
Abstract
Introduction
Choreiform movement disorders are characterized by involuntary, rapid, irregular, and unpredictable movements of the limbs, face, neck, and trunk. These movements often initially go unnoticed by the affected individuals and may blend together with seemingly intended, random motions. Choreiform movements can occur both at rest and during voluntary movements. They typically increase in intensity with stress and physical activity and essentially cease during deep sleep stages. In particularly in advanced stages of Huntington disease (HD), choreiform hyperkinesia occurs alongside with dystonic postures of the limbs or trunk before they typically decrease in intensity.
Summary or definition of the topic
The differential diagnosis of HD can be complex. Here, the authors aim to provide guidance for the diagnostic process. This guidance was prepared for the German Neurological Society (DGN) for German-speaking countries.
Recommendations
Hereditary (inherited) and non-hereditary (non-inherited) forms of chorea can be distinguished. Therefore, the family history is crucial. However, even in conditions with autosomal-dominant transmission such as HD, unremarkable family histories do not necessarily rule out a hereditary form (e.g., in cases of early deceased or unknown parents, uncertainties in familial relationships, as well as in offspring of parents with CAG repeats in the expandable range (27–35 CAG repeats) which may display expansions into the pathogenic range).
Conclusions
The differential diagnosis of chorea can be challenging. This guidance prepared for the German Neurological Society (DGN) reflects the state of the art as of 2023.
Similar content being viewed by others


Introduction
This guideline is an abridged and translated short version of the guidance on chorea and Huntington’s Disease prepared for the German Neurological Society, covering diagnostic aspects as well as symptomatic treatment options. A complete version of this guideline (in German) can be found on the website of the Deutsche Gesellschaft für Neurologie (www.dgn.org/leitlinien) and the AWMF (Arbeitsgemeinschaft wissenschaftlicher Medizinischer Gesellschaften).
AWMF registry No. Is 030/028; level of guideline: S2k; last update: November 24th, 2022; Valid until: November 23th, 2027; source: https://register.awmf.org/de/leitlinien/detail/030-028
This guideline has been approved by the German Neurological Society (DGN) and the German Association for Psychiatry, Psychotherapy, and Psychosomatics; German Society of Human Genetics; Swiss Neurological Society; Austrian Society of Neurology, and was reviewed by the German HD Association (Deutsche Huntington-Hilfe e.V.)
What´s new?
The differential diagnosis of chorea can be complex. Taking a family history can be helpful in many cases. A variety of new mutations causing chorea as a symptom has been described. For sporadic cases, it can be helpful to differentiate based on: course, age at disease onset, symmetry, additional neurological, clinical and paraclinical findings such as concurrent ataxia, polyneuropathy, cutaneous or scleral telangiectasias, liver pathology, laboratory abnormalities or specific MRI findings.
Guidelines in detail
The recommendations of this guideline were established through a Delphi process (strength of consensus > 75–95% for all recommendations), achieved over two rounds of voting. All statements for which consensus strength is not specified were met with a consensus of > 95%.
Hereditary disease entities presenting with chorea or featuring choreiform movements
-
C9orf72 expansion mutation (frontotemporal dementia, motor neuron disease and movement disorders, probably frequent phenocopy of HD [43])
-
Spinocerebellar ataxia 17 (corresponds to Huntington‘s-disease-like-4; HDL4 [95])
-
Spinocerebellar ataxia types 3, 2, 1 and 7 [78]
-
Spinocerebellar ataxia type 8 [95]
-
Spinocerebellar ataxia type 48/SCAR 16 due to mutations in the STUB1 gene [61, 91]
-
Dentato-rubro-pallido-Luysian atrophy (mainly Japan, DRPLA; [78])
-
Ataxia telangiectasia and ataxia telangiectasia like disease (serum alpha-fetoprotein ↑ [14])
-
Ataxia with oculomotor apraxia (AOA1 (serum albumin ↓) and AOA2 (serum alpha-fetoprotein↑; now also called SCAN2 [2, 14, 57])
-
McLeod syndrome (CK↑, ± acanthocytes in blood smear↑, myocardial abnormalities, striatal atrophy, analysis of the Kell/Kx blood group phenotype or mutations in XK gene [25])
-
Chorea-acanthocytosis (CK↑, ± acanthocytes in blood smear↑, striatal atrophy, ChoreinWestern blot (LMU, Munich), detection of mutation in the CHAC gene (VPS13A [25, 95])
-
Huntington’s disease-like 2 (HDL2; predominantly in patients of African origin [14, 95])
-
Benign hereditary chorea (including thyroid transcription factor 1 gene, TITF1/NKX2-1; L-Dopa or methylphenidate therapy might be helpful [32, 106])
Other rare inherited disease entities
-
Huntington’s disease-like 1 and 3, only described in individual families [14]
-
HDL1 with prion protein (PrP) gene mutations (PRNP) and rapid progression [14, 95]
-
HDL3, a family [47]
-
RNF216 mutation (autosomal recessive, leukoencephalopathic lesions and possibly Serum gonadotropin ↓ [93])
-
ANO3 mutations [52]
-
FRRS1L mutations (Saudi Arabia; also epilepsy [95])
