
Abstract
Background
The microbiome of many insects consists of a diverse community of microorganisms that can play critical roles in the functioning and overall health of their hosts. Although the microbial communities of insects have been studied thoroughly over the past decade, little is still known about how biotic interactions affect the microbial community structure in and on the bodies of insects. In insects that are attacked by parasites or parasitoids, it can be expected that the microbiome of the host insect is affected by the presence of these parasitic organisms that develop in close association with their host. In this study, we used high-throughput amplicon sequencing targeting both bacteria and fungi to test the hypothesis that parasitism by the endoparasitoid Cotesia glomerata affected the microbiome of its host Pieris brassicae. Healthy and parasitized caterpillars were collected from both natural populations and a laboratory culture.
Results
Significant differences in bacterial community structure were found between field-collected caterpillars and laboratory-reared caterpillars, and between the external and the internal microbiome of the caterpillars. Parasitism significantly altered the internal microbiome of caterpillars, but not the external microbiome. The internal microbiome of all parasitized caterpillars and of the parasitoid larvae in the caterpillar hosts was dominated by a Wolbachia strain, which was completely absent in healthy caterpillars, suggesting that the strain was transferred to the caterpillars during oviposition by the parasitoids.
Conclusion
We conclude that biotic interactions such as parasitism have pronounced effects on the microbiome of an insect host and possibly affect interactions with higher-order insects.
Similar content being viewed by others


Introduction
Although the past decade has witnessed an enormous rise in studies characterizing the microbiome of insects [1, 2], little is still known about how microbial communities assemble in and on the bodies of insects. Although not necessarily true for all insects [3, 4], microorganisms can play a critical role in the fitness and overall health of insects [5,6,7,8] or provide protection against pathogens and support detoxification of pesticides or harmful plant secondary metabolites [9,10,11]. Gut microbial communities often deliver metabolic benefits to their hosts by the production of vitamins and providing digestive enzymes that improve nutrient uptake [1, 12]. The composition of insect gut microbial communities varies extensively between insect taxa and it is associated with the environment, diet, developmental stage, and phylogeny of the host [1, 13, 14]. Several hundreds of bacterial phylotypes have been described in termites [15, 16] and over a few tens in Lepidoptera [17, 18], while there is an almost complete absence of bacteria in aphid guts [19]. In contrast to bacteria, only very little is known about fungi, possibly because fungi are particularly associated with insects feeding on wood or detritus [1, 20]. Similarly, the external surfaces of animals, including the exoskeleton of insects, are commonly colonized by microorganisms [21]. Although only little is known about their ecological role, external microbes are assumed to play a major role in body odour [22, 23] and contribute to increased defence against predators and survival [24]. Besides, by altering body odours external microbes may also signal the presence of suitable prey or hosts [25, 26].
Recent studies that have compared the internal and external component of the insect microbiome revealed higher diversity of the external microbial community [27,28,29]. In general, insect guts are colonized by bacteria ingested with food that are able to survive and thrive in the gut [27, 30]. Furthermore, in most insects a substantial part of the internal microbiome consists of specialized gut symbionts that are obtained by vertical transmission, resulting in a gut microbiome consisting of several resident “core” microbiota [31]. In contrast, the external microbiome is often composed of microorganisms that commonly occur in the environment, and has been shown to vary significantly with geographic location and habitat [29], suggesting that local environmental conditions and local availability of microbes strongly determine the external microbial community on insects.
In insects that are attacked by parasites or parasitoids (i.e. insects whose larvae live as parasites in other insects and eventually kill their hosts), it can be expected that the microbiome of the host insect is to some extent affected by the presence of these parasitic organisms that develop in intimate association with their host. Furthermore, insect parasitoids harbour their own microbial communities, including symbionts, that may be transferred to the next generation and also affect the host microbiome [21, 32]. Parasites like helminths and protozoa residing in the insect gut have been shown to alter the composition of the gut microbiome, and may thereby strongly impact host immunity and gut homeostasis [33]. Likewise, larvae of endoparasitoids that feed on host's tissues and/or hemolymph may impact the internal microbial community of host insects, while having less or no impact on the external community. However, at present very little is known about how parasites or parasitoids affect the microbiome of their host insects (but see [34]).
In this study, we used high-throughput amplicon sequencing targeting both bacteria and fungi to test the hypothesis that parasitism by endoparasitoids affects the microbiome of host insects. Specifically, we compared the internal and external microbiome of healthy and parasitized caterpillars of the large cabbage white Pieris brassicae (Lepidoptera: Pieridae) and one of its main parasitoids, Cotesia glomerata (Hymenoptera: Braconidae). Additionally, we assessed the internal and external microbiome of the developing parasitoid larvae in parasitized caterpillars. Caterpillars were collected from both natural populations and a laboratory culture to identify whether parasitoids consistently alter the microbiome of host caterpillars across origin of the caterpillars.
Results
Bacterial and fungal diversity
After quality filtering, removal of rare sequences and rarefying, a total of 4,287 bacterial zOTUs and 707 fungal OTUs were retained for further analysis (Additional file 1: Table S1 and S2). Rarefaction curves approached saturation, indicating that our sequencing depth was sufficient to cover the microbial diversity (Additional file 2: Fig. S1). Alpha diversity comparisons of the bacterial communities on and in the caterpillars revealed significant (F1,196 = 122.370; p < 0.001) differences between field-collected and lab-reared caterpillars (Fig. 1A, B; Additional file 1: Table S3). Overall, on average 75.6 (range: 1–288) bacterial zOTUs were associated with field-collected caterpillars, while only 8.9 zOTUs (range: 1–95) were found in the lab-reared caterpillars. Shannon diversity was also significantly higher (F1,196 = 309.586; p < 0.001) in the field-collected caterpillars compared to the lab-reared caterpillars (mean Shannon diversity: 2.7 and 0.4, respectively) (Fig. 1B; Additional file 1: Table S3 and S4). The external microbiome of the caterpillars was also significantly (F1,196 = 88.817; p < 0.001) more diverse in terms of bacteria than the internal microbiome (Fig. 1A, B; Additional File 1: Table S3). This was especially the case for field-collected caterpillars, having a mean zOTU richness of 107.3 (range: 7–288) and a mean Shannon diversity of 3.5 (range: 0.5–5.3) for the external samples, compared to 43.9 (range:1–287) and 1.8 (range: 0–5.2) for the internal samples, respectively (Fig. 1A, B; Additional file 1: Table S4). Bacterial communities of the external microbiome of the parasitoid larvae were also more diverse (F1,81 = 42.610; p < 0.001) than those of the internal microbiome, but differences in diversity between parasitoid larvae from field-collected and lab-reared caterpillars were small (Fig. 1A, B). On average, 20.6 zOTUs (range: 1–64) were found on the outside of the parasitoid larvae, while 3.9 zOTUs were found inside (range: 1–27) (Fig. 1A, B; Additional file 1: Table S4).

Boxplots showing alpha diversity comparisons of the different caterpillars (Pieris brassicae) and parasitoid larvae (Cotesia glomerata) samples studied. Samples were divided in different subgroups according to habitat, health status and origin for caterpillars, and habitat and origin for parasitoid larvae. The upper and lower whiskers correspond to the first and third quartiles, with the bar in the middle marking the median value. Alpha diversity was measured by the number of observed of (z)OTUs (top panels) and Shannon index (bottom panels) for bacteria (A, B) and fungi (C, D). Abbreviations used: H = healthy; P = parasitized; E = external; and I = internal
Fungal communities showed less variation in diversity compared to bacteria, with no or little variation between field-collected and lab-reared caterpillars and between the external and internal microbiome (Fig. 1C, D; Additional file 1: Table S3). On average, 15 fungal OTUs (range: 3–32) were found in the external microbiome of the caterpillars, while the internal microbiome comprised on average 17 fungal OTUs (range: 4–30) with only little variation between parasitized and healthy caterpillars and between field-collected and lab-reared individuals (Fig. 1C; Additional file 1: Table S5). The average Shannon diversity of the fungal communities from the external compartment of the caterpillars was 2.0 (range: 0.1–3.4), and 2.3 for the internal compartment (range: 0.6–3.2) (Fig. 1D; Additional file 1: Table S5). Similar values were obtained for the parasitoid larvae (Fig. 1C, D; Additional file 1: Table S5).
