
Abstract
Background
Autophagy plays essential roles in the development and pathogenesis of mesial temporal lobe epilepsy (mTLE). In this research, we aim to identify and validate the autophagy-related genes associated with mTLE through bioinformatics analysis and experimental validations.
Methods
We obtained the dataset GSE143272 and high-throughput sequencing results of mTLE from public databases. Potential differentially expressed autophagy-related genes related to mTLE were identified using R software. Subsequently, genomes pathway enrichment analysis, protein-protein interactions (PPIs), and the gene ontology (GO) enrichment were performed for the selected autophagy-related genes. The mRNA expression profiles of hub genes were then used to establish a least absolute shrinkage and selection operator (LASSO) model. Finally, seven hub candidate autophagy-related genes were confirmed in hippocampus using the lithium-pilocarpine chronic epilepsy model.
Results
A total of 40 differential expression genes (DEGs) among the core autophagy-related genes were identified. The analysis results of PPI revealed that interactions among these DEGs. KEGG pathway and GO analysis of selected candidate autophagy-related genes indicated that those enriched terms mainly focused on macroautophagy, regulation of autophagy, cellular response to extracellular stimulus and mitochondrion disassembly. The results suggested that SQSTM1, VEGFA, BNIP and WIPI2 were consistent with the bioinformatics analysis. The expression levels of SQSTM1 and VEGFA in epilepsy model samples were significantly higher than those in normal control, while BNIP and WIPI2 expression levels were notably decreased. The final hub gene-based LASSO regression model accurately predicted the occurrence of epilepsy (AUC = 0.88).
Conclusions
Through bioinformatics analysis of public data, we identified 40 candidate autophagy-related genes associated with mTLE. SQSTM1, VEGFA, BNIP and WIPI2 may play significant roles in autophagy, influencing the onset and development of mTLE by regulating autophagy pathway. These findings deepen our understanding of mTLE, and may serve as sensitive and valuable indicators for the prognosis and diagnosis of this condition.
Similar content being viewed by others


Background
Epilepsy is a chronic nervous system disorder characterized by recurrent and unprovoked seizures [1]. Temporal lobe epilepsy (TLE) is one of the most common types of epilepsy in adults, often originating in childhood. Mesial temporal lobe epilepsy (mTLE) is usually associated with hippocampal sclerosis (HS), which is the most prevalent neuropathological discovery [2]. Despite being considered as a polygenic and complex disorder, the pathological mechanisms underlying mTLE with HS remain largely unknown. Autophagy, an intracellular catabolic process, plays a crucial role in removing protein aggregates and damaged intracellular organelles by transporting them to lysosomes [3], thereby maintaining cell health and cellular homeostasis. However, abnormal autophagy can lead to various neurological disorders, including epilepsy. Notably, studies have suggested a correlation between autophagy and epilepsy [4], with autophagic impairment being implicated in conditions, like focal cortical dysplasia, tuberous sclerosis complex and Lafora disease [5,6,7]. Experimental models have shown that suppressing autophagy can induce epilepsy, while rescuing autophagy can prevent it [8, 9]. Interestingly, epilepsy itself can lead to dysregulated autophagy. Xiao et al. observed increased apoptosis and decreased mitochondrial autophagy in the hippocampus of epileptic rats induced lithium chloride pilocarpine injection [10]. Moreover, studies have reported several potential mechanisms through which autophagy may influence epileptogenesis, including aberrant substrate accumulation, the mammalian target of rapamycin (mTOR) pathway, and the formation of epileptogenic networks [11, 12]. However, the role of autophagy in the pathogenesis of epilepsy is still unclear, highlighting the need for further investigation. Therefore, this study aims to identify autophagic candidate genes that may play an important role in the pathogenesis of mTLE, and potentially serving as sensitive indicators for mTLE.
Methods
Genes associated with autophagy and microarray data
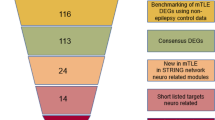
The research workflow is illustrated in Fig. 1. Candidate autophagy-related genes were identified by searching the Human Autophagy Database. Subsequently, we utilized GeneCards: the Human Gene Database to extract epilepsy medical subject headings (MeSH) terms and other epilepsy-related textual terms. These results were then combined with the previous public high-throughput sequencing data of mTLE to track the expression profiles of mTLE-related autophagy genes [13]. The dataset comprised temporal cortical samples from 75 neurologically healthy individuals and 85 patients with confirmed neuropathological mTLE + HS. Exon and gene-level transcriptome analysis were performed, residual methods were applied to alter the data, and the false detection rate (FDR) threshold was set at 5%.

