vries M, Nedeljkovic I, van der Plaat DA, Zhernakova A, Lahousse L, Brusselle GG, et al. DNA methylation is associated with lung function in never smokers. Respir Res. 2019;20(1):268.
Saccone NL, Schwantes-An TH, Wang JC, Grucza RA, Breslau N, Hatsukami D et al. Multiple cholinergic nicotinic receptor genes affect nicotine dependence risk in African and European Americans. Genes, Brain and Behavior. 2010;9(7):741 – 50.
Vogt J, Harrison BJ, Spearman H, Cossins J, Vermeer S, ten Cate LN, et al. Mutation analysis of CHRNA1, CHRNB1, CHRND, and RAPSN genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet. 2008;82(1):222–7.
McMillin MJ, Below JE, Shively KM, Beck AE, Gildersleeve HI, Pinner J, et al. Mutations in ECEL1 cause distal arthrogryposis type 5D. Am J Hum Genet. 2013;92(1):150–6.
Zhu Z, Li J, Si J, Ma B, Shi H, Lv J et al. A large-scale genome-wide association analysis of lung function in the chinese population identifies novel loci and highlights shared genetic aetiology with obesity. Eur Respir J. 2021;58(4).
Villanueva S, Burgos J, López-Cayuqueo KI, Lai K-MV, Valenzuela DM, Cid LP, et al. Cleft palate, moderate lung developmental retardation and early postnatal lethality in mice deficient in the Kir7. 1 inwardly rectifying K + channel. PLoS ONE. 2015;10(9):e0139284.
Yin W, Kim H-T, Wang S, Gunawan F, Wang L, Kishimoto K, et al. The potassium channel KCNJ13 is essential for smooth muscle cytoskeletal organization during mouse tracheal tubulogenesis. Nat Commun. 2018;9(1):1–13.
Liu B, Xu K, Jiang Y, Li X. Aberrant expression of Per1, Per2 and Per3 and their prognostic relevance in non-small cell lung cancer. Int J Clin Exp Pathol. 2014;7(11):7863.
Xiang R, Cui Y, Wang Y, Xie T, Yang X, Wang Z, et al. Circadian clock gene Per2 downregulation in non–small cell lung cancer is associated with tumour progression and metastasis. Oncol Rep. 2018;40(5):3040–8.
Papagiannakopoulos T, Bauer MR, Davidson SM, Heimann M, Subbaraj L, Bhutkar A, et al. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metabol. 2016;24(2):324–31.
Song Y, Zhang W, Zhang J, You Z, Hu T, Shao G, et al. TWIST2 inhibits EMT and induces oxidative stress in lung cancer cells by regulating the FGF21-mediated AMPK/mTOR pathway. Exp Cell Res. 2021;405(1):112661.
Liu D, Xing HR, Liu Y, Sun Z, Ye T, Li J, et al. Asymmetric division gene Neurl2 mediates Twist2 regulation of self-renewal of mouse lewis lung cancer stem cells. J Cancer. 2019;10(15):3381.
Ding N, Liu D, Duan X, Zhang J, Ma S, Chen Y. Twist2 reduced NLRP3-Induced inflammation of Infantile Pneumonia via Regulation of mitochondrial permeability transition by FOXO1. Int Arch Allergy Immunol. 2022:1–16.
White MJ, Risse-Adams O, Goddard P, Contreras MG, Adams J, Hu D, et al. Novel genetic risk factors for asthma in african american children: Precision Medicine and the SAGE II study. Immunogenetics. 2016;68(6):391–400.
Ren Z, Liu J, Yao L, Li J, Qi Z, Li B. Glutamate receptor ionotropic, kainate 1 serves as a novel tumor suppressor of colorectal carcinoma and predicts clinical prognosis. Experimental and Therapeutic Medicine. 2020;20(6):1.
Nazarian A, Kulminski AM. Genome-wide analysis of sex disparities in the Genetic Architecture of Lung and colorectal cancers. Genes. 2021;12(5):686.
Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell. 2019;178(2):316–29. e18.