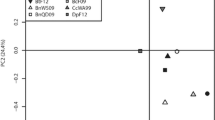
Non-metric multidimensional scaling (NMDS) ordination of the Bray–Curtis distances of Hellinger-transformed relative abundance data of the bacterial communities revealed clear differences between individuals of field-collected and lab-reared populations and between external and internal samples from healthy and parasitized caterpillars. While the external samples of healthy and parasitized caterpillars grouped together (Fig. 2B), the corresponding internal samples were clearly separated (Fig. 2C). NMDS ordination also separated field-collected and lab-reared caterpillars (Fig. 2A–C). In contrast, samples from the parasitoid larvae (both internal and external microbiomes) clustered together, along with the internal samples of the parasitized caterpillars (Fig. 2A). Nonparametric multivariate analysis of variance showed significant differences in bacterial community composition between caterpillars from natural populations and those reared in the laboratory (F1,196 = 82.136; p < 0.001), and between healthy and parasitized caterpillars (F1,196 = 15.839; p < 0.001) (Table 1). Significance of the interaction term was low (F1,196 = 2.244; p = 0.045), indicating that effects of parasitism were not strongly affected by habitat. There was also a strong significant difference between the internal and external microbiome (F1,196 = 16.950; p < 0.001), and this difference depended strongly on the health status (F1,196 = 15.337; p < 0.001) and habitat (F1,196 = 5.981; p < 0.001) of the caterpillars. Likewise, there was a three-way interaction effect, but significance was low (F1,196 = 2.122; p = 0.045) (Table 1). A significant difference was found between bacterial communities from the internal microbiome of healthy and parasitized caterpillars (F1,98 = 37.405; p < 0.001), while no significant difference was found for the external microbiome (F1,98 = 0.891; p = 0.476) (Table 1). For fungi the NMDS did not show such clear patterns as for bacteria, but fungal communities from field-collected caterpillars also diverged from the lab-reared caterpillars (F1,183 = 17.987; p < 0.001) (Table 1).

Non-metric multidimensional scaling (NMDS) ordination plots based on Bray–Curtis distances of Hellinger-transformed relative abundance data of the bacterial (A–C) and fungal communities (D–F) of the different caterpillars (Pieris brassicae) and parasitoid larvae (Cotesia glomerata) samples studied. Results for all samples are shown in A (stress = 0.181) and D (stress = 0.282). Results for the external samples are shown in B (stress = 0.170) and E (stress = 0.239), and for the internal samples in C (stress = 0.181) and F (stress = 0.266). Abbreviations used: Cat = caterpillars; H = healthy; P = parasitized; FC = field-collected; and LR = lab-reared
Bacterial and fungal density
Significantly higher amounts of bacteria (F1,72 = 136.116 p < 0.001) were found on and in lab-reared caterpillars compared to the caterpillars from the field. No significant differences were observed between healthy and parasitized caterpillars, nor between internal and external samples (Fig. 3A; Additional file 1: Table S3). On average, the internal and external microbiome of lab-reared caterpillars contained 1.23 × 106 and 3.30 × 105 16S rRNA gene copy numbers per μL of DNA, respectively, while this was 7.51 × 103 and 1.74 × 103 for the field-collected caterpillars, respectively (Additional file 1: Table S6). By contrast, no differences were found in bacterial 16S rRNA gene copy numbers between parasitoid larvae from field-collected and lab-reared caterpillar populations (Fig. 3A; Additional file 1: Table S6). Fungal densities were highly comparable between field-collected and lab-reared caterpillars (Fig. 3B), having an overall average of 1.70 × 103 and 1.14 × 103 ITS copies per µL DNA for the internal compartment and 5.57 × 103 and 3.94 × 103 for the external compartment of the microbiome, respectively (Additional file 1: Table S7). Similar fungal densities were also found in the parasitoid larvae. Among the field-collected caterpillars, the internal microbiome contained fewer fungal ITS copy numbers in healthy caterpillars (mean: 4.86 × 102) than in parasitized caterpillars (mean: 2.67 × 103). There was no difference in the number of ITS copy numbers between the external microbiome of healthy (mean: 5.02 × 103) and parasitized caterpillars (mean: 6.12 × 103) (Fig. 3B) (Additional file 1: Table S7).

Boxplots showing the numbers of bacterial 16S rRNA gene (A) and fungal ITS copies (B) for the different caterpillars (Pieris brassicae) and parasitoid larvae (Cotesia glomerata) samples studied. Samples were divided in different subgroups according to habitat, organism, health status and origin for caterpillars, and habitat and origin for parasitoid larvae. The upper and lower whiskers correspond to the first and third quartiles, with the bar in the middle marking the median value. For each subgroup, ten random samples were analyzed. Abbreviations used: H = healthy; P = parasitized; E = external; and I = internal
Microbial community composition
Bacteria found in and on the caterpillars investigated represented several environmental and insect-associated species belonging to diverse phyla, among which the most abundant were Proteobacteria, Firmicutes and Actinobacteria (Additional file 1: Table S1). Irrespective of health status, the external microbiome of field-collected caterpillars was composed of diverse bacteria from different phyla occurring at relatively low abundances. By contrast, lab-reared insects showed bacterial communities (both external and internal) that were characterized by two highly abundant zOTUs, i.e. an unidentified Enterobacteriaceae member (zOTU2) and Acinetobacter sp. (zOTU5). zOTU2 was detected in almost every lab-reared caterpillar at an overall average relative abundance of 79.6% (calculated based on the entire dataset), while zOTU5 occurred in about half of the caterpillars at an overall average relative abundance of 9.4%. In contrast, both zOTUs were absent or occurred at lower relative densities in the natural populations (Fig. 4). Interestingly, the internal samples of all parasitized caterpillars studied, including both field-collected and lab-reared caterpillars, as well as all samples taken from the parasitoid larvae were dominated by one particular bacterium (zOTU1), the insect symbiont Wolbachia pipientis (Fig. 4). Relative abundance of this symbiont ranged from 3.2% up to 97.6% (mean: 62.7%) in the internal microbiome of parasitized caterpillars, with a lower relative abundance in the lab-reared insects (mean: 31.9%) compared to field-collected caterpillars (mean: 79.3%). Further, the bacterium was found in and on all parasitoid larvae. For parasitoid larvae from field-collected caterpillars, it occurred at a mean relative abundance of 73.7% (4.8–100%) in the external microbiome and 97.6% (79.6–100%) in the internal microbiome. Similarly, in parasitoid larvae from lab-reared insects it was present at a relative abundance of 76.1% (1.9–97.7%) and 99.4% (95.8–100%) in the external and internal samples, respectively. In addition to Wolbachia, the external samples of the parasitoid larvae contained a huge variety of other microorganisms, which were, especially for the field-collected caterpillars, also found in the internal microbiome of parasitized and unparasitized caterpillars (Fig. 4). In contrast, the Wolbachia zOTU was completely absent in samples from healthy caterpillars, and was also not found in the external samples from parasitized caterpillars (Fig. 4), as was also confirmed by a specific PCR targeting Wolbachia DNA (Additional file 1: Table S8).