Workflow analysis
Abbreviations: LASSO: Least absolute shrinkage and selection operator; mTLE: Mesial temporal lobe epilepsy; PPI: Protein-protein interaction; ROC: Receiver operating characteristic
Functional enrichment analysis of autophagy-related genes
The candidate autophagy-related genes in mTLE underwent KEGG pathway enrichment and GO functional enrichment analysis using R software. The GO analysis included cellular component (CC), biological process (BP), and molecular function (MF).
PPI construction and identification of hub module
PPI networks were constructed using the STRING database. Subsequently, the cytoHubba plugin of the Cytoscape software was employed to identify the hub module. The cytoHubba plugin ranks nodes according to their properties in the network, facilitating the identification of key targets and sub-networks within complex networks.
Screening of differential blood and brain autophagy-related mRNA expression profiles in mTLE
Peripheral blood mRNA expression profiles were acquired from the public database GEO (GSE143272). Total RNA samples were initially collected from peripheral blood samples of 34 drug-naive epilepsy patients and 57 patients exhibiting differential response to anti-seizure medication monotherapy, as well as 50 healthy individuals as controls. Subsequently, the peripheral blood expression profiles of patients with epilepsy receiving or not receiving anti-seizure medication monotherapy and their responses to the treatment were acquired. By comparing the gene expression profiles of drug-naive epilepsy patients with those of healthy controls in the GSE143272 database, mRNA expression biomarkers associated with epilepsy and the anti-seizure medication response were identified. These expression profiles exhibited significant differences and potential differential expression genes (DEGs) among the above three groups of microarray data and were used for further analysis.
Correlation analysis of the candidate DEGs associated with autophagy
Spearman correlation analysis was conducted using the “corrplot” package in R software to determine the correlations among the identified DEGs associated with autophagy.
LASSO-Cox regression model analysis
The mRNA expression profiles of the hub genes were extracted to construct a LASSO model. Using the R software package “glmnet”, we integrated the time of onset, the presence of epilepsy, and gene expression data, applying the LASSO-Cox method for regression analysis. A 5-fold cross-validation was implemented in the LASSO-Cox regression model to identify the optimal values.
Establishment of pilocarpine-induced chronic seizures model
A total of 20 Sprague-dawley (SD) rats, aged 8–9 weeks, male, weighing 160–200 g, were obtained from Beijing Vitleyhua Experimental Animal Co., Ltd. All rats were provided with ad libitum access to food and water. Before the experiment, the rats were adapted to the environment for at least 3 days. All procedures are in line with the “Regulations on Experimental Animal Management”. On the first day of the experiment, the rats were injected with lithium chloride. Then after 24 h, they received an injection of methylbromine (1 mg/kg, Macklin S835305), followed by an abdominal injection of pilocarpine (initial dose of 30 mg/kg, TargetMol T0804) for 30 min. Additional doses of pilocarpine were injected every 30 min at 10 mg/kg increments until the rats reached IV (bilateral forelimb clonus with rearing and falling) or status epileptics (SE) – without obvious intervals, with the maximum dose of 60 mg/ kg. Two hours following the onset of, epileptic seizures, diazepam was administered to terminate the seizures. The symptoms of seizures in rats were graded according to the Racine grading criteria. Subsequently, all rats exhibiting spontaneous recurrent seizures were designated as chronic epilepsy animals and were randomly selected for EEG recording.
RNA isolation and quantitative reverse-transcription polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the RNA extraction kit (DP501, TIANGEN, China) following the manufacturer’s protocol for RNA RT-qPCR. RNA quantity was determined using Nanodrop (Thermo-Fisher). First-strand cDNA synthesis was carried out with the cDNA synthesis kit (KR118, TIANGEN, China) according to the manufacturer’s instructions. The qPCRs were conducted in a Quant-studio 6 flex Real-Time PCR system (Applied Biosystems) with SYBR Green kit (FP209, TIANGEN, China). The primers used were: 5’− GCTGGTTCGTGGTGGACTTCATC − 3’ and 5’− TGCTCTGGCGGTCTTGTAAACTTC − 3’ (Sangon Biotech, China). β-actin primers were used as a reference (B661202 Sangon Biotech). The relative expression levels were calculated using the 2−ΔΔCt method.
Statistical analysis
The t-test was performed using GraphPad Prism 9 software. For differential gene expression analysis, a criteria of |log2FC| > 0.5, and a P-value less than 0.05 was considered to be statistically significant.