Jiang P, Li F, Liu Z, Hao S, Gao J, Li S. BTB and CNC homology 1 (Bach1) induces lung cancer stem cell phenotypes by stimulating CD44 expression. Respir Res. 2021;22(1):1–11.
Wiel C, Le Gal K, Ibrahim MX, Jahangir CA, Kashif M, Yao H, et al. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell. 2019;178(2):330–45. e22.
Chillappagari S, Garapati V, Mahavadi P, Naehrlich L, Schmeck BT, Schmitz ML, et al. Defective BACH1/HO-1 regulatory circuits in cystic fibrosis bronchial epithelial cells. J Cyst Fibros. 2021;20(1):140–8.
Niu L, Guo W, Song X, Song X, Xie L. Tumor-educated leukocytes mRNA as a diagnostic biomarker for non‐small cell lung cancer. Thorac cancer. 2021;12(6):737–45.
Guo M, Du Y, Gokey JJ, Ray S, Bell SM, Adam M, et al. Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat Commun. 2019;10(1):1–16.
Lu B, Su Y, Das S, Liu J, Xia J, Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129(2):371–83.
Lee Y, Yoon K-A, Joo J, Lee D, Bae K, Han J-Y, et al. Prognostic implications of genetic variants in advanced non-small cell lung cancer: a genome-wide association study. Carcinogenesis. 2013;34(2):307–13.
Turkowski K, Herzberg F, Günther S, Brunn D, Weigert A, Meister M, et al. Fibroblast growth Factor—14 Acts as Tumor suppressor in lung adenocarcinomas. Cells. 2020;9(8):1755.
Nan Y, Jin F, Yang S, Tian Y, Xie Y, Fu E, et al. Discovery of a set of biomarkers of human lung adenocarcinoma through cell-map proteomics and bioinformatics. Med Oncol. 2010;27(4):1398–406.
Brehm JM, Man Tse S, Croteau-Chonka DC, Forno E, Litonjua AA, Raby BA, et al. A genome-wide association study of post-bronchodilator lung function in children with asthma. Am J Respir Crit Care Med. 2015;192(5):634–7.
Wijk SC, Prabhala P, Michaliková B, Sommarin M, Doyle A, Lang S, et al. Human primary airway basal cells display a continuum of molecular phases from health to disease in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2021;65(1):103–13.
Polineni D, Dang H, Gallins PJ, Jones LC, Pace RG, Stonebraker JR, et al. Airway mucosal host defense is key to genomic regulation of cystic fibrosis lung disease severity. Am J Respir Crit Care Med. 2018;197(1):79–93.
Valette K, Li Z, Bon-Baret V, Chignon A, Bérubé J-C, Eslami A, et al. Prioritization of candidate causal genes for asthma in susceptibility loci derived from UK Biobank. Commun biology. 2021;4(1):1–15.
Wei W, Zhao X, Liu J, Zhang Z. Downregulation of LINC00665 suppresses the progression of lung adenocarcinoma via regulating miR-181c-5p/ZIC2 axis. Aging. 2021;13(13):17499.
Liu A, Xie H, Li R, Ren L, Yang B, Dai L, et al. Silencing ZIC2 abrogates tumorigenesis and anoikis resistance of non-small cell lung cancer cells by inhibiting Src/FAK signaling. Mol Therapy-Oncolytics. 2021;22:195–208.
Güre AO, Stockert E, Scanlan MJ, Keresztes RS, Jäger D, Altorki NK et al. Serological identification of embryonic neural proteins as highly immunogenic tumor antigens in small cell lung cancer. Proceedings of the National Academy of Sciences. 2000;97(8):4198 – 203.
Sabater L, Bataller L, Suárez-Calvet M, Saiz A, Dalmau J, Graus F. ZIC antibodies in paraneoplastic cerebellar degeneration and small cell lung cancer. J Neuroimmunol. 2008;201:163–5.
Liu B, Yang L, Huang B, Cheng M, Wang H, Li Y, et al. A functional copy-number variation in MAPKAPK2 predicts risk and prognosis of lung cancer. Am J Hum Genet. 2012;91(2):384–90.