Bacterial community profiles of the different caterpillars (Pieris brassicae) and parasitoid larvae (Cotesia glomerata) samples studied. Bacterial taxa represent the most prevalent taxa in the different subgroups based on origin and health status for caterpillars and origin for parasitoid larvae (present at a mean relative abundance > 0.5% in at least one subgroup). For each zOTU, the average relative abundance for each subgroup is given in the box as a percentage, whereas the color indicates prevalence (white is absent). zOTUs are identified by a BLAST search against type materials in GenBank. When no significant similarity was found with type materials, the BLAST analysis was performed against entire GenBank (indicated with and asterisk). Identifications were performed at genus level; when identical scores were obtained for different genera, identifications were performed at family level. When identity percentages were lower than 99%, the percentage of sequence identity with the GenBank entry is given between brackets. Hits with uncultured bacteria are indicated as unidentified bacterium. Abbreviations used: H = healthy; P = parasitized; E = external; and I = internal
The fungal community composition was homogeneous among all samples studied, with fewer notable differences between field-collected and lab-reared caterpillars (Additional file 2: Fig. S2). Fungal communities were dominated by two Alternaria species, OTU6 and OTU8, and one Sporobolomyces sp. (OTU10), occurring in 74.4, 83.9 and 73.3% of all samples studied, respectively. Additionally, Malassezia sp. (OTU16) was commonly detected and occurred in 37.4% of the samples studied (Additional file 1: Table S2).
Discussion
Although the microbiome of insects has been studied thoroughly over the past decade [35, 36], including Lepidoptera [17, 37], little is still known about how microbial communities assemble in and on the bodies of insects, and to which extent insect microbiomes are affected by the presence of parasites [38] or parasitoids (but see [34]). Our taxonomic analysis revealed that the bacterial microbiome of P. brassicae caterpillars was mainly composed of Proteobacteria, Firmicutes and Actinobacteria. These phyla represent the most common phyla in lepidopteran species, including Pieris spp. [18, 34, 39,40,41]. In contrast to other studies that found very low bacterial abundances in P. brassicae caterpillars [42], estimation of the bacterial abundance by qPCR suggested higher bacterial densities in our samples. This was confirmed by plating a selection of samples on trypticase soy agar supplemented with 0.5 g/L cycloheximide (up to104 bacterial colony forming units (cfu) per specimen) (Additional file 1: Table S9). Members of the Ascomycota and Basidiomycota were the most common fungi found in our dataset, including several environmental fungi that commonly occur on cultivated plants like Alternaria, Cladosporium and Sporobolomyces. Fungal symbionts of insects have been mostly studied in insects feeding on wood or detritus [1, 15], but have been recently studied in Lepidoptera as well [43]. Although fungi may contribute to the provision of nutrients and regulation of host defenses in insects, the exact functions that most fungi play in associations with insects are yet to be discovered [2].
Our results further showed that caterpillars from natural populations harbour a more diverse and evenly distributed bacterial microbiome than lab-reared caterpillars, both at the outside and the inside of the insect. While on average 75.6 bacterial zOTUs were found in the field-collected caterpillars, the microbiome of lab-reared caterpillars comprised an average of only 8.9 zOTUs. Two bacteria were dominant in and on the bodies of lab-reared caterpillars, while they only sporadically occurred in natural populations. These bacteria were identified as an Acinetobacter species and a member of the family Enterobacteriaceae. Acinetobacter species and Enterobacteriaceae are ubiquitous in nature, and occur in diverse habitats, including soil, plants and insect guts [1, 37, 41, 44, 45]. The reason why they occur more abundantly in lab-reared insects compared to wild-collected insects is not yet clear, but similar patterns in diverging microbiome composition between natural and lab-reared insect populations have been observed in other caterpillars (Spodoptera spp.) [46], house flies (Musca domestica) [29], fruit flies (Drosophila spp.) [27] and leafhoppers (Psammotettix alienus) [47]. Factors like diet and ecological and environmental differences between natural habitats and artificial rearing environments seem to play a major role, while parental effects are less important [1, 14, 27, 41, 46] Larvae of most butterfly species largely mirror the bacterial community composition of their diets, suggesting passive acquisition of their bacterial gut inhabitants through food ingestion rather than active selection [41]. Indeed, previous studies have shown that caterpillars lack resident gut symbionts, and mainly harbor transient environmental microorganisms that are present on host plants, including microorganisms that originate from the soil and are transferred through the plant [48, 49]. The stable environmental conditions when rearing insects in the laboratory or the limited pool of bacteria present in the rearing facilities may have favored the growth of particular fast-growing species that outcompeted or reduced the growth of other species. This may also explain the higher bacterial concentrations in samples from lab-reared caterpillars compared to field-collected caterpillars. While less clear, also for fungi differences were found in fungal diversity between field-collected and lab-reared caterpillar populations. Nevertheless, it has to be noted that fungal density was rather low in our samples; in general ITS copy numbers varied between 102 and 104 copies per µL DNA. Given the fact that fungi can possess more than 100 ITS copies per genome [50], these values thus represent low densities, which was also confirmed by plating a subset of samples on yeast potato dextrose agar with 0.5 g/L chloramphenicol (up to 102 fungal cfu per specimen (Additional file 1: Table S9)).
The external bacterial microbiome of the caterpillars was significantly more diverse than its internal counterpart. Our results show that parasitism altered the internal microbiome of caterpillars, but not the external microbiome. The internal samples of all parasitized caterpillars as well as all samples taken from the parasitoid larvae were dominated by one particular bacterial strain, the insect symbiont Wolbachia pipientis (zOTU1), while it was completely absent in healthy caterpillars. In some parasitoid larvae W. pipientis was the only bacterium detected. This pattern was present in both wild and lab-reared caterpillars, although its relative abundance was higher in parasitized caterpillars from the field (79.3%) compared to parasitized caterpillars from the lab (31.9%). The factors driving this difference in relative abundance are not yet clear, but it may be possible that caterpillars reared in the laboratory and collected in the field have developed different immune responses to Wolbachia [51]. Also, the microbiome of lab-reared caterpillars was strongly dominated by a member of the family Enterobacteriaceae, which occurred to a much lesser extent in field-collected caterpillars, and which may have inhibited the Wolbachia strain from excessive reproduction in the lab-reared caterpillars. Furthermore, caution must be taken with the biological interpretation of relative abundance data, since inter-sample differences in cell density are not considered [52].
Wolbachia is a genus of well-studied intracellular endosymbionts that are commonly found in arthropods and that are able to manipulate host reproduction to favor its own maternal transmission [53, 54]. However, Wolbachia is often mutualistic for many insects, as it provides its host resistance against viruses, insecticides or plant defenses, and contributes to nutritional provisioning [55, 56]. Wolbachia is estimated to be present in about 80% of lepidopteran species, including species belonging to the Pieridae family [57]. Interestingly, in addition to the Wolbachia strain dominating parasitized P. brassicae caterpillars, we also found another Wolbachia strain (zOTU101) in a few healthy (four samples) and parasitized field-collected caterpillars (8 samples), while it did not occur in any lab-reared caterpillar, suggesting a rather limited distribution of this strain (Fig. 4; Additional file 1: Table S1). At the nucleotide level, both Wolbachia strains shared 95.6% 16S rRNA gene sequence identity on a total of 248 bp. Wolbachia is also commonly reported in parasitoids [58], including C. glomerata [32, 59]. PCR analyses targeting the 16S rRNA gene of Wolbachia confirmed its presence in the C. glomerata laboratory culture that was used to infect the lab-reared caterpillars in this study (Additional file 1: Table S8). Since it was not found in healthy caterpillars, but was highly present in the internal compartment of parasitized caterpillars as well as in the parasitoid larvae and adults, it is reasonable to assume that the parasitoids transferred Wolbachia into the caterpillars during oviposition after which it established and replicated, explaining its high relative abundance in parasitized caterpillars. This is in line with previous studies showing that parasitoids may transfer Wolbachia into their host during oviposition, as seen in whiteflies [60]. However, surprisingly Wolbachia, being an intracellular bacterium, was also abundantly found on the outside of the parasitoid larvae. One plausible explanation may be that Wolbachia-containing host tissues were damaged by larval feeding or during the dissection leading to contamination of the outside of the parasitoid larvae. Likewise, tissue damage and/or gut disruption may explain the high microbial diversity in the external parasitoid samples and explain why they represented a community similar to the internal host samples. Further research is needed to exclude this scenario.