-
Primary Familial Brain Calcification (formerly “Fahr’s disease”, cMRI/CCT helpful (SLC20A2-,PDGFB, PDGFRB or XPR1 gene [95])
-
POLG gene mutations (dystonia, myoclonus, discrete chorea [101])
-
Leigh's disease [63]
-
SETX mutation (with motor neuron disease [94])
-
Laurence-Moon-Biedl-Bardet syndrome [65]
-
Friedreich ataxia [41]
-
NBIA “neurodegeneration with brain iron accumulation” (umbrella term for e.g. Pantothenate kinase-associated neurodegeneration (PKAN 2), neuroferritinopathies (FTL), Aceruloplasminemia (CP), phospholipase-associated neurodegeneration (PLAN), Betapropeller protein-associated neurodegeneration (WDR45), infantile neuroaxonal dystrophy (PLA2G6), C19orf12, C2orf37, FA2H, ATP13A2, COASY and DCAF17 mutations—more likely no chorea)) with iron deposits in the basal ganglia as a typical MRI finding [3, 14, 82, 95, 103, 113])
-
TAR DNA binding protein variation (TARDBP; with frontotemporal dementia [51])
-
Niemann-Pick type C [46]
-
Cereoid lipofuscinosis [72]
-
Lipidoses, aminoacidosis and carbohydrate metabolism disorders [69, 96]
-
Phenylketonuria [44]
-
Paroxysmal kinesiogenic dyskinesia (PKD; paroxysmal kinesiogenic choreoathetosis; dystonia 10 [14, 50], weekly parenteral doses of vitamins and minerals might be helpful [10])
-
Paroxysmal non-kinesiogenic dyskinesia (PNKD); dystonia type 8 [14, 50]
-
Paroxysmal choreoathetosis with infantile febrile convulsions; ICCA [14]
-
Tuberous brain sclerosis [116]
-
FUS-related ALS [34]
-
Mutations in iron-responsive element-binding protein 2; IREB2 [20]
-
18p deletions syndrome [22]
-
X-linked Dystonia-Parkinsonism; Lubag; Dyt3 [31]
-
FXTAS [56]
-
Dopamine D2 receptor variant (also dystonia [107])
-
GLRB mutations (GlyR β-subunit; with hyperekplexia [29])
-
CAMK4 variant (with dystonia, autism, developmental delay, later chorea [120])
-
Eukaryotic translation elongation factor (EEF1A2) mutations (also associated with epilepsy, autism, intellectual impairment, sudden onset of chorea [55])
-
ERCC4 gene mutations in xeroderma pigmentosa (with ataxia [15])
-
Replication factor complex subunit 1 (RFC1) mutations (cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) and leading cause of late-onset ataxia, 11% Chorea [105])
-
Polynucleotide kinase phosphatase (PNKP) mutation (rather benign course, early onset, with microcephaly, epilepsy, developmental delay, ataxia with oculomotor apraxia (AOA type 4) and polyneuropathy [12])
-
“Benign chorea type 2” (with onset of the disease around the age of 40; Japan [97])
Hereditary chorea primarily presenting in children and adolescents
-
NKX2-1, ADCY-5, FOXG1, GNAO1, GPR88, SLC2A1, SQSTM1, ATP8A2 or SYT-1 Mutations [5]
-
Hereditary disorders of glycosylation (CDG; in children [71])
-
ELAC2 gene mutations (rare mitochondrial disease with cardiomyopathy, children with developmental delay, possibly acanthocytes [76])
-
SCN2A mutation (neonatal, early childhood epilepsy, developmental disorders, possibly autism and episodic ataxia [115])
-
PDE10A mutations (MRI with bilateral striatal lesions [68])
-
KCNQ2 mutations (associated with fever [27])
-
ATP1A3 mutations (alternating hemiplegia of childhood (AHC), rapid-onset dystonia, parkinsonism, CAPOS (cerebellar ataxia, areflexia, pes cavus, optic atrophy, sensorineural hearing loss [100])
-
ATP1A2 mutations (regression, hemiplegia, epilepsy [11])
-
SUCLA2 mutation in mitochondrial DNA (hypotonia, Dyston/Leigh-like syndrome, deafness, but also myopathy, ataxia [36])
-
Glutaric aciduria (AR [44])
-
ARX loss (intellectual impairment [87])
Autoimmune and paraneoplastic choreiform syndromes
Sydenham's chorea (chorea minor, post-streptococcal infection disease): anti-streptolysin O (ASL), anti-DNAse B (anti-streptodornase B, ASNB). The relevance of anti-basal ganglia antibodies (immunohistochemical analysis [38] and dopamine D2 receptor Ab (cell-based testing is unclear; so far laboratory analysis established in one centre; specificity not clearly proven [24].
Systemic lupus erythematosus (SLE); antiphospholipid antibody syndrome (APS); Chorea gravidarum (often SLE, APS or other autoimmune diseases are discussed as the underlying cause for chorea gravidarum [14, 86]).
Behçet's disease [40].
Chorea in synaptic (idiopathic and paraneoplastic) autoimmune encephalitis is possible, but rarely isolated. GAD65-AK; CASPR2-Ab; NMDA-R-AK; CRMP-5 IgG [108], N-type or P/Q-type calcium channel antibodies [17, 70]; Anti-SOX1-Ab [117], LGI-1-Ab (30–60 high-frequency, short daily brachiofacial dystonic seizures per day preceding or paralleling limbic encephalitis), and probably also other autoimmune, partly post-infectious or -vaccine encephalitis, e.g. B. IgLON5-Ab (usually with sleep disorders and stridor); [39, 60, 74, 88, 110].
Very rare: paraneoplastic chorea with antibodies against onconeural (intracellular) antigens, most of these cases are progressive and multisystemic syndromes with subacute onset (anti-CV2/CRMP-5, Anti-Hu, Anti-Yo, Anti-Ma2 [14, 21, 30].
Phosphodiesterase 10A antibody [6]; Takayasu vasculitis [62]; Rasmussen syndrome [35]; celiac disease with anti-gliadin antibodies [112]; steroid responsive Encephalopathy in Autoimmune Thyroiditis (SREAT [102]); Microscopic polyangiitis (MPA [45]).