Qian F, Deng J, Wang G, D Ye R, Christman W. Pivotal role of mitogen-activated protein kinase-activated protein kinase 2 in inflammatory pulmonary diseases. Curr Protein Pept Sci. 2016;17(4):332–42.
Turner J, Gonzalez-Juarrero M, Ellis DL, Basaraba RJ, Kipnis A, Orme IM, et al. In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J Immunol. 2002;169(11):6343–51.
Lyon H, Lange C, Lake S, Silverman EK, Randolph AG, Kwiatkowski D, et al. IL10 gene polymorphisms are associated with asthma phenotypes in children. Genetic Epidemiology: The Official Publication of the International Genetic Epidemiology Society. 2004;26(2):155–65.
Vahl JM, Friedrich J, Mittler S, Trump S, Heim L, Kachler K, et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br J Cancer. 2017;117(11):1644–55.
Wang R, Lu M, Chen H, Chen S, Luo X, Qin Y, et al. Increased IL-10 mRNA expression in tumor-associated macrophage correlated with late stage of lung cancer. J Experimental Clin Cancer Res. 2011;30(1):62.
Goodale BC, Rayack EJ, Stanton BA. Arsenic alters transcriptional responses to Pseudomonas aeruginosa infection and decreases antimicrobial defense of human airway epithelial cells. Toxicol Appl Pharmcol. 2017;331:154–63.
Richmond BW, Brucker RM, Han W, Du R-H, Zhang Y, Cheng D-S, et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat Commun. 2016;7(1):1–12.
Chaubey S, Nader YM, Shah D, Kumova OK, Prahaladan V, Carey AJ et al. α1, 3-Fucosyltransferase-IX, an enzyme of pulmonary endogenous lung stem cell marker SSEA-1, alleviates experimental bronchopulmonary dysplasia. Pediatr Res. 2020:1–10.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (Methodol). 1995;57(1):289–300.
Kostric M, Milger K, Krauss-Etschmann S, Engel M, Vestergaard G, Schloter M et al. Development of a stable lung microbiome in healthy neonatal mice. Microb Ecol. 2017:1–14.
Chiba E, Tomosada Y, Vizoso-Pinto MG, Salva S, Takahashi T, Tsukida K, et al. Immunobiotic Lactobacillus rhamnosus improves resistance of infant mice against respiratory syncytial virus infection. Int Immunopharmacol. 2013;17(2):373–82.
Villena J, Chiba E, Tomosada Y, Salva S, Marranzino G, Kitazawa H, et al. Orally administered Lactobacillus rhamnosus modulates the respiratory immune response triggered by the viral pathogen-associated molecular pattern poly (I: C). BMC Immunol. 2012;13(1):1–15.
Racedo S, Villena J, Medina M, Agüero G, Rodríguez V, Alvarez S. Lactobacillus casei administration reduces lung injuries in a Streptococcus pneumoniae infection in mice. Microbes Infect. 2006;8(9–10):2359–66.
Ezendam J, van Loveren H. Lactobacillus casei Shirota administered during lactation increases the duration of autoimmunity in rats and enhances lung inflammation in mice. Br J Nutr. 2008;99(1):83–90.
Forsythe P, Inman MD, Bienenstock J. Oral treatment with live Lactobacillus reuteri inhibits the allergic airway response in mice. Am J Respir Crit Care Med. 2007;175(6):561–9.
Tomosada Y, Chiba E, Zelaya H, Takahashi T, Tsukida K, Kitazawa H, et al. Nasally administered Lactobacillus rhamnosus strains differentially modulate respiratory antiviral immune responses and induce protection against respiratory syncytial virus infection. BMC Immunol. 2013;14(1):40.
Beisswenger C, Coyne CB, Shchepetov M, Weiser JN. Role of p38 MAP kinase and transforming growth factor-β signaling in transepithelial migration of invasive bacterial pathogens. J Biol Chem. 2007;282(39):28700–8.
Gräb J, Rybniker J. The expanding role of p38 mitogen-activated protein kinase in programmed host cell death. Microbiol insights. 2019;12:1178636119864594.
Wu Y, Cheng X, Jiang G, Tang H, Ming S, Tang L, et al. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. npj Biofilms and Microbiomes. 2021;7(1):1–9.