The presence of Wolbachia in adult parasitoids could have a positive effect on the wasps by enhancing host-searching ability and oviposition frequency [61]. On the other hand, it has also been suggested that Wolbachia can have negative effects on parasitoid populations as it can increase the susceptibility to hyperparasitism by hyperparasitoids, i.e. parasitic wasps that attack the larvae and pupae of primary parasitoids [62, 63]. Hyperparasitoids strongly rely on herbivore-induced plant volatiles (HIPVs) to locate potential hosts [64, 65], but also use other cues such as changes in the body odors of parasitized herbivores to locate their host from a short distance [66]. Although the underlying mechanisms are still unclear, it can be hypothesized that microorganisms may be involved in mediating body odor changes [67], as was recently demonstrated for honey bees [68], and/or may act synergistically with other agents like polyDNAvirus and venom affecting HIPV emission and revealing the presence of parasitoid hosts to its hyperparasitoids [69]. Whether and to which extent Wolbachia is involved in this process requires further research.
Conclusions
Together, our results show that the microbiome of caterpillars from natural populations harbored a much more diverse bacterial microbiome than lab-reared caterpillars. The external microbiome of the caterpillars was also significantly more diverse than its internal counterpart. Fungal communities were less diverse and showed less variation. Our results also clearly show that parasitism significantly altered the internal microbiome of the caterpillars, but not the external microbiome. The internal microbiome of all parasitized caterpillars and of the parasitoid larvae was dominated by a Wolbachia strain, while this bacterium was completely absent in healthy caterpillars. Further research is needed to elucidate the possible role of this endosymbiont in the interaction between the host caterpillar, the parasitoid, and higher trophic levels.
Materials and methods
Study system
In this study, caterpillars of the large cabbage white Pieris brassicae (Lepidoptera: Pieridae) and one of its main parasitoids, Cotesia glomerata (Hymenoptera: Braconidae), were used as study organisms. Pieris brassicae is an important cosmopolitan pest species of many crops belonging to the family Brassicaceae such as cabbage, cauliflower, brussels sprouts and rape. Cotesia glomerata is a gregarious koinobiont wasp that parasitizes a wide range of caterpillars of pierid butterflies, but P. brassicae and Pieris rapae are its main hosts. The wasp lays approximately 20–40 eggs inside first or second instar caterpillars where the larvae will hatch and consume the body from the inside, while the caterpillars are still alive and continue feeding themselves. Typically, after 15 to 20 days the parasitoid larvae emerge from their caterpillar host, which ultimately kills the caterpillar [70].
Sample collection
A total of 102 fifth-instar caterpillars of P. brassicae were used in this study, including 59 non-parasitized caterpillars and 43 caterpillars parasitized by C. glomerata (Additional file 1: Table S10). Among these, 63 caterpillars (35 non-parasitized and 28 parasitized) were collected from the field. Thirty-nine lab-reared individuals (24 non-parasitized and 15 parasitized) were included for the sake of comparison. Field-collected individuals were obtained between July and September 2019 from three organic farms growing cauliflower (Brassica oleracea L. var. botrytis) (Field 1 and 2, both located in Bornem, Belgium) or white cabbage (Brassica oleracea L. var. capitata) (Field 3, Randwijk, The Netherlands). To minimize collection of sibling larvae, each field-collected caterpillar was retrieved from a different plant, and sampling was performed over a three months period.
With regard to the lab-reared insects, lab cultures from P. brassicae and C. glomerata were used that both originated from agricultural fields near Wageningen University, The Netherlands. The P. brassicae culture was reared and maintained on Brussels sprouts plants (Brassica oleracea L. var. gemmifera cv. Cyrus) in a large cage in a greenhouse compartment (21 ± 1 °C, 25–35% RH, 16:8 h light/dark). Male and female butterflies were allowed to freely mate in the cage and lay their eggs on different plants. Adults were fed with a saturated sugar solution. Cotesia. glomerata was reared in another cage on P. brassicae under the same conditions. When C. glomerata larvae had pupated, pupae were collected and transferred to a smaller cage with no plants and emerged parasitoids were provided with honey and water until they were used in the experiments. When P. brassicae larvae had hatched from our rearing, multiple cohorts of early first instar larvae from different egg-clutches were collected. To minimize a priori variation between healthy and parasitized caterpillars, for each egg-clutch hatchlings were subjected to two treatments: half of the caterpillars were parasitized by C. glomerata and the other half was left untreated. In order to parasitize the larvae, caterpillars were put into a clean plastic cage (one cage for hatchlings from the same egg-clutch) with mated C. glomerata females and exposed to parasitism for five minutes. For each egg-clutch both groups of caterpillars were then put on Brussels sprouts in two separate cages (one cage for parasitized caterpillars and one for non-parasitized caterpillars; different cages were used for hatchlings from different egg-clutches) within the same greenhouse compartment, until the caterpillars were collected for further analysis. Caterpillars used in the experiment were randomly picked from each cage, and represented individuals from different egg-clutches. All caterpillars were collected using a pair of tweezers that was sterilized by applying 70% ethanol before the collection of each caterpillar. Additionally, gloves were worn that were sterilized with ethanol before a caterpillar was collected. When caterpillars were collected, they were placed individually in empty plastic sterile containers (12 cm diameter; 5 cm height) with a pierced lid, which in case of field-collected caterpillars were transported to the laboratory in a cooling box. Subsequently, caterpillars were left starving overnight at room temperature to allow the insects to empty their gut content, while minimizing contact with their own frass.