Results
Genes associated with autophagy in mTLE
A total of 233 autophagy-related genes were initially identified from the HADb. Subsequently, 40 autophagy-related genes were selected based on differential expression mTLE, comprising 23 upregulated genes and 17 downregulated genes. The summary of these findings is presented in Fig. 2a, with detailed information available in Supplement Table 1.

Identification of autophagy-related genes and functional enrichment analysis. a Screening autophagy-related DEGs in mTLE. b GO enrichment analysis of genes associated with autophagy in mTLE. c Chord diagram illustrating top 10 KEGG enrichment analysis results
Functional and pathway enrichment analysis
The 40 differentially expressed autophagy-related genes associated with mTLE were included in KEGG pathway and GO term analysis. The top enriched BP included cellular response to extracellular stimulus, regulation of autophagy, macroautophagy, and mitochondrion disassembly. GO terms related to CC focused on phagophore assembly site, autophagosome, and plasma membrane bounded cell projection cytoplasm. GO terms related to MF included protein serine/threonine kinase activity, ubiquitin protein ligase binding, and protease binding. Moreover, KEGG pathway enrichment analysis identified FoxO signaling pathway, autophagy, apoptosis and mTOR signaling pathway (Fig. 2b and c).
Identification of the hub module of mTLE autophagy-related genes
With the application of STRING database, PPI network of autophagy related genes was constructed. Next, the MCODE plugin was applied for module analysis. The PPI network was primarily divided into three main modules (Fig. 3a and b). Among these modules, a hub module consisting of 20 genes with high connectivity throughout the PPI network was identified and may play a crucial role in physiological processes (Fig. 3c; Table 1).

PPI network of autophagy related candidate genes and central module in mTLE. a Autophagy-related genes associated with mTLE were categorized into three major modules. b The PPI network of mTLE autophagy-related genes depicted red dots representing up-regulated genes, green dots representing down-regulated genes, and black lines indicating interactions between the encoded proteins. c The PPI network of hub module genes
Screening of differential blood and brain autophagy-related gene expression profiles and correlation analysis of the DEGs
Firstly, the public dataset GSE143272 was normalized using the “normalize between arrays” function of the limma package. Subsequent analysis of this dataset with R software, unveiled 3399 autophagy related genes comprising 1571 upregulated genes and 1828 downregulated genes, demonstrating differential expression between the epilepsy and control groups in blood, visually displayed in the form of volcano plots (Fig. 4a). A Venn diagram depicted the common DEGs in blood and brain tissue (Fig. 4b). Moreover, violin plots and heatmaps showing the expression of 14 autophagy-related DEGs in peripheral blood samples from individuals with mTLE compared to controls (Fig. 4c and d). Figure 4e presented the correlation of these 14 DEGs of autophagy within the GSE143272 dataset.

DEGs of autophagy and Spearman correlation analysis of the 14 hub genes. a A Volcano plot of the 3399 autophagy-related DEGs in the blood of patients with epilepsy. b Venn diagrams of the screened dysregulated genes in epilepsy and the hub genes. The blue part indicates the epilepsy-related genes in blood and brain tissue, while the yellow part indicates the hub genes. c A heatmap of the 14 autophagy-related DEGs in mTLE and healthy controls. d The violin plots of 14 autophagy-related DEGs in blood of epilepsy patients and controls. e Spearman correlation analysis of the 14 autophagy-related DEGs
Potential predictive marker of epilepsy in LASSO model
As the lambda increases, the coefficients of less important variables tend to shrink to zero, while the coefficients of more important variables are more likely to be retained. Seven genes, namely SQSTM1, BNIP3, FOXO3, WIPI2, SERPINA1, ERBB2, and VEGFA, were identified with non-zero regression coefficients, and the value of lambda.min = 0.0925551197393762 was chosen. The gene-based model index was established using the following formula: Index = 0.913938338631802*SQSTM1–0.451510329994845*BNIP3 + 0.271284377754037*FOXO3 + 0.241742532558963*WIPI2 + 0.730859248215233*SERPINA1–0.861673960375642*ERBB2 + 0.0451370867086589*VEGFA (Fig. 5; Table 2)

The model for predicting epilepsy. a LASSO regression model. b ROC curves analysis of dataset. c ROC curves analysis of seven genes
Data validation using qPCR
To confirm the reliability of our analysis results, the mRNA expression levels of seven autophagy-related DEGs were further validated by qPCR in samples from our animal model. The results indicated that SQSTM1 and VEGFA exhibited significantly higher expression levels in epilepsy rats compared to the control group. Conversely, the mRNA expression levels of BNIP and WIPI2 were notably decreased. No significant differences was observed in the expression levels of SERPINA1, ERBB2, and FOXO3 (Fig. 6).