Tzeng A, Sangwan N, Jia M, Liu C-C, Keslar KS, Downs-Kelly E, et al. Human breast microbiome correlates with prognostic features and immunological signatures in breast cancer. Genome Med. 2021;13(1):1–17.
Yabumoto Y, Watanabe M, Ito Y, Maemura K, Otsuki Y, Nakamura Y, et al. Expression of GABAergic system in pulmonary neuroendocrine cells and airway epithelial cells in GAD67-GFP knock-in mice. Med Mol Morphol. 2008;41(1):20–7.
Barrios J, Kho AT, Aven L, Mitchel JA, Park J-A, Randell SH, et al. Pulmonary neuroendocrine cells secrete γ-aminobutyric acid to induce goblet cell hyperplasia in primate models. Am J Respir Cell Mol Biol. 2019;60(6):687–94.
Danielsson J, Zaidi S, Kim B, Funayama H, Yim PD, Xu D, et al. Airway epithelial cell release of GABA is regulated by protein kinase A. Lung. 2016;194(3):401–8.
Sui P, Wiesner DL, Xu J, Zhang Y, Lee J, Van Dyken S, et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science. 2018;360(6393):eaan8546.
Murillo-Rincon AP, Klimovich A, Pemöller E, Taubenheim J, Mortzfeld B, Augustin R, et al. Spontaneous body contractions are modulated by the microbiome of Hydra. Sci Rep. 2017;7(1):1–9.
Ishii J, Sato H, Sakaeda M, Shishido-Hara Y, Hiramatsu C, Kamma H, et al. POU domain transcription factor BRN 2 is crucial for expression of ASCL 1, ND 1 and neuroendocrine marker molecules and cell growth in small cell lung cancer. Pathol Int. 2013;63(3):158–68.
Taylor SC, Laperriere G, Germain H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: from variable nonsense to publication quality data. Sci Rep. 2017;7(1):1–8.
Taylor SC, Carbonneau J, Shelton DN, Boivin G. Optimization of Droplet Digital PCR from RNA and DNA extracts with direct comparison to RT-qPCR: clinical implications for quantification of Oseltamivir-resistant subpopulations. J Virol Methods. 2015;224:58–66.
Manichaikul A, Palmer AA, Sen S, Broman KW. Significance thresholds for quantitative trait locus mapping under selective genotyping. Genetics. 2007;177(3):1963–6.
Humphrey PT, Whiteman NK. Insect herbivory reshapes a native leaf microbiome. Nat Ecol Evol. 2020;4(2):221–9.
Nilsson AC, Björkman P, Welinder-Olsson C, Widell A, Persson K. Clinical severity of Mycoplasma pneumoniae (MP) infection is associated with bacterial load in oropharyngeal secretions but not with MP genotype. BMC Infect Dis. 2010;10(1):1–8.
Regalado J, Lundberg DS, Deusch O, Kersten S, Karasov T, Poersch K, et al. Combining whole-genome shotgun sequencing and rRNA gene amplicon analyses to improve detection of microbe–microbe interaction networks in plant leaves. ISME J. 2020;14(8):2116–30.
Rello J, Lisboa T, Lujan M, Gallego M, Kee C, Kay I, et al. Severity of pneumococcal pneumonia associated with genomic bacterial load. Chest. 2009;136(3):832–40.
Tkacz A, Hortala M, Poole PS. Absolute quantitation of microbiota abundance in environmental samples. Microbiome. 2018;6(1):1–13.
Stämmler F, Gläsner J, Hiergeist A, Holler E, Weber D, Oefner PJ, et al. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome. 2016;4(1):1–13.
Sibila O, Laserna E, Shoemark A, Keir HR, Finch S, Rodrigo-Troyano A, et al. Airway bacterial load and inhaled antibiotic response in bronchiectasis. Am J Respir Crit Care Med. 2019;200(1):33–41.
Rydell-Törmänen K, Johnson JR. The applicability of mouse models to the study of human disease. Mouse Cell Culture: Methods and Protocols. 2019:3–22.
Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, et al. Conducting a microbiome study. Cell. 2014;158(2):250–62.