Infectious causes
HIV encephalopathy [84]; viral encephalitis (mumps, measles, varicella-zoster virus, herpes simplex virus, ECHO group viruses); new variant of Creutzfeldt-Jakob disease; diphtheria; bacterial endocarditis; neurobrucellosis; neurosyphilis; neuroborreliosis; other bacterial encephalitides; cerebral toxoplasmosis; CNS cryptococcosis; neurocysticercosis [14]; Whipple's disease (including ataxia, vertical gaze palsy, oculomasticatory myorhythmias, cognitive impairment [79]); subacute sclerosing panencephalitis (SSPE [98]); Influenza A encephalopathy [81]; SARS-CoV-2 encephalitis [42].
Structural lesions of the basal ganglia
Ischemic or hemorrhagic infarcts; neoplasms; abscessing lesions (incl. toxoplasmosis abscesses and tuberculomas); demyelinating lesions; central pontine/extrapontine myelinolysis; neurosarcoidosis [4, 14]; cavernoma [83]; structural lesions, may also cause hemichorea. Vascular encephalopathy, lacunar infarcts may also cause intermittent chorea [92].
Metabolic, endocrine and toxic causes
Nonketotic hyperglycemia in diabetes mellitus (T1-weighted MRI sequences often show localized hyperintensity, especially in the putamen [18]).
Hypoglycemia; hypo/hypernatremia; hypocalcemia; hypoparathyroidism (sometimes presenting as hemichorea and with calcification of the basal ganglia [26]; hyperthyroidism; acute intermittent porphyria; liver failure including chronic hepatocerebral degeneration; kidney failure; carbon monoxide; manganese; mercury; thallium; organophosphates, 3-NP [14]; vitamin B12 deficiency [28]; Wernicke encephalopathy [89]; 3-Hydroxy-sobutyryl-CoAHydrolase (HIBCH) deficiency (children, sometimes only paroxysmal chorea, e.g. during stress [99]), Tay-Sachs disease [59].
Medication and drug-induced chorea
Dopamine receptor antagonists (e.g. phenothiazine, butyrophenone, benzamide) including antiemetics (metoclopramide); medications for the treatment of Parkinson's disease (such as L-dopa, dopamine agonists, anticholinergics); antiepileptic drugs (e.g. phenytoin, carbamazepine, valproic acid, gabapentin, lamotrigine, pregabalin [85], levetiracetam [118]; calcium channel blockers (cinnarizine, flunarizine, verapamil); lithium; tricyclic antidepressants; SSRI [54]; anti-malarial drugs; steroids; oral contraceptives; antihistamines (H1 and H2); psychostimulants (methylphenidate, amphetamines, pemoline, cocaine); baclofen, digoxin, cyclosporine, theophylline [14]; buphrenorphine, hydromorphone and other opiates [66]; ceftriaxone [119]; memantine [9].
Other causes
Polycythaemia vera [67]; essential thrombocythaemia [109]; post-pump chorea after cardiac surgery [90]; superficial siderosis [58]; Moyamoya disease [16]; chorea gravidarum (idiopathic or secondary,see under: “Autoimmune and paraneoplastic choreiform syndromes” [49]; Covid-19 vaccination [7]; intoxications with wood or plant protection products, e.g. propiconazole [77].
Important differential diagnoses for the syndrome chorea include
-
1.
Focal epilepsy [33]. Chorea can sometimes be mistaken for focal epilepsy.
-
2.
Tic Disorders: Unlike chorea, tic disorders are characterized by typical premonitory urges and the ability to suppress tics for a short time [8].
-
3.
Akathisia: It can be challenging to differentiate akathisia from chorea, especially since many antidopaminergic drugs including presynaptic dopamine depletors, such as tetrabenazine, can induce akathisia. If akathisia is suspected, a reduction in antihyperkinetic medication may be indicated. Propranolol, anticholinergics, benzodiazepines, and postsynaptic serotonin-5-HT2a receptor antagonists like ritanserin, cyproheptadine, trazodone, mianserin, or mirtazapine can be helpful in managing drug-induced akathisia [80].
-
4.
Myoclonus-dystonia disorders: Conditions like epsilon-sarcoglycan (SGCE) DYT11 gene mutations (often alcohol-sensitive) or VPS16 gene mutations can present as myoclonus-dystonia disorders [75]. Additionally, many other conditions discussed above may exhibit a myoclonus-dystonia phenotype (e.g., benign chorea or PKAN2).
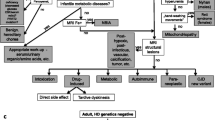
Suggested work-up in a patient with chorea
Please refer to Table 1. Medical history, particularly family history, medication history, presence of other relevant medical conditions (as mentioned above).
Neurological assessment to determine if chorea is the only presenting movement disorder and if additional neurological or systemic signs are present. In general, chorea is often more noticeable during conversations, especially when emotionally charged, such as during discussions of stressful topics or during neuropsychological testing. However, during the actual neurological examination, chorea may be less pronounced [8].
Cerebral imaging studies (MRI, if contraindicated CT scan, asking for focal lesions or caudate and/or cortical atrophy. Note that for the determination of caudate atrophy, coronal sections should be available. Signal changes in T2-weighted imaging? Contrast-enhancing lesions? Hypointensities suggesting iron deposition on iron-sensitive magnetic resonance images in order to rule out symptomatic causes or provide evidence for pathognomonic alterations (e.g. "Eye of the tiger" sign for PKAN2; hypointensities in the basal ganglia, particularly in bilateral globus pallidus and substantia nigra for NBIAs; "Face of the giant panda" sign for Wilson's disease; cerebellar or pontine atrophy for hereditary ataxia).
Depending on the findings and circumstances: Comprehensive laboratory testing, including cerebrospinal fluid analysis to explore the differential diagnoses mentioned above. FDG-PET-CT or FDG-PET-MRI for tumor screening in suspected paraneoplastic etiology. Heavy Metal Assessment (mercury, manganese, thallium), "drug test" in serum and/or urine.