mRNA expression of seven autophagy-related genes
Discussion
Structural, genetic, infectious, metabolic, and immune factors, either singly or in combination, can contribute to epileptogenesis in mTLE [14]. Moreover, the etiologies of mTLE vary among individuals. Accumulating evidence suggests the involvement of autophagy in epileptogenesis related to mTLE. For instance, a previous study demonstrated increased levels of autophagy markers, such as p-mTOR/m-TOR ratio, LC3-II, and phospho-Akt/Akt ratios, along with the occurrence of epileptiform discharge originating from the hippocampus or limbic cortical regions following kainic acid (KA) administration in mice, while establishing temporal lobe epilepsy models [15]. Additionally, another study revealed a sudden increasing in autophagy markers, namely beclin-1 and LC3II/LC3I in pilocarpine epilepsy model [16]. Nevertheless, further comprehensive validation is essential to enhance our understanding of the role of autophagy in the initiation and progression of mTLE.
Recently, a growing number of researches have focused on the relationship between autophagy (including autophagy-related factors or marker proteins) and mTLE. Aronica et al. identified miRNA-146a (miR-146a) as a potential regulator of astrocyte inflammation in TLE, observed in both rats and human subjects [17]. Another study highlighted a significant increase in MAP1LC3, phospho-mTOR/mTOR, and Beclin1 levels following seizure induction in rat models, implying a role of autophagic factors in the pathogenesis of TLE [18]. In addition, emerging evidence has indicated that regulation of Beclin1, a key autophagy-related molecule, could represent a therapeutic approach for managing epilepsy [19]. However, research in this area remains limited and warrants further investigation.
Currently, a multitude of studies have explored autophagy-related genes, such as WDR45 [9, 20], CYLD [21], and 4E-BP2 [22], which have been linked with epilepsy development. Despite this, bioinformatics analysis focusing on autophagy genes in mTLE has been scarce untill now. This study has identified 40 candidate autophagy-related genes in mTLE by bioinformatics analysis, with ATG16L1 being among the identified genes. A study reported that increasing ATG16L1 levels through antagomir-223 treatment alleviated epilepsy in KA-treated mice, suggesting the microRNA-223/ATG16L1 pathway may offer a novel treatment option for TLE [23]. Additionally, Castaneda-Cabral et al. found an elevation of VEGF-A in microvasculature of patients with TLE, which indicating a connection to the blood-brain barrier dysfunction [24].
Our KEGG analysis has revealed that the identified target genes are predominantly enriched in autophagy, apoptosis, mTOR signaling pathway, and FoxO signaling pathway. In mTLE with HS, there is a loss of neuronal populations and gliosis in the hippocampal region, whereas the temporal cortex exhibits fewer alterations, whose pathogenesis and progression are closely related to autophagy. Gao et al. demonstrated that electroacupuncture treatment for TLE can promote autophagy via the AKT/mTOR signaling pathway [25]. Wen et al. showed that miR-421, which targets MYD88, can suppress autophagy in hippocampal neurons of epileptic mice by down-regulating the TLR/MYD88 pathway [26]. In summary, autophagy plays a key role in neurological disorders, especially epilepsy. Thus, it is essential to reveal the potential biological functions of these DEGs related to autophagy.
The qRT-PCR validation of the expression levels of seven genes, including SERPINA1, BNIP, ERBB2, SQSTM1, WIPI2, FOXO3, and VEGFA, corroborated the analysis results. We found that SQSTM1 and VEGFA were significantly upregulated, while BNIP and WIPI2 were significantly downregulated in epilepsy model.