Microbiome sampling
Both the microbiota associated with the surface of the cuticle and the interior of the insect body were sampled. The external microbiota of the caterpillars were obtained by putting each caterpillar in a 2 mL microcentrifuge tube containing 1 mL of phosphate-buffered saline with 0.01% Tween80 (PBS-T), and vortexing it for 20 s. The washing solution was then used as a sample from the external microbiome. Subsequently, to remove potential residual external microbes, the caterpillar was placed into another tube containing 1 mL of sodium hypochlorite (2.5%) and vortexed again for 20 s, followed by two final washing steps in PBS-T [29]. Application of 2.5% bleach has been shown to be very effective in removing externally contaminating DNA [71, 72]. Each caterpillar was then dissected under sterile conditions to confirm whether or not the caterpillars had been parasitized, and collect the parasitoid larvae. To this end, caterpillars were pinned to a sterile dissection plate with flame-sterilized needles (one in the head and one at the posterior end) and cut open along the entire length of the caterpillar. Next, after opening the insect and pinning both sides with two additional needles, some drops of sterile water were applied on the dissected body in order to ease isolation of the parasitoid larvae (parasitoid larvae float in water). To avoid contamination of the parasitoid larvae with host microbes, the dissection was performed very carefully, aiming to not disrupt the host gut or any other tissues. For each parasitized caterpillar, all parasitoid larvae found inside the caterpillar’s body were removed using a sterilized pair of tweezers and put together in a clean microcentrifuge tube. In general, between 20 and 40 late stage parasitoid larvae (close to egression (ca. 3–4 mm)) were retrieved from all parasitized caterpillars (Fig. 5). The whole body remainder of the caterpillars was then homogenized with a Bead Ruptor Elite (Omni international, Kennesaw, USA) in 1 mL PBS-T and a mixture of glass beads of different sizes (three beads of 2 mm and two beads of 5 mm in diameter) using two cycles of 10 s at a speed of 5.5 m/s with a 10 s break in between. The resulting homogenate was used as a sample reflecting the internal microbiota. Additionally, for the parasitized caterpillars, the external and internal microbiomes from the pool of parasitoid larvae were also collected following the same protocol, with the exception for the working volume used (300 µL instead of 1 mL). PCR screening [73] of the parasitoid larvae confirmed that all parasitized field-collected caterpillars were infested with C. glomerata, which is in agreement with the high number of parasitoid larvae found in the caterpillars. Although there are also other parasitic wasps than C. glomerata that can attack P. brassicae, most of them (if not all of them) are solitary wasps injecting only one egg per caterpillar [74]. Moreover, in contrast to other parasitoids such as Cotesia rubecula and Hyposoter ebeninus, that interrupt development of P. brassicae caterpillars at the 3rd instar, caterpillars parasitized by C. glomerata can develop until the 5th larval instar [66], indicating that the parasitized caterpillars collected from the field were only parasitized by C. glomerata.

A Dissection of Cotesia glomerata larvae from Pieris brassicae caterpillars. Each caterpillar contained between 20 and 40 late stage parasitoid larvae that were close to egression (ca. 3–4 mm). B Close-up picture of some larvae
DNA extraction and molecular analysis
Genomic DNA was isolated from all external and internal samples (300 µL) using the PowerPro Soil Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions, with one modification: in the second step of the protocol the use of a vortex adapter was replaced by two cycles of 30 s (with a 10 s break in between) in the Bead Ruptor Elite at a speed of 5.5 m/s. Additionally, two negative controls in which the sample material was replaced by sterile, DNA-free water was included to confirm absence of reagent contamination. DNA samples were then subjected to PCR amplification of the hypervariable region of the bacterial 16S rRNA gene (primers 515 F and 806 R) [75] and the fungal ITS1 region (primers BITS and B58S3) [76] using Illumina barcoded primers, designed according to Kozich et al. (2013) [75] (dual-index sequencing strategy; Additional file 1: Table S11 and S12). In each run, two negative PCR controls (in which DNA template was replaced by DNA-free water) and a DNA mock community sample (one for bacteria and one for fungi) were included. Both mock communities were composed of a number of species that were likely to occur in insects [29] (Additional file 1: Table S13). PCR amplification was performed in a reaction volume of 40 µL, consisting of 2 µL DNA, 0.5 µM of each primer, 150 µM of each dNTP, 1 × Titanium Taq PCR buffer and 1 × Titanium Taq DNA polymerase (Takara Bio, Saint-Germain-en-Laye, France). The reactions were initiated by denaturation at 94 °C for 120 s, followed by 35 cycles of 45 s at 95 °C, 45 s at 59 °C and 45 s at 72 °C, and a final elongation step of 10 min at 72 °C. All samples were successfully amplified for bacteria, while fungal amplicons were only obtained for 273 out of 289 samples. For the negative DNA extraction and PCR controls, very faint to no bands were obtained after gel electrophoresis. Amplicons from positive insect samples as well as from the different controls were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter Genomics GmbH, South Plainfield, UK) following the manufacturer’s instructions. Subsequently, a Qubit high sensitivity fluorometer (Invitrogen, Carlsbad, USA) was used to measure the concentration of the purified amplicons, and each sample was then pooled in equimolar concentrations into two libraries, one pool of bacterial V4 amplicons and one pool of fungal ITS1 amplicons. Next, following ethanol precipitation, the amplicon pools were loaded onto a 1.5% agarose gel, and the bands corresponding to the expected fragment length were excised from the gel and purified using a QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Following gel extraction, the concentration of the libraries was measured again, diluted to 2 nM, and then sent for sequencing at the Center for Medical Genetics (University of Antwerp, Antwerp, Belgium) using an Illumina MiSeq sequencer with a v2 500-cycle reagent kit (Illumina, San Diego, USA). Additionally, for a subset of randomly selected samples (10 per group), bacterial and fungal densities were quantified by determining total bacterial 16S rRNA gene and fungal ITS1 copy numbers using qPCR with the same primers pairs as those used for the sequencing approach (but without barcodes) (for details, see [77]). Furthermore, the same samples were subjected to a PCR analysis targeting the 16S rRNA gene of Wolbachia (wspec primers) as previously described [78]. Both PCR analyses were performed in duplicate.
Illumina sequences were received as a demultiplexed FASTQ file, with barcodes and primer sequences removed. For the V4 sequences, paired-end reads were merged using USEARCH (v11.0.667) to form consensus sequences [79] with not more than 10 mismatches allowed in the overlap region. For the ITS sequences, only forward reads were retained. Subsequently, sequences were truncated at the 248th base, and reads shorter than 248 bp or reads with a total expected error threshold above 0.2 and 1 for the V4 and ITS regions, respectively, were discarded using USEARCH (v11.0.667). Next, Mothur’s (v1.39.3) commands ‘classify.seqs’ and ‘remove.lineage’ or ‘get.lineage’ in combination with the Silva database (v1.38, for bacteria) and UNITE database (v6, for fungi), respectively, were used to identify and remove potential mitochondrial, chloroplast or other non-target sequences. Bacterial sequences were classified into zero-radius operational taxonomic units (zOTUs [80]; also known as amplicon sequence variants (ASVs) [81]) by the UNOISE3 algorithm as implemented in USEARCH [82]. Only zOTUs with a minimum abundance of eight reads were kept and chimeric sequences identified by the algorithm were removed. Fungal sequences were clustered into operational taxonomic units (OTUs) based on a 3% sequence dissimilarity cut-off. The advantage of zOTUs is that they enable resolution of closely related taxa that would be incorporated into the same OTU when applying a 3% dissimilarity cut-off. However, given that many fungal species house intraspecific and intragenomic variations in ITS1 [83] fungal diversity is still commonly assessed by the use of 97% OTUs as fungal species proxies [84]. OTU clustering was performed using the UPARSE greedy algorithm in USEARCH, during which chimeric sequences were also removed [79], as were global singletons (i.e. OTUs with only 1 sequence represented in the entire data set). Next, both the bacterial and fungal data sets were analyzed in R (v3.5.2) using microDecon (v1.2.0) [85] to control for the presence of contaminants based on (z)OTU prevalence in the insect samples versus the mean of the two PCR control samples [86, 87]. At the same time, the DNA extraction controls were removed from the dataset since they yielded only very low sequence numbers (less than 100). Additionally, (z)OTUs occurring below a 0.1% and OTUs occurring below 1% relative abundance threshold per sample were discarded from further analysis (which was in accordance with the thresholds defined by the mocks communities). Finally, the number of sequences was rarefied to 2500 sequenced for bacteria and 1000 sequences for fungi. The taxonomic origin of each bacterial zOTU and fungal OTU was determined with the SINTAX algorithm as implemented in USEARCH based on the SILVA Living Tree Project v123 for bacteria and the UNITE database v6 for fungi. Further, the identity of the most important zOTUs and OTUs was verified with a BLAST search in GenBank against type materials. When no significant similarity was found with type materials (< 97% identity), the BLAST analysis was performed against entire GenBank. For fungi, for which less type strain sequences are available, the BLAST search was performed against both type strains and the GenBank database excluding uncultured and environmental sample sequences. Analysis of the mock communities demonstrated that only the expected taxa were found, indicating that the experimental conditions were met to achieve robust data.