Additional tests in selected cases: Positron Emission Tomography (e.g., FDG-PET) to detect reduced glucose utilization/hypometabolism in basal ganglia (e.g., in HD and other neurodegenerative disorders causing chorea) or increased glucose utilization/hypermetabolism in Sydenham's chorea or autoimmune encephalitis including SLE with cerebral involvement, associated with hypometabolism in the prefrontal and premotor cortex [53, 114], hypermetabolism in the hippocampus and orbitofrontal cortex [64], and potentially parieto-occipital hypometabolism in patients with neuropsychiatric disorders [23], contralateral striatal hypoperfusion in patients with non-ketotic hyperglycemia.
If Huntington’s disease is suspect
Molecular genetic testing (determination of CAG repeats in the Huntingtin gene, Chromosome 4p) after informed consent following the Genetic Diagnosis Act (GenDG).
Unified Huntington’s Disease Rating Scale (UHDRS’99) total motor score.
Neuropsychological or Behavioral Neurological Assessment (psychomotor slowing, frontal-executive dysfunction, memory impairment, decreased speech fluency, spatial-visual disturbances, formal cognitive testing according to UHDRS).
Psychiatric Examination (personality changes, changes in motivation, irritability, aggression, depression, suicidal ideation, delusions, hallucinations, obsessive–compulsive disorders, and sexual disorders; it is recommended to use the "Problem Behaviour Assessment" scale (PBA-s), which is also used in the ENROLL-HD observational study).
Availability of data and materials
Not applicable.
References
Adler, C. H., & Wrabetz, L. (1996). Lesch-Nyhan variant: Dystonia, ataxia, near-normal intelligence, and no self-mutilation. Movement Disorders, 11(5), 583–584. https://doi.org/10.1002/mds.870110519
Anheim, M., Monga, B., Fleury, M., Charles, P., Barbot, C., Salih, M., & Koenig, M. (2009). Ataxia with oculomotor apraxia type 2: Clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain, 132(Pt 10), 2688–2698. https://doi.org/10.1093/brain/awp211
Arber, C. E., Li, A., Houlden, H., & Wray, S. (2016). Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories. Neuropathology and Applied Neurobiology, 42(3), 220–241. https://doi.org/10.1111/nan.12242
Ataya, A., & Harman, E. (2015). Images in clinical medicine. Hemichoreoathetosis in neurosarcoidosis. The New England Journal of Medicine, 372(21), e27. https://doi.org/10.1056/NEJMicm1407763
Baizabal-Carvallo, J. F., & Cardoso, F. (2020). Chorea in children: Etiology, diagnostic approach and management. Journal of Neural Transmission (Vienna), 127(10), 1323–1342. https://doi.org/10.1007/s00702-020-02238-3
Balint, B., & Bhatia, K. P. (2021). Autoimmune movement disorders with neuronal antibodies—An update. Current Opinion in Neurology, 34(4), 565–571. https://doi.org/10.1097/WCO.0000000000000956
Batot, C., Chea, M., Zeidan, S., Mongin, M., Pop, G., Mazoyer, J., & Degos, B. (2022). Clinical and radiological follow-up of a pfizer-BioNTech COVID-19 vaccine-induced hemichorea-hemiballismus. Tremor and Other Hyperkinetic Movements, 12, 1–6. https://doi.org/10.5334/tohm.688
Bonomo, R., Latorre, A., Balint, B., Smilowska, K., Rocchi, L., Rothwell, J. C., & Bhatia, K. P. (2020). Voluntary inhibitory control of chorea: A case series. Movement Disorders Clinical Practice, 7(3), 308–312. https://doi.org/10.1002/mdc3.12907
Borges, L. G., & Bonakdarpour, B. (2017). Memantine-induced chorea and dystonia. Practical Neurology, 17(2), 133–134. https://doi.org/10.1136/practneurol-2016-001470
Bruton, A., & Fuller, L. (2019). Paroxysmal kinesigenic dyskinesia symptoms markedly reduced with parenteral vitamins and minerals: A case report. Perm Journal. https://doi.org/10.7812/TPP/19.036
Calame, D. G., Houck, K., Lotze, T., Emrick, L., & Parnes, M. (2021). A novel ATP1A2 variant associated with severe stepwise regression, hemiplegia, epilepsy and movement disorders in two unrelated patients. European Journal of Paediatric Neurology, 31, 21–26. https://doi.org/10.1016/j.ejpn.2021.01.004
Caputi, C., Tolve, M., Galosi, S., Inghilleri, M., Carducci, C., Angeloni, A., & Leuzzi, V. (2019). PNKP deficiency mimicking a benign hereditary chorea: The misleading presentation of a neurodegenerative disorder. Parkinsonism & Related Disorders, 64, 342–345. https://doi.org/10.1016/j.parkreldis.2019.03.012
Carapito, R., Paul, N., Untrau, M., Le Gentil, M., Ott, L., Alsaleh, G., & Bahram, S. (2015). A de novo ADCY5 mutation causes early-onset autosomal dominant chorea and dystonia. Movement Disorders, 30(3), 423–427. https://doi.org/10.1002/mds.26115
Cardoso, F., Seppi, K., Mair, K. J., Wenning, G. K., & Poewe, W. (2006). Seminar on choreas. Lancet Neurology, 5(7), 589–602.