Vascular endothelial growth factor A (VEGFA) is a dimeric glycoprotein secreted by neural tube cells during brain development. Serving as a crucial co-factor between the vascular and nervous systems, VEGFA plays a significant role in endothelial cell proliferation angiogenesis, migration, and neural stem cells proliferation, exerting its biological function by binding to VEGFR2 in the nervous system [27]. It was first isolated and purified from cultured bovine pituitary astrocytes by Ferrara et al. in 1989 [28]. Castaneda-Cabral et al. found elevated VEGFA levels in the neocortex of patients with drug-resistant TLE (DR-TLE) [29], underscoring its potential involvement in TLE pathogenesis. In the rat model of TLE, VEGFA has been observed to be up-regulated early in neurons and glial cells in the hippocampus after SE [30, 31]. Furthermore, several studies have highlighted the antiepileptic effects of VEGFA in pilocarpine-induced SE rats. VEGFA administration has been shown to reduce neuronal apoptosis post-seizure induction, promote survival and, proliferation of neural stem cells, and facilitate nerve repair. These findings suggest a potential neuroprotective role for VEGFA following SE, positioning it as a promising neurovascular molecular target [32,33,34]. However, VEGFA also have a dual role in epilepsy, as it has been linked to blood-brain barrier disruption and epileptogenic inflammation, contributing to the development of epilepsy [35, 36].
WIPI2, a human homolog of yeast ATG18, containing three WD-repeats and a seven-bladed b-propeller structure with conserved motifs that facilitates its interaction with other proteins, plays a vital role in phagophore formation, the initial step of autophagy [37]. By interacting with ATG16L1, WIPI2b recruits the ATG12–ATG5–ATG16L1 complex to the phagophore, which is required for LC3 lipidation and autophagosome formation [38, 39]. Dooley et al. discovered that WIPI2 mutants fail to bind ATG16L1, leading to the inhibition of LC3 lipidation and the formation of LC3 puncta [40]. In a case report involving three siblings, it was noted that individuals with homozygous missense WIPI2 variants exhibited early central nervous system (CNS) involvement, presenting with recurrent and refractory generalized myoclonic and tonic seizures. Furthermore, their EEG results revealed bilateral temporal epileptiform activity [41]. These observations provide evidence linking mutation in WIPI2, a major autophagy gene, to the development of epilepsy.
BNIP3, an atypical BH3-only protein share homology with BCL2, plays a dual role in regulating programmed cell death [42] and serving as a potent inducer of autophagy in various cell types [43, 44]. The functional impact of BNIP3-mediated autophagy differs according to the cell type and context, but it is evidently implicated in the pathogenesis of epilepsy. In PTZ-induced epileptic models, the knock-out of TRPM2 has shown efficacy in ameliorating epilepsy-induced hippocampal pathological damages, probably via the PARP1 downstream signaling pathway involving BNIP3 [45]. Neuronal ceroid lipofuscinoses (NCLs) associated with mutations in the CLN8 gene can present as progressive epilepsy, with BNIP3 identified as potential protein partners of CLN8, further underlining its role in epileptogenesis [46].
SQSTM1 (p62) is a scaffold protein with PB1 and UBA domains, as well as a TRAF6 binding sequence [47]. In 1998, Shin noticed the unique cytoplasmic punctate structure formed by p62 and created the name sequestosome1 (SQSTM1) based on this characteristic [48]. SQSTM1 is implicated in cell signaling, differentiation, and particularly in clearance of toxic protein aggregates [47]. Depletion of p62 in conjunction with autophagy inhibition has been shown to prevent the accumulation of ubiquitin-positive protein aggregates, suggesting the essential role of p62 in basal autophagy [49]. The significance of p62 as a selective autophagy adaptor for the aggregation and clearance of misfolded proteins is reflected in the epileptogenesis. He et al. found that p62 expression was remarkably high in patients with TLE and in the epileptic hippocampus of mice [23]. In a mouse model of myoclonic epilepsy of Lafora, the absence of p62 impaired glycogen aggregation, exacerbated pathology, and increased susceptibility to epilepsy [50]. These findings provide valuable insights for the development of novel treatment strategies for epilepsy.
In addition, we extracted the mRNA expression profiles of candidate genes to construct the LASSO model, which identified seven genes with non-zero regression coefficients. Among these genes, some have been previously reported to be associated with epilepsy. Analysis of the ROC curve indicated that the LASSO model exhibits a high AUC value, suggesting its potential utility as a biomarker for epilepsy.
In this study, we have unveiled the potential significance of SQSTM1, VEGFA, BNIP, and WIPI2 in autophagy and their subsequent impact on the initiation and progression of mTLE. We propose that these genetic markers could function as diagnostic and therapeutic indicators for clinical application in epileptic patients. Particularly, the impaired degradation of the upregulated SQSTM1 by autophagy leads to its accumulation [51], contributing to heightened seizure susceptibility [52]. Similarly, BNIP has emerged as a reliable indicator of mTLE, with alterations in its gene expression levels causing autophagy dysfunction and consequently elevating the risk of epilepsy [53]. By detecting and analyzing the expression levels of these genes, we anticipate more precise epilepsy risk prediction in patients. This predictive capability could pave the way for the development of targeted and personalized treatment strategies, ultimately improving patient outcomes.