Data analysis
For each sample, a rarefaction curve was generated to see whether (z)OTU richness reached an asymptote. Rarefaction curves were created using the Phyloseq package in R showing the number of observed (z)OTUs as a function of the number of sequences [87, 88]. Subsequently, observed (z)OTU richness and Shannon diversity were calculated for each sample with the phyloseq package in R. The Shannon diversity index is an index that is commonly used to characterize species diversity in a community and accounts for both abundance and evenness of the species present. Higher scores indicate high diversity, while scores close to 0 indicate low diversity [89]. Samples were grouped according to habitat (field-collected vs. lab-reared caterpillars), organism (caterpillar vs. parasitoid larvae), origin (external vs. internal) and health status (healthy vs. parasitized). For the caterpillars, a three-way analysis of variance (ANOVA) was used to assess whether habitat, origin and health status affected species richness, Shannon diversity and microbial densities. All two-way interactions and three-way interaction were included in the model as well. Based on the Hellinger transformed relative abundance data of the observed bacteria and fungi in each of the sampled individuals, the bacterial and fungal community composition was visualized by non‐metric multidimensional scaling (NMDS) using the Bray–Curtis coefficient as distance measure in the R software package vegan [90]. To test the hypothesis that caterpillars bacterial and fungal communities differed between habitats, origin and health status, permutational analysis of variance (PERMANOVA) [91] was performed using the “adonis” function in the software package vegan [90]. All factors and their interactions were included as fixed factors in the analysis. Significance was tested using 1,000 permutations. All analyses were performed for bacteria and fungi separately.
Availability of data and materials
The sequences obtained in this study were deposited in the Sequence Read Archive (SRA) at NCBI under Bioproject PRJNA700075.
References
Engel P, Moran NA. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol Rev. 2013;37:699–735.
Gurung K, Wertheim B, Salles JF. The microbiome of pest insects: it is not just bacteria. Entomol Exp Appl. 2019;167:156–70.
Moran NA, Ochman H, Hammer TJ. Evolutionary and ecological consequences of gut microbial communities. Annu Rev Ecol Evol Syst. 2019;50:451–75.
Hammer TJ, Sanders JG, Fierer N. Not all animals need a microbiome. FEMS Microbiol Lett. 2019;366:fnz117.
Dharampal PS, Carlson C, Currie CR, Steffan SA. Pollen-borne microbes shape bee fitness. Proc R Soc B Biol Sci. 2019;286:20182894.
Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24:392–400.
Koch H, Schmid-Hempel P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc Natl Acad Sci. 2011;108:19288–92.
Pozo MI, van Kemenade G, van Oystaeyen A, Aledón-Catalá T, Benavente A, Van den Ende W, Wäckers F, Jacquemyn H. The impact of yeast presence in nectar on bumble bee behavior and fitness. Ecol Monogr. 2020;90:e01393.
McLean AHC, Parker BJ, Hrček J, Henry LM, Godfray HCJ. Insect symbionts in food webs. Philos Trans R Soc B Biol Sci. 2016;371:20150325.
Frago E, Dicke M, Godfray HCJ. Insect symbionts as hidden players in insect-plant interactions. Trends Ecol Evol. 2012;27:705–11.
van den Bosch TJM, Welte CU. Detoxifying symbionts in agriculturally important pest insects. Microb Biotechnol. 2017;10:531–40.
Jing T-Z, Qi F-H, Wang Z-Y. Most dominant roles of insect gut bacteria: digestion, detoxification, or essential nutrient provision? Microbiome. 2020;8:38.
Dillon RJ, Dillon VM. The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol. 2004;49:71–92.
Yun J-H, Roh SW, Whon TW, Jung M-J, Kim M-S, Park D-S, Yoon C, Nam Y-D, Kim Y-J, Choi J-H, Kim J-Y, Shin N-R, Kim S-H, Lee W-J, Bae J-W. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl Environ Microbiol. 2014;80:5254–64.
Brune A, Dietrich C. The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annu Rev Microbiol. 2015;69:145–66.
Hongoh Y, Deevong P, Inoue T, Moriya S, Trakulnaleamsai S, Ohkuma M, Vongkaluang C, Noparatnaraporn N, Kudo T. Intra- and interspecific comparisons of bacterial diversity and community structure support coevolution of gut microbiota and termite host. Appl Environ Microbiol. 2005;71:6590–9.
Pinto-Tomás A, Sittenfeld A, Uribe L, Chavarría F, Mora M, Janzen D, Goodman R, Simon H. Comparison of midgut bacterial diversity in tropical caterpillars (Lepidoptera: Saturniidae) fed on different diets. Environ Entomol. 2011;40:1111–22.
Robinson CJ, Schloss P, Ramos Y, Raffa K, Handelsman J. Robustness of the bacterial community in the cabbage white butterfly larval midgut. Microb Ecol. 2010;59:199–211.
Grenier A-M, Duport G, Pagès S, Condemine G, Rahbé Y. The phytopathogen Dickeya dadantii (Erwinia chrysanthemi 3937) is a pathogen of the pea aphid. Appl Environ Microbiol. 2006;72:1956–65.
Genta F, Dillon R, Terra W, Ferreira C. Potential role for gut microbiota in cell wall digestion and glucoside detoxification in Tenebrio molitor larvae. J Insect Physiol. 2006;52:593–601.
Douglas AE. Multiorganismal insects: diversity and function of resident microorganisms. Annu Rev Entomol. 2015;60:17–34.
Davis TS, Crippen TL, Hofstetter RW, Tomberlin JK. Microbial volatile emissions as insect semiochemicals. J Chem Ecol. 2013;39:840–59.
Mogilnicka I, Bogucki P, Ufnal M. Microbiota and malodour—etiology and management. Int J Mol Sci. 2020;21:2886.
Stockley P, Bottell L, Hurst JL. Wake up and smell the conflict: odour signals in female competition. Philos Trans R Soc B Biol Sci. 2013;368:20130082.
Busula AO, Takken W, de Boer JG, Mukabana WR, Verhulst NO. Variation in host preferences of malaria mosquitoes is mediated by skin bacterial volatiles. Med Vet Entomol. 2017;31:320–6.
Verhulst NO, Qiu YT, Beijleveld H, Maliepaard C, Knights D, Schulz S, Berg-Lyons D, Lauber CL, Verduijn W, Haasnoot GW, Mumm R, Bouwmeester HJ, Claas FHJ, Dicke M, van Loon JJA, Takken W, Knight R, Smallegange RC. Composition of human skin microbiota affects attractiveness to malaria mosquitoes. PLoS ONE. 2011;6:e28991.
Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A. Bacterial communities of diverse Drosophila species: ecological context of a host–microbe model system. PLOS Genet. 2011;7:e1002272.
Deguenon JM, Travanty N, Zhu J, Carr A, Denning S, Reiskind MH, Watson DW, Roe RM, Ponnusamy L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci Rep. 2019;9:20365.
Park R, Dzialo MC, Spaepen S, Nsabimana D, Gielens K, Devriese H, Crauwels S, Tito RY, Raes J, Lievens B, Verstrepen KJ. Microbial communities of the house fly Musca domestica vary with geographical location and habitat. Microbiome. 2019;7:147.
Gomes SIF, Kielak AM, Hannula SE, Heinen R, Jongen R, Keesmaat I, De Long JR, Bezemer TM. Microbiomes of a specialist caterpillar are consistent across different habitats but also resemble the local soil microbial communities. Anim Microbiome. 2020;2:37.