Carre, G., Marelli, C., Anheim, M., Geny, C., Renaud, M., Rezvani, H. R., & Tranchant, C. (2017). Xeroderma pigmentosum complementation group F: A rare cause of cerebellar ataxia with chorea. Journal of the Neurological Sciences, 376, 198–201. https://doi.org/10.1016/j.jns.2017.03.021
Cavallieri, F., Zedde, M., Assenza, F., & Valzania, F. (2021). Steroid-responsive acute left-arm chorea as a presenting symptom of moyamoya disease. Canadian Journal of Neurological Sciences, 48(2), 287–289. https://doi.org/10.1017/cjn.2020.155
Chang, K., Lwanga, A., Kaur, T., & Helgason, C. (2018). P/Q and N-type voltage-gated calcium channel binding antibodies associated with paraneoplastic chorea and mixed invasive ductal and lobular carcinoma of the breasts in an elderly patient. Cureus, 10(8), e3097. https://doi.org/10.7759/cureus.3097
Chang, K. H., Tsou, J. C., Chen, S. T., Ro, L. S., Lyu, R. K., Chang, H. S., & Chen, C. J. (2010). Temporal features of magnetic resonance imaging and spectroscopy in non-ketotic hyperglycemic chorea-ballism patients. European Journal of Neurology, 17(4), 589–593. https://doi.org/10.1111/j.1468-1331.2009.02867.x
Cincotta, M., & Walker, R. H. (2022). One side of the story; clues to etiology in patients with asymmetric chorea. Tremor Other Hyperkinet Movement (N Y), 12, 3. https://doi.org/10.5334/tohm.675
Costain, G., Ghosh, M. C., Maio, N., Carnevale, A., Si, Y. C., Rouault, T. A., & Yoon, G. (2019). Absence of iron-responsive element-binding protein 2 causes a novel neurodegenerative syndrome. Brain, 142(5), 1195–1202. https://doi.org/10.1093/brain/awz072
Crespo-Burillo, J. A., Hernando-Quintana, N., Ruiz-Palomino, P., & Martin-Martinez, J. (2015). Chorea secondary to striatal encephalitis due to anti-CV2/CRMP5 antibodies. Case description and review of the literature. Neurología, 30(7), 451–453. https://doi.org/10.1016/j.nrl.2013.10.007
Crosiers, D., Blaumeiser, B., & Van Goethem, G. (2019). Spectrum of movement disorders in 18p deletion syndrome. Movement Disorders Clinical Practice, 6(1), 70–73. https://doi.org/10.1002/mdc3.12707
Curiel, R., Akin, E. A., Beaulieu, G., DePalma, L., & Hashefi, M. (2011). PET/CT imaging in systemic lupus erythematosus. Annals of the New York Academy of Sciences, 1228, 71–80. https://doi.org/10.1111/j.1749-6632.2011.06076.x
Dale, R. C., Merheb, V., Pillai, S., Wang, D., Cantrill, L., Murphy, T. K., & Brilot, F. (2012). Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain, 135(Pt 11), 3453–3468. https://doi.org/10.1093/brain/aws256
Danek, A. (2002). Progress in molecular chorea diagnosis. McLeod syndrome and chorea acanthocytosis. Der Nervenarzt, 73(6), 564–569. https://doi.org/10.1007/pl00020831
Desai, K., Walzade, P., Ravat, S. H., & Agarwal, P. A. (2019). Adult-onset isolated hemichorea revealing iatrogenic hypoparathyroidism and bilateral basal ganglia calcification. Annals of Indian Academy of Neurology, 22(4), 496–499. https://doi.org/10.4103/aian.AIAN_123_18
Dhamija, R., Goodkin, H. P., Bailey, R., Chambers, C., & Brenton, J. N. (2017). A case of KCNQ2-associated movement disorder triggered by fever. Journal of Child Neurology, 32(14), 1123–1124. https://doi.org/10.1177/0883073817736702
Edvardsson, B., & Persson, S. (2011). Chorea associated with vitamin B12 deficiency. European Journal of Neurology, 18(10), e138-139. https://doi.org/10.1111/j.1468-1331.2011.03478.x
Estevez-Fraga, C., Magrinelli, F., Latorre, A., Cordivari, C., Houlden, H., Tinazzi, M., & Bhatia, K. P. (2020). A new family with GLRB-related hyperekplexia showing chorea in homo- and heterozygous variant carriers. Parkinsonism & Related Disorders, 79, 97–99. https://doi.org/10.1016/j.parkreldis.2020.08.016
Etemadifar, M., Salari, M., Badiee, H., & Mirmosayyeb, O. (2017). Anti-ma2 receptor encephalitis mimicking Huntington chorea. Journal of Research in Medical Sciences: The Official Journal of Isfahan University of Medical Sciences, 22, 31. https://doi.org/10.4103/1735-1995.202148
Evidente, V. G. H. (1993). X-linked dystonia-parkinsonism. In M. P. Adam, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. W. Gripp, & A. Amemiya (Eds.), GeneReviews((R)). Seattle (WA).