Rausch P, Künzel S, Suwandi A, Grassl GA, Rosenstiel P, Baines JF. Multigenerational influences of the Fut2 gene on the dynamics of the gut microbiota in mice. Front Microbiol. 2017;8:991.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(D1):D633–D42.
McMurdie PJ, Holmes S. Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8(4):e61217.
Neu AT, Allen EE, Roy K. Defining and quantifying the core microbiome: Challenges and prospects. Proceedings of the National Academy of Sciences. 2021;118(51):e2104429118.
Cougoul A, Bailly X, Vourc’h G, Gasqui P. Rarity of microbial species: in search of reliable associations. PLoS ONE. 2019;14(3):e0200458.
Risely A, Gillingham MA, Béchet A, Brändel S, Heni AC, Heurich M et al. Phylogeny-and abundance-based metrics allow for the consistent comparison of core gut microbiome diversity indices across host species. Front Microbiol. 2021:1128.
Bates D, Maechler M, Bolker B, Walker S, Christensen RHB, Singmann H, et al. Package ‘lme4’. Vienna, Austria: CRAN R Foundation for Statistical Computing; 2012.
Bartoń K, MuMIn. Multi-Model Inference. 2015. R package version. 2015;1:17.
Gatti DM, Svenson KL, Shabalin A, Wu L-Y, Valdar W, Simecek P et al. Quantitative trait locus mapping methods for diversity outbred mice. G3: Genes, Genomes, Genetics. 2014;4(9):1623-33.
Cheng R, Abney M, Palmer AA, Skol AD. QTLRel: an R package for genome-wide association studies in which relatedness is a concern. BMC Genet. 2011;12(1):1–3.
Acknowledgements
We thank Sven Künzel for assistance in lab work and mouse breeding; Anastasia Vock and Anika Jonas for their help in the animal facility; Marie Vallier for helpful discussions; and Shauni Doms for assistance in bioinformatic analyses.
Funding
This work was supported by the German Science Foundation (DFG) grants EXC2167 “Precision Medicine in Chronic Inflammation (PMI)”, CRC 1182, and the Leibniz Science Campus Evolutionary Medicine of the Lung (EvoLUNG).
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
JFB and SI developed and designed the study and acquired funding. YG designed the pipeline for QTL mapping. CJC conducted the study experiments and was responsible for methodologies and investigations, data analyses and visualization, and software implementation. CJC, BMH, MB, and JFB contributed to formal data interpretation and study validation. Supervision was provided by JFB and MB. Original draft preparation was by CJC, BMH, and JFB with input from MB. All authors contributed to final review and edits.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The analysis of G15 intercross mice was approved by the “Ministerium für Energiewende, Landwirtschaft, Umwelt und ländliche Räume des Landes Schleswig- Holstein” in Kiel, Germany (reference number: V 312– 72241. 122–5 (12 − 2/09)). The analysis of Il10 KO and wildtype C57BL/6 mice was performed according to approved animal protocols and institutional guidelines of the Max Planck Institute for Evolutionary Biology in Plön. Mice were maintained and handled in accordance with FELASA guidelines and German animal welfare law (Tierschutzgesetz § 11, permit from Veterinäramt Kreis Plön: 1401–144/PLÖ–004697).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1: Figure S1.
Lung bacterial community composition of AIL mice at ASV level.
Additional file 2: Table S1-3.
Summary statistics of CMM-RA, CMM-QMP, and ddPCR-DNA/RNA traits, respectively. Table S4-6. Sources of variation for CMM-RA, CMM-QMP, and ddPCR-DNA/RNA traits, respectively.
Additional file 3: Table S7.
QTL mapping statistics for CMM-RA, CMM-QMP, and ddPCR-DNA/RNA with significant and suggestive associations.
Additional file 4: Table S8.
Average number of reads from each step of the DADA2 pipeline.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chung, C.J., Hermes, B.M., Gupta, Y. et al. Genome-wide mapping of gene-microbe interactions in the murine lung microbiota based on quantitative microbial profiling. anim microbiome 5, 31 (2023). https://doi.org/10.1186/s42523-023-00250-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-023-00250-y