Bright M, Bulgheresi S. A complex journey: transmission of microbial symbionts. Nat Rev Microbiol. 2010;8:218–30.
Dicke M, Cusumano A, Poelman E. Microbial symbionts of parasitoids. Annu Rev Entomol. 2020;65:171–90.
Fredensborg BL, Kálvalíð IFÍ, Johannesen TB, Stensvold CR, Nielsen HV, Kapel CMO. Parasites modulate the gut-microbiome in insects: a proof-of-concept study. PLoS ONE. 2020;15:e0227561.
Cavichiolli de Oliveira N, Cônsoli FL. Beyond host regulation: changes in gut microbiome of permissive and non-permissive hosts following parasitization by the wasp Cotesia flavipes. FEMS Microbiol Ecol. 2020;96:fiz206.
Broderick NA, Raffa KF, Goodman RM, Handelsman J. Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl Environ Microbiol. 2004;70:293–300.
Garofalo C, Milanovic V, Cardinali F, Aquilanti L, Clementi F, Osimani A. Current knowledge on the microbiota of edible insects intended for human consumption: a state-of-the-art review. Food Res Int. 2019;125:108527.
Paniagua Voirol LR, Frago E, Kaltenpoth M, Hilker M, Fatouros NE. Bacterial symbionts in Lepidoptera: their diversity, transmission, and impact on the host. Front Microbiol. 2018;9:556.
Vicente CSL, Ozawa S, Hasegawa K. Composition of the cockroach gut microbiome in the presence of parasitic nematodes. Microbes Environ. 2016;31:314–20.
Gao X, Li W, Luo J, Zhang L, Ji J, Zhu X, Wang L, Zhang S, Cui J. Biodiversity of the microbiota in Spodoptera exigua (Lepidoptera: Noctuidae). J Appl Microbiol. 2019;126:1199–208.
Wang Y, Zhu J, Fang J, Shen L, Ma S, Zhao Z, Yu W, Jiang W. Diversity, composition and functional inference of gut microbiota in Indian cabbage white Pieris canidia (Lepidoptera: Pieridae). Life. 2020;10:254.
Phalnikar K, Kunte K, Agashe D. Dietary and developmental shifts in butterfly-associated bacterial communities. R Soc Open Sci. 2018;5:171559.
Paniagua Voirol LR, Weinhold A, Johnston PR, Fatouros NE, Hilker M. Legacy of a butterfly’s parental microbiome in offspring performance. Appl Environ Microbiol. 2020;86:e00596-e620.
Ravenscraft A, Berry M, Hammer T, Peay K, Boggs C. Structure and function of the bacterial and fungal gut microbiota of Neotropical butterflies. Ecol Monogr. 2019;89:e01346.
Álvarez-Pérez S, Lievens B, Jacquemyn H, Herrera CM. Acinetobacter nectaris sp. nov. and Acinetobacter boissieri sp. nov., isolated from floral nectar of wild Mediterranean insect-pollinated plants. Int J Syst Evol Microbiol. 2013;63:1532–9.
Kim PS, Shin N-R, Kim JY, Yun J-H, Hyun D-W, Bae J-W. Acinetobacter apis sp. Nov., isolated from the intestinal tract of a honey bee, Apis mellifera. J Microbiol Seoul Korea. 2014;52:639–45.
Martínez-Solís M, Collado MC, Herrero S. Influence of diet, sex, and viral infections on the gut microbiota composition of Spodoptera exigua caterpillars. Front Microbiol. 2020;11:753.
Wang H, Wu N, Liu Y, Kundu J, Liu W, Wang X. Higher bacterial diversity of gut microbiota in different natural populations of leafhopper vector does not influence WDV Transmission. Front Microbiol. 2019;10:1144.
Hammer TJ, Janzen DH, Hallwachs W, Jaffe SP, Fierer N. Caterpillars lack a resident gut microbiome. Proc Natl Acad Sci USA. 2017;114:9641–6.
Hannula SE, Zhu F, Heinen R, Bezemer TM. Foliar-feeding insects acquire microbiomes from the soil rather than the host plant. Nat Commun. 2019;10:1254.
Lofgren LA, Uehling JK, Branco S, Bruns TD, Martin F, Kennedy PG. Genome-based estimates of fungal rDNA copy number variation across phylogenetic scales and ecological lifestyles. Mol Ecol. 2019;28:721–30.
Jiang H, Vilcinskas A, Kanost MR. Immunity in lepidopteran insects. In: Söderhäll K, editor. Invertebrate immunity. Advances in experimental medicine and biology, vol. 708. New York: Springer; 2010. p. 181–204.
Widder S, Allen RJ, Pfeiffer T, Curtis TP, Wiuf C, Sloan WT, Cordero OX, Brown SP, Momeni B, Shou W, Kettle H, Flint HJ, Haas AF, Laroche B, Kreft JU, Rainey PB, Freilich S, Schuster S, Milferstedt K, van der Meer JR, Groβkopf T, Huisman J, Free A, Picioreanu C, Quince C, Klapper I, Labarthe S, Smets BF, Wang H. Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. 2016;10:2557–68.
Sanaei E, Charlat S, Engelstädter J. Wolbachia host shifts: routes, mechanisms, constraints and evolutionary consequences. Biol Rev. 2021;96:433–53.
Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–51.
Berticat C, Boquien G, Raymond M, Chevillon C, Berticat C, Boquien G, Raymond M, Chevillon C. Insecticide resistance genes induce a mating competition cost in Culex pipiens mosquitoes. Genet Res. 2002;79:41–7.
Hedges L, Brownlie J, O’Neill S, Johnson K. Wolbachia and virus protection in insects. Science. 2008;322:702.
Ahmed MZ, Araujo-Jnr EV, Welch JJ, Kawahara AY. Wolbachia in butterflies and moths: geographic structure in infection frequency. Front Zool. 2015;12:16.
Stouthamer R, Breeuwer JA, Hurst GD. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol. 1999;53:71–102.
Rattan R, Hadapad A, Reineke A, Gupta PR, Zebitz C. Molecular evidence for the presence of the endosymbiontic bacteria Wolbachia in Cotesia populations (Hymenoptera: Braconidae). J Asia-Pac Entomol. 2011;14:183–5.
Ahmed MZ, Li S-J, Xue X, Yin X-J, Ren S-X, Jiggins FM, Greeff JM, Qiu B-L. The intracellular bacterium Wolbachia uses parasitoid wasps as phoretic vectors for efficient horizontal transmission. PLOS Pathog. 2015;11(2):e1004672.
Furihata S, Hirata M, Matsumoto H, Hayakawa Y. Bacteria endosymbiont, Wolbachia, promotes parasitism of parasitoid wasp Asobara japonica. PLoS ONE. 2015;10:e0140914.
Sullivan DJ, Volkl W. HYPERPARASITISM: multitrophic ecology and behavior. Annu Rev Entomol. 1999;44(1):291–315.
van Nouhuys S, Kohonen M, Duplouy A. Wolbachia increases the susceptibility of a parasitoid wasp to hyperparasitism. J Exp Biol. 2016;219:2984–90.
Poelman EH, Bruinsma M, Zhu F, Weldegergis BT, Boursault AE, Jongema Y, van Loon JJA, Vet LEM, Harvey JA, Dicke M. Hyperparasitoids use herbivore-induced plant volatiles to locate their parasitoid host. PLoS Biol. 2012;10:e1001435.
Zhu F, Broekgaarden C, Weldegergis BT, Harvey JA, Vosman B, Dicke M, Poelman E. Parasitism overrides herbivore identity allowing hyperparasitoids to locate their parasitoid host using herbivore-induced plant volatiles. Mol Ecol. 2015;24:2886–99.