Farrenburg, M., & Gupta, H. V. (2020). Levodopa-responsive chorea: A review. Annals of Indian Academy of Neurology, 23(2), 211–214. https://doi.org/10.4103/aian.AIAN_221_19
Fasano, A., Di Bonaventura, C., Bove, F., Espay, A. J., Morgante, F., Fabbrini, G., & Berardelli, A. (2019). Movement disorders phenomenology in focal motor seizures. Parkinsonism & Related Disorders, 61, 161–165. https://doi.org/10.1016/j.parkreldis.2018.10.021
Flies, C. M., & Veldink, J. H. (2020). Chorea is a pleiotropic clinical feature of mutated fused-in-sarcoma in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener, 21(3–4), 309–311. https://doi.org/10.1080/21678421.2020.1733021
Gambardella, A., Andermann, F., Shorvon, S., Le Piane, E., & Aguglia, U. (2008). Limited chronic focal encephalitis: Another variant of Rasmussen syndrome? Neurology, 70(5), 374–377. https://doi.org/10.1212/01.wnl.0000298723.96653.99
Garone, C., Gurgel-Giannetti, J., Sanna-Cherchi, S., Krishna, S., Naini, A., Quinzii, C. M., & Hirano, M. (2017). A novel SUCLA2 mutation presenting as a complex childhood movement disorder. Journal of Child Neurology, 32(2), 246–250. https://doi.org/10.1177/0883073816666221
Ghosh, R., Roy, D., Dubey, S., Das, S., & Benito-Leon, J. (2022). Movement disorders in multiple sclerosis: An update. Tremor Other Hyperkinet Movement (N Y), 12, 14. https://doi.org/10.5334/tohm.671
Gupta, H. V., Barnes, H., Radhi, F. A., & Jassam, Y. (2020). Chorea and Parkinsonism with elevated striational antibody. Annals of Indian Academy of Neurology, 23(2), 223–224. https://doi.org/10.4103/aian.AIAN_364_19
Hacohen, Y., Wright, S., Waters, P., Agrawal, S., Carr, L., Cross, H., & Lim, M. J. (2013). Paediatric autoimmune encephalopathies: Clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. Journal of Neurology, Neurosurgery and Psychiatry, 84(7), 748–755. https://doi.org/10.1136/jnnp-2012-303807
Hamid, M., Adan, K., Satte, A., & Bourazza, A. (2021). Chorea in neuro-Behcet’s disease. Cureus, 13(10), e19039. https://doi.org/10.7759/cureus.19039
Hanna, M. G., Davis, M. B., Sweeney, M. G., Noursadeghi, M., Ellis, C. J., Elliot, P., & Marsden, C. D. (1998). Generalized chorea in two patients harboring the Friedreich’s ataxia gene trinucleotide repeat expansion. Movement Disorders, 13(2), 339–340. https://doi.org/10.1002/mds.870130223
Hassan, M., Syed, F., Ali, L., Rajput, H. M., Faisal, F., Shahzad, W., & Badshah, M. (2021). Chorea as a presentation of SARS-CoV-2 encephalitis: A clinical case report. Journal of Movement Disorders. https://doi.org/10.14802/jmd.20098
Hensman Moss, D. J., Poulter, M., Beck, J., Hehir, J., Polke, J. M., Campbell, T., & Tabrizi, S. J. (2014). C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology, 82(4), 292–299. https://doi.org/10.1212/WNL.0000000000000061
Hermann, A., & Walker, R. H. (2015). Diagnosis and treatment of chorea syndromes. Current Neurology and Neuroscience Reports, 15(2), 514. https://doi.org/10.1007/s11910-014-0514-0
Iftikhar, S. (2018). Chorea and calcifications: Atypical presentation of microscopic polyangiitis. Mayo Clinic Proceedings, 93(7), 961–962. https://doi.org/10.1016/j.mayocp.2018.05.023
Josephs, K. A., Van Gerpen, M. W., & Van Gerpen, J. A. (2003). Adult onset Niemann-Pick disease type C presenting with psychosis. Journal of Neurology, Neurosurgery and Psychiatry, 74(4), 528–529. https://doi.org/10.1136/jnnp.74.4.528
Kambouris, M., Bohlega, S., Al-Tahan, A., & Meyer, B. F. (2000). Localization of the gene for a novel autosomal recessive neurodegenerative Huntington-like disorder to 4p15.3. American Journal of Human Genetics, 66(2), 445–452. https://doi.org/10.1086/302744
Kaur, J., Parveen, S., Shamim, U., Sharma, P., Suroliya, V., Sonkar, A. K., & Faruq, M. (2020). Investigations of Huntington’s disease and Huntington’s disease-like syndromes in Indian choreatic patients. Journal of Huntington’s Disease, 9(3), 283–289. https://doi.org/10.3233/JHD-200398
Kim, A., Choi, C. H., Han, C. H., & Shin, J. C. (2009). Consecutive pregnancy with chorea gravidarum associated with moyamoya disease. Journal of Perinatology, 29(4), 317–319. https://doi.org/10.1038/jp.2008.183
Klein, C. (2014). Genetics in dystonia. Parkinsonism & Related Disorders, 20(Suppl 1), S137-142. https://doi.org/10.1016/S1353-8020(13)70033-6
Kovacs, G. G., Murrell, J. R., Horvath, S., Haraszti, L., Majtenyi, K., Molnar, M. J., & Spina, S. (2009). TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Movement Disorders, 24(12), 1843–1847. https://doi.org/10.1002/mds.22697
Koya Kutty, S., Mulroy, E., Magrinelli, F., Di Lazzaro, G., Latorre, A., & Bhatia, K. P. (2021). Huntington disease-like phenotype in a patient with ANO3 mutation. Parkinsonism & Related Disorders, 90, 120–122. https://doi.org/10.1016/j.parkreldis.2021.02.022
Krakauer, M., & Law, I. (2009). FDG PET brain imaging in neuropsychiatric systemic lupus erythematosis with choreic symptoms. Clinical Nuclear Medicine, 34(2), 122–123. https://doi.org/10.1097/RLU.0b013e318192c4d2