Although several studies have revealed the relationship between autophagy and epilepsy, the precise molecular mechanism remains elusive. Therefore, future research endeavors should delve deeper into elucidating how these genes influence the autophagy process, ultimately leading to the manifestation of epilepsy. This comprehensive exploration should encompass the complex regulation of gene expression, protein-protein interactions, and signal transduction pathways.
This study acknowledges certain limitations. We identified seven candidate autophagy-related genes associated with mTLE through bioinformatics analysis and the mRNA expression levels of seven DEGs were validated in hippocampus from lithium-pilocarpine chronic epilepsy model. However, there was no significant difference in the expression levels of SERPINA1, ERBB2, and FOXO3, which may be attributed to the limited sample size. Furthermore, while the research demonstrated that bioinformatics analysis can reveal critical insights into molecular pathways underlying epilepsy, the candidate key pathways and genes identified through bioinformatics analysis and molecular experiments require further validation. It is essential to determine to what extent downregulation of those genes contributes to the development of epilepsy.
Conclusions
In conclusion, our study identified 40 potential autophagy-related genes in mTLE were identified through bioinformatics analysis. SQSTM1, VEGFA, BNIP, and WIPI2 may affect the onset and progression of mTLE by regulating autophagy. These findings may deepen our understanding of mTLE and provide valuable biomarkers for epilepsy.
Availability of data and materials
Authors approved the data and materials availability.
Abbreviations
- BP:
-
Biological process
- CC:
-
Cellular component
- CNS:
-
Central nervous system
- DEGs:
-
Differential expression genes
- DR-TLE:
-
Drug-resistant temporal lobe epilepsy
- FDR:
-
False detection rate
- GO:
-
Gene ontology
- HS:
-
Hippocampal sclerosis
- KA:
-
Kainic acid
- LASSO:
-
Least absolute shrinkage and selection operator
- mTLE:
-
Mesial temporal lobe epilepsy
- mTOR:
-
Mammalian target of rapamycin
- MeSH:
-
Medical subject headings
- MF:
-
Molecular function
- NCLs:
-
Neuronal ceroid lipofuscinoses
- PPIs:
-
Protein-protein interactions
- ROC:
-
Receiver operating characteristic
- SE:
-
Status epileptics
- SQSTM1:
-
Sequestosome1
- TLE:
-
Temporal lobe epilepsy
- VEGFA:
-
Vascular endothelial growth factor A
References
Zhang S, Chen F, Zhai F, Liang S. Role of HMGB1/TLR4 and IL-1beta/IL-1R1 signaling pathways in epilepsy. Front Neurol. 2022;13:904225.
Manna I, Fortunato F, De Benedittis S, Sammarra I, Bertoli G, Labate A, et al. Non-coding RNAs: new biomarkers and therapeutic targets for temporal lobe epilepsy. Int J Mol Sci. 2022;23(6):3063.
Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–22.
Giorgi FS, Biagioni F, Lenzi P, Frati A, Fornai F. The role of autophagy in epileptogenesis and in epilepsy-induced neuronal alterations. J Neural Transm (Vienna). 2015;122(6):849–62.
Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035.
Knecht E, Criado-Garcia O, Aguado C, Gayarre J, Duran-Trio L, Garcia-Cabrero AM, et al. Malin knockout mice support a primary role of autophagy in the pathogenesis of Lafora disease. Autophagy. 2012;8(4):701–3.
Marsan E, Baulac S. Review: Mechanistic target of rapamycin (mTOR) pathway, focal cortical dysplasia and epilepsy. Neuropathol Appl Neurobiol. 2018;44(1):6–17.
McMahon J, Huang X, Yang J, Komatsu M, Yue Z, Qian J, et al. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J Neurosci. 2012;32(45):15704–14.
Carvill GL, Liu A, Mandelstam S, Schneider A, Lacroix A, Zemel M, et al. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia. 2018;59(1):e5-13.
Xiao D, Lv J, Zheng Z, Liu Y, Zhang Y, Luo C, et al. Mechanisms of microRNA142 in mitochondrial autophagy and hippocampal damage in a rat model of epilepsy. Int J Mol Med. 2021;47(6):98.
Bockaert J, Marin P. mTOR in brain physiology and pathologies. Physiol Rev. 2015;95(4):1157–87.