Zhu F, Weldegergis B, Lhie B, Harvey J, Dicke M, Poelman E. Body odors of parasitized caterpillars give away the presence of parasitoid larvae to their primary hyperparasitoid enemies. J Chem Ecol. 2014;40:986–95.
James AG, Austin CJ, Cox DS, Taylor D, Calvert R. Microbiological and biochemical origins of human axillary odour. FEMS Microbiol Ecol. 2013;83:527–40.
Vernier CL, Chin IM, Adu-Oppong B, Krupp JJ, Levine J, Dantas G, Ben-Shahar Y. The gut microbiome defines social group membership in honey bee colonies. Sci Adv. 2020;6:eabd3431.
Zhu F, Cusumano A, Bloem J, Weldegergis BT, Villela A, Fatouros NE, van Loon JJA, Dicke M, Harvey JA, Vogel H, Poelman E. Symbiotic polydnavirus and venom reveal parasitoid to its hyperparasitoids. Proc Natl Acad Sci. 2018;115:5205–10.
Brodeur J, Geervliet JBF, Vet LEM. The role of host species, age and defensive behaviour on ovipositional decisions in a solitary specialist and gregarious generalist parasitoid (Cotesia species). Entomol Exp Appl. 1996;81:125–32.
Greenstone MH, Weber DC, Coudron TA, Payton ME, Hu JS. Removing external DNA contamination from arthropod predators destined for molecular gut-content analysis. Mol Ecol Resour. 2012;12:464–9.
Binetruy F, Dupraz M, Buysse M, Duron O. Surface sterilization methods impact measures of internal microbial diversity in ticks. Parasit Vectors. 2019;12:268.
Traugott M, Zangerl P, Juen A, Schallhart N, Pfiffner L. Detecting key parasitoids of lepidopteran pests by multiplex PCR. Biol Contr. 2006;39:39–46.
van Nouhuys S, Shaw MR, Stefanescu C. Parasitoids of European butterflies. Ecology of butterflies of Europe. Cambridge: Cambridge University Press; 2009. p. 130–56.
Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the Miseq Illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20.
Bokulich NA, Mills DA. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl Environ Microbiol. 2013;79:2519–26.
Borremans A, Crauwels S, Vandeweyer D, Smets R, Verreth C, Van Der Borght M, Lievens B, Van Campenhout L. Comparison of six commercial meat starter cultures for the fermentation of yellow mealworm (Tenebrio molitor) paste. Microorganisms. 2019;7:540.
Doudoumis V, Tsiamis G, Wamwiri F, Brelsfoard C, Alam U, Aksoy E, Dalaperas S, Abd-Alla A, Ouma J, Takac P, Aksoy S, Bourtzis K. Detection and characterization of Wolbachia infections in laboratory and natural populations of different species of tsetse flies (genus Glossina). BMC Microbiol. 2012;12:S3.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
Edgar RC, UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv. 2016;081257.
Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–43.
Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31:3476–82.
De Filippis F, Laiola M, Blaiotta G, Ercolini D. Different amplicon targets for sequencing-based studies of fungal diversity. Appl Environ Microbiol. 2017;83.
Bossaert S, Winne V, Van Opstaele F, Buyse J, Verreth C, Herrera-Malaver B, Van Geel M, Verstrepen KJ, Crauwels S, De Rouck G, Lievens B. Description of the temporal dynamics in microbial community composition and beer chemistry in sour beer production via barrel ageing of finished beers. Int J Food Microbiol. 2020;339:109030.
McKnight DT, Huerlimann R, Bower DS, Schwarzkopf L, Alford RA, Zenger KR. microDecon: a highly accurate read-subtraction tool for the post-sequencing removal of contamination in metabarcoding studies. Environ DNA. 2019;1:14–25.
Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226.
R Core Team. R: a language and environment for statistical computing (Version 3.5.2, R foundation for statistical computing, Vienna, Austria) 2018. https://www.R-project.org
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217.
Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27(3):379–423.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin P, O’Hara R, Simpson G, Solymos P, Stevens MHH, Wagner H. Vegan: community ecology package. R package vegan, vers. 2.2-1. 2015.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
Acknowledgements
The authors would like to thank the different growers involved in this study for letting us sample their fields. Furthermore, we thank Pieter Rouweler and André Gidding for rearing of the insects. Two anonymous referees provided constructive feedback on a previous version of this manuscript.
Funding
This work was supported by the Flemish Fund for Scientific Research (FWO; G.0961.19 N) and the Dutch Research Council, NWO (ALW Open Programme ALWOP.343 to M.D. and ALW Open Programme ALWOP.368 to E.H.P).
Author information
Authors and Affiliations
Contributions
GG, MD, EP, HJ and BL conceived the study. MB, CV and SB aided in the execution of the study. GG, MB and LW collected samples. GG performed most of the laboratory work and analyzed the data with the help of HJ. SC aided in the analysis of the sequence data. Pictures were made by MB. GG, HJ and BL led the writing of the manuscript. All authors contributed critically to the drafts and approved publication of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
zOTUs bacteria. Identification of bacterial zero radius operational taxonomic units (ZOTUs) according to the Silva v1.23 database and distribution over the investigated samples. Table S2. OTUs fungi. Identification of fungal operational taxonomic units (OTUs) according to the UNITE v6 database and distribution over the investigated samples. Table S3. 3 way ANOVA. Results of three way ANOVA of observed richness, Shannon diversity and densities for bacteria and fungi (caterpillars only). Table S4. Diversity bacteria. Diversity metrics for the bacterial communities. Table S5. Diversity fungi. Diversity metrics for the fungal communities. Table S6. Copy number bacteria. Determination of bacterial 16S rRNA gene copy numbers using qPCR. Table S7. Copy number fungi. Determination of fungal ITS copy numbers using qPCR. Table S8. Wolbachia detection. Occurrence of Wolbachia in studied samples, as determined using a specific Wolbachia PCR. Table S9. Plating results. Plating results: log number of colony forming units (cfu) per caterpillar (n = 2 per subgroup of health status). Table S10. Sample details. Sample details, including number of caterpillars used in this study. Table S11. Illumina primers V4. Primer design and sample-specific barcodes for the bacterial V4 region of the 16S rRNA gene. Table S12. Illumina primers ITS. Primer design and sample-specific barcodes for the fungal ITS region. Table S13. Mocks. Composition of mock communities.
Additional file 2: Fig. S1.
Rarefaction curves for the different samples studied, based on the bacterial V4 dataset (A) and the fungal ITS dataset (B). Rarefaction curves approached saturation, indicating that our sequencing depth was sufficient to cover the microbial diversity. Fig. S2. Fungal community profiles of the different caterpillars (Pieris brassicae) and parasitoid larvae (Cotesia glomerata) samples studied. Fungal taxa represent the most prevalent taxa in the different subgroups based on origin and health status for caterpillars and origin for parasitoid larvae (present at a mean relative abundance > 1% in at least one subgroup). For each OTU, the average relative abundance for each subgroup is given in the box as a percentage, whereas the color indicates prevalence (white is absent). OTUs are identified by a BLAST search against GenBank excluding uncultured/environmental sample sequences. Identifications were performed at genus level; when identical scores were obtained for different genera, identifications were performed at a higher taxonomic level. When identity percentages were lower than 99%, the percentage of sequence identity with the GenBank entry is given between brackets. Abbreviations used: H = healthy; P = parasitized; E = external; and I = internal.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gloder, G., Bourne, M.E., Verreth, C. et al. Parasitism by endoparasitoid wasps alters the internal but not the external microbiome in host caterpillars. anim microbiome 3, 73 (2021). https://doi.org/10.1186/s42523-021-00135-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-021-00135-y