Laban, T., Larroche, C., Comparon, C., Dhote, R., & Degos, B. (2021). Fluoxetine-induced chorea. Revue Neurologique (Paris), 177(8), 1010–1011. https://doi.org/10.1016/j.neurol.2020.12.007
Lance, E. I., Kronenbuerger, M., Cohen, J. S., Furmanski, O., Singer, H. S., & Fatemi, A. (2018). Successful treatment of choreo-athetotic movements in a patient with an EEF1A2 gene variant. SAGE Open Medical Case Reports, 6, 2050313X18807622. https://doi.org/10.1177/2050313X18807622
Lapostolle, A., Delion, T., Arnaud, S., Manceau, P., & Degos, B. (2021). FXTAS patient presenting as Huntington-like generalized chorea. Revue Neurologique (Paris), 177(4), 445–446. https://doi.org/10.1016/j.neurol.2020.08.008
Le Ber, I., Moreira, M. C., Rivaud-Pechoux, S., Chamayou, C., Ochsner, F., Kuntzer, T., & Durr, A. (2003). Cerebellar ataxia with oculomotor apraxia type 1: Clinical and genetic studies. Brain, 126(Pt 12), 2761–2772. https://doi.org/10.1093/brain/awg283
Le Rhun, E., Soto Ares, G., Pecheux, N., Destee, A., & Defebvre, L. (2008). Superficial hemosiderosis of the central nervous system improved by corticosteroids. Revue Neurologique (Paris), 164(3), 264–270. https://doi.org/10.1016/j.neurol.2007.08.010
Lefter, S., O’Mahony, O., Sweeney, B., & Ryan, A. M. (2021). Late-onset Tay-Sachs disease in an Irish family. Movement Disorders Clinical Practice, 8(1), 106–110. https://doi.org/10.1002/mdc3.13096
Leypoldt, F., Armangue, T., & Dalmau, J. (2015). Autoimmune encephalopathies. Annals of the New York Academy of Sciences, 1338(1), 94–114. https://doi.org/10.1111/nyas.12553
Lieto, M., Riso, V., Galatolo, D., De Michele, G., Rossi, S., Barghigiani, M., & Silvestri, G. (2020). The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. European Journal of Neurology, 27(3), 498–505. https://doi.org/10.1111/ene.14094
Lopes, A. S., Clemente, G., Len, C. A., Masruha, M. R., & Terreri, M. T. (2015). Chorea: A rare manifestation of Takayasu’s arteritis. Revista Brasileira de Reumatologia, 55(4), 384–386. https://doi.org/10.1016/j.rbr.2013.09.003
Macaya, A., Munell, F., Burke, R. E., & De Vivo, D. C. (1993). Disorders of movement in Leigh syndrome. Neuropediatrics, 24(2), 60–67. https://doi.org/10.1055/s-2008-1071515
Mackay, M., Tang, C. C., Volpe, B. T., Aranow, C., Mattis, P. J., Korff, R. A., & Eidelberg, D. (2015). Brain metabolism and autoantibody titres predict functional impairment in systemic lupus erythematosus. Lupus Science and Medicine, 2(1), e000074. https://doi.org/10.1136/lupus-2014-000074
Malik, G. M., Mubarik, M., Khan, M. D., Lone, B. A., Kadla, S. A., & Bhat, F. A. (1995). Laurence-Moon-Biedl-Bardet syndrome with chorea. Journal of the Association of Physicians of India, 43(4), 295–296.
Martin, E. J., Vaughan, C. L., Atayee, R., Hirst, J. M., O’Donnell, K., & Edmonds, K. P. (2018). Hydromorphone-induced chorea as an atypical presentation of opioid neurotoxicity: A case report and review of the literature. Palliative Medicine, 32(9), 1529–1532. https://doi.org/10.1177/0269216318786861
Marvi, M. M., & Lew, M. F. (2011). Polycythemia and chorea. Handbook of Clinical Neurology, 100, 271–276. https://doi.org/10.1016/B978-0-444-52014-2.00019-7
Mencacci, N. E., Kamsteeg, E. J., Nakashima, K., R’Bibo, L., Lynch, D. S., Balint, B., & Bhatia, K. P. (2016). De novo mutations in PDE10A cause childhood-onset chorea with bilateral striatal lesions. American Journal of Human Genetics, 98(4), 763–771. https://doi.org/10.1016/j.ajhg.2016.02.015
Mizuguchi, M., & Kamoshita, S. (1993). Movement disorders in miscellaneous disorders–inherited metabolic diseases. Nihon Rinsho, 51(11), 2919–2923.
Morgan, T. T., Armitage, A., Stone, B., & Benge, J. (2019). Non paraneoplastic immune-mediated calcium channel chorea. Proceedings (Baylor University Medical Center), 32(2), 281–282. https://doi.org/10.1080/08998280.2019.1581318
Mostile, G., Barone, R., Nicoletti, A., Rizzo, R., Martinelli, D., Sturiale, L., & Zappia, M. (2019). Hyperkinetic movement disorders in congenital disorders of glycosylation. European Journal of Neurology, 26(9), 1226–1234. https://doi.org/10.1111/ene.14007
Nass, R., Petito, C., Stoner, E., & New, M. (1986). Neuronal ceroid lipofuscinosis with hypergonadotropic hypogonadism. Journal of Child Neurology, 1(2), 142–144. https://doi.org/10.1177/088307388600100209
Nguyen, Q. T. R., Ortigoza Escobar, J. D., Burgunder, J. M., Mariotti, C., Saft, C., Hjermind, L. E., & Bachoud-Levi, A. C. (2022). Corrigendum: Combining literature review with a ground truth approach for diagnosing huntington’s disease phenocopy. Frontiers in Neurology, 13, 891800. https://doi.org/10.3389/fneur.2022.891800
O’Toole, O., Lennon, V. A., Ahlskog, J. E., Matsumoto, J. Y., Pittock, S. J., Bower, J., & McKeon, A. (2013). Autoimmune chorea in adults. Neurology, 80(12), 1133–1144. https://doi.org/10.1212/WNL.0b013e3182886991