Gan J, Qu Y, Li J, Zhao F, Mu D. An evaluation of the links between microRNA, autophagy, and epilepsy. Rev Neurosci. 2015;26(2):225–37.
Guelfi S, Botia JA, Thom M, Ramasamy A, Perona M, Stanyer L, et al. Transcriptomic and genetic analyses reveal potential causal drivers for intractable partial epilepsy. Brain. 2019;142(6):1616–30.
You J, Huang H, Chan CTY, Li L. Pathological targets for treating temporal lobe epilepsy: discoveries from microscale to macroscale. Front Neurol. 2021;12:779558.
Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett. 2007;414(1):57–60.
Cao L, Xu J, Lin Y, Zhao X, Liu X, Chi Z. Autophagy is upregulated in rats with status epilepticus and partly inhibited by vitamin E. Biochem Biophys Res Commun. 2009;379(4):949–53.
Aronica E, Fluiter K, Iyer A, Zurolo E, Vreijling J, van Vliet EA, et al. Expression pattern of miR-146a, an inflammation-associated microRNA, in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2010;31(6):1100–7.
Rami A, Benz A. Exclusive activation of caspase-3 in mossy fibers and altered dynamics of autophagy markers in the mice hippocampus upon status epilepticus induced by kainic acid. Mol Neurobiol. 2018;55(5):4492–503.
Yang M, Lin P, Jing W, Guo H, Chen H, Chen Y, et al. Beclin1 deficiency suppresses epileptic seizures. Front Mol Neurosci. 2022;15:807671.
Cong Y, So V, Tijssen MAJ, Verbeek DS, Reggiori F, Mauthe M. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy. 2021;17(12):3908–23.
Dobson-Stone C, Hallupp M, Shahheydari H, Ragagnin AMG, Chatterton Z, Carew-Jones F, et al. CYLD is a causative gene for frontotemporal dementia - amyotrophic lateral sclerosis. Brain. 2020;143(3):783–99.
Sharma V, Sood R, Lou D, Hung TY, Levesque M, Han Y, et al. 4E-BP2-dependent translation in parvalbumin neurons controls epileptic seizure threshold. Proc Natl Acad Sci U S A. 2021;118(15):e2025522118.
He Z, Chen H, Zhong Y, Yang Q, Wang X, Chen R, et al. MicroRNA 223 targeting ATG16L1 affects microglial autophagy in the kainic acid model of temporal lobe epilepsy. Front Neurol. 2021;12:704550.
Castaneda-Cabral JL, Colunga-Duran A, Urena-Guerrero ME, Beas-Zarate C, Nunez-Lumbreras MLA, Orozco-Suarez S, et al. Expression of VEGF- and tight junction-related proteins in the neocortical microvasculature of patients with drug-resistant temporal lobe epilepsy. Microvasc Res. 2020;132:104059.
Gao D, Ma L, Xie Y, Xiao B, Xue S, Xiao W, et al. Electroacupuncture promotes autophagy by regulating the AKT/mTOR signaling pathway in temporal lobe Epilepsy. Neurochem Res. 2022;47(8):2396–404.
Wen X, Han XR, Wang YJ, Wang S, Shen M, Zhang ZF, et al. MicroRNA-421 suppresses the apoptosis and autophagy of hippocampal neurons in epilepsy mice model by inhibition of the TLR/MYD88 pathway. J Cell Physiol. 2018;233(9):7022–34.
Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, et al. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet. 2004;36(8):827–35.
Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. 1989. Biochem Biophys Res Commun. 2012;425(3):540–7.
Castaneda-Cabral JL, Beas-Zarate C, Rocha-Arrieta LL, Orozco-Suarez SA, Alonso-Vanegas M, Guevara-Guzman R, et al. Increased protein expression of VEGF-A, VEGF-B, VEGF-C and their receptors in the temporal neocortex of pharmacoresistant temporal lobe epilepsy patients. J Neuroimmunol. 2019;328:68–72.
Nicoletti JN, Shah SK, McCloskey DP, Goodman JH, Elkady A, Atassi H, et al. Vascular endothelial growth factor is up-regulated after status epilepticus and protects against seizure-induced neuronal loss in hippocampus. Neuroscience. 2008;151(1):232–41.
Rigau V, Morin M, Rousset MC, de Bock F, Lebrun A, Coubes P, et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130(Pt 7):1942–56.