Park, J., Reilaender, A., Petry-Schmelzer, J. N., Stobe, P., Cordts, I., Harmuth, F., & Haack, T. B. (2022). Transcript-specific loss-of-function variants in VPS16 are enriched in patients with dystonia. Neurol Genetics, 8(1), e644. https://doi.org/10.1212/NXG.0000000000000644
Paucar, M., Pajak, A., Freyer, C., Bergendal, A., Dory, M., Laffita-Mesa, J. M., & Svenningsson, P. (2018). Chorea, psychosis, acanthocytosis, and prolonged survival associated with ELAC2 mutations. Neurology, 91(15), 710–712. https://doi.org/10.1212/WNL.0000000000006320
Paul, B. S., Paul, G., Kaur, J., & Singh, G. (2017). Chorea as unusual complication of fungicide poisoning. Journal of Postgraduate Medicine, 63(1), 53–54. https://doi.org/10.4103/0022-3859.198156
Pedroso, J. L., de Freitas, M. E., Albuquerque, M. V., Saraiva-Pereira, M. L., Jardim, L. B., & Barsottini, O. G. (2014). Should spinocerebellar ataxias be included in the differential diagnosis for Huntington’s diseases-like syndromes? Journal of the Neurological Sciences, 347(1–2), 356–358. https://doi.org/10.1016/j.jns.2014.09.050
Peregrin, J., & Malikova, H. (2015). Primary whipple disease of the brain: Case report with long-term clinical and MRI follow-up. Neuropsychiatric Disease and Treatment, 11, 2461–2469. https://doi.org/10.2147/Ndt.S92066
Poyurovsky, M., & Weizman, A. (2020). Treatment of antipsychotic-induced akathisia: Role of serotonin 5-HT(2a) receptor antagonists. Drugs, 80(9), 871–882. https://doi.org/10.1007/s40265-020-01312-0
Prasuhn, J., Royl, G., Wandinger, K. P., Bruggemann, N., Neumann, A., & Munte, T. F. (2018). Transient generalized chorea in influenza A encephalopathy. Tremor Other Hyperkinet Movement (N Y), 8, 591. https://doi.org/10.7916/D8F495TP
Prohaska, R., Sibon, O. C., Rudnicki, D. D., Danek, A., Hayflick, S. J., Verhaag, E. M., & Walker, R. H. (2012). Brain, blood, and iron: Perspectives on the roles of erythrocytes and iron in neurodegeneration. Neurobiology of Diseases, 46(3), 607–624. https://doi.org/10.1016/j.nbd.2012.03.006
Qiu, J., Cui, Y., Sun, L., Guo, Y., & Zhu, Z. (2018). Hemichorea associated with cavernous angioma and a small errhysis: A case report and literature review. Medicine (Baltimore), 97(43), e12889. https://doi.org/10.1097/MD.0000000000012889
Rajakaruna, G. K., Italiano, C. M., John, M., & Nolan, D. (2020). Chorea associated with persistent low-level viremia in a patient living with HIV: A case report. Journal of Virus Eradication, 6(1), 27–29.
Rissardo, J. P., & Caprara, A. L. F. (2020). Pregabalin-associated movement disorders: A literature review. Brain Circulation, 6(2), 96–106. https://doi.org/10.4103/bc.bc_57_19
Robottom, B. J., & Weiner, W. J. (2011). Chorea gravidarum. Handbook of Clinical Neurology, 100, 231–235. https://doi.org/10.1016/B978-0-444-52014-2.00015-X
Rodgers, J., Calvert, S., Shoubridge, C., & McGaughran, J. (2021). A novel ARX loss of function variant in female monozygotic twins is associated with chorea. European Journal of Medical Genetics, 64(11), 104315. https://doi.org/10.1016/j.ejmg.2021.104315
Sabater, L., Gaig, C., Gelpi, E., Bataller, L., Lewerenz, J., Torres-Vega, E., & Graus, F. (2014). A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurology, 13(6), 575–586. https://doi.org/10.1016/S1474-4422(14)70051-1
Sabatini, J. S., Schutz-Pereira, G. L., Feltrin, F., Teive, H. A. G., & Camargo, C. H. F. (2016). Wernicke’s encephalopathy with chorea: Neuroimaging findings. Dement Neuropsychol, 10(4), 370–372. https://doi.org/10.1590/s1980-5764-2016dn1004020
Saft, C., Reber, D., Streuer, M., & Andrich, J. (2011). Post pump chorea in a 77-year-old male. Neurological Sciences, 32(4), 699–701. https://doi.org/10.1007/s10072-011-0583-7
Saft, C., Skodda, S., Nguyen, H. P., Park, J., & Haack, T. B. (2021). Teaching video neuroimage: New STUB1 variant causes chorea, tremor, dystonia, myoclonus, ataxia, depression, cognitive impairment, epilepsy, and superficial siderosis. Neurology, 97(17), E1749–E1750. https://doi.org/10.1212/Wnl.0000000000012264
Salgado, P., Taipa, R., Domingos, J., Dias, D., Pires, M. M., & Magalhaes, M. (2017). Vascular pathology causing late onset generalized chorea: A clinico-pathological case report. Movement Disorders Clinical Practice, 4(6), 819–823. https://doi.org/10.1002/mdc3.12528
Santens, P., Van Damme, T., Steyaert, W., Willaert, A., Sablonniere, B., De Paepe, A., & Dermaut, B. (2015). RNF216 mutations as a novel cause of autosomal recessive Huntington-like disorder. Neurology, 84(17), 1760–1766. https://doi.org/10.1212/WNL.0000000000001521
Saracchi, E., Castelli, M., Bassi, M. T., Brighina, E., Cereda, D., Marzorati, L., & Brighina, L. (2014). A novel heterozygous SETX mutation in a patient presenting with chorea and motor neuron disease. Amyotroph Lateral Scler Frontotemporal Degener, 15(1–2), 138–140. https://doi.org/10.3109/21678421.2013.865751
Schneider, S. A., & Bird, T. (2016). Huntington’s disease, Huntington’s disease look-alikes, and benign