Nicoletti JN, Lenzer J, Salerni EA, Shah SK, Elkady A, Khalid S, et al. Vascular endothelial growth factor attenuates status epilepticus-induced behavioral impairments in rats. Epilepsy Behav. 2010;19(3):272–7.
Lenzer-Fanara JR, Li T, Salerni EA, Payen F, Croll SD. VEGF treatment during status epilepticus attenuates long-term seizure-associated alterations in astrocyte morphology. Epilepsy Behav. 2017;70(Pt A):33–44.
Han W, Jiang L, Song X, Li T, Chen H, Cheng L. VEGF modulates neurogenesis and microvascular remodeling in epileptogenesis after status epilepticus in immature rats. Front Neurol. 2021;12:808568.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–96.
van Vliet EA, da Costa Araujo S, Redeker S, van Schaik R, Aronica E, Gorter JA. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain. 2007;130(Pt 2):521–34.
Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–7.
Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–22.
Dooley HC, Wilson MI, Tooze SA. WIPI2B links PtdIns3P to LC3 lipidation through binding ATG16L1. Autophagy. 2015;11(1):190–1.
Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55(2):238–52.
Maroofian R, Gubas A, Kaiyrzhanov R, Scala M, Hundallah K, Severino M, et al. Homozygous missense WIPI2 variants cause a congenital disorder of autophagy with neurodevelopmental impairments of variable clinical severity and disease course. Brain Commun. 2021;3(3):fcab183.
Park CW, Hong SM, Kim ES, Kwon JH, Kim KT, Nam HG, et al. BNIP3 is degraded by ULK1-dependent autophagy via MTORC1 and AMPK. Autophagy. 2013;9(3):345–60.
Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14(1):146–57.
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–81.
Zheng Q, Zhu T, Hu H, Zhao Y, Ying Y, Luo X, et al. TRPM2 ion channel is involved in the aggravation of cognitive impairment and down regulation of epilepsy threshold in pentylenetetrazole-induced kindling mice. Brain Res Bull. 2020;155:48–60.
Passantino R, Cascio C, Deidda I, Galizzi G, Russo D, Spedale G, et al. Identifying protein partners of CLN8, an ER-resident protein involved in neuronal ceroid lipofuscinosis. Biochim Biophys Acta. 2013;1833(3):529–40.
Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32(2):95–100.
Shin J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res. 1998;21(6):629–33.
Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45.
Pellegrini P, Hervera A, Varea O, Brewer MK, Lopez-Soldado I, Guitart A, et al. Lack of p62 impairs glycogen aggregation and exacerbates Pathology in a mouse model of myoclonic epilepsy of Lafora. Mol Neurobiol. 2022;59(2):1214–29.
Li T, Zhang H, Wang Z, Gao S, Zhang X, Zhu H, et al. The regulation of autophagy by the miR-199a-5p/p62 axis was a potential mechanism of small cell lung cancer cisplatin resistance. Cancer Cell Int. 2022;22(1):120.
Ying C, Ying L, Yanxia L, Le W, Lili C. High mobility group box 1 antibody represses autophagy and alleviates hippocampus damage in pilocarpine-induced mouse epilepsy model. Acta Histochem. 2020;122(2):151485.
Cho Y, Jeong YJ, Song KH, Chung IK, Magae J, Kwon TK, et al. 4-O-Methylascochlorin-mediated BNIP-3 expression controls the balance of apoptosis and autophagy in cervical carcinoma cells. Int J Mol Sci. 2022;23(23):15138.
Acknowledgements
We acknowledge HA and GEO database for providing their platforms and contributors for uploading their meaningful datasets.
Funding
This work was funded by grants from the National Natural Science Foundation of China (82071447, 81571266, 81771405) and the Sanming Project of Medicine in Shenzhen (No. SZSM201911003). The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
LZ and SC conceived the idea and made revisions to this paper; MY, YL and XL collected and analyzed the data and drafted the paper; SZ, LL and QZ participated in the information registration and performed the statistical analysis. YZ and YF contributed to the draft of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Animal Care and Use Committee, Sun Yat-Sen University (Approval No. SYSU-IACUC-2022-B1339).
Consent for publication
Not applicable.
Competing interests
Author Liemin Zhou is the member of the Editorial Board for Acta Epileptologica, who was not involved in the journal’s review of, or decisions related to this manuscript.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, M., Li, Y., Liu, X. et al. Autophagy-related genes in mesial temporal lobe epilepsy: an integrated bioinformatics analysis. Acta Epileptologica 6, 16 (2024). https://doi.org/10.1186/s42494-024-00160-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42494-024-00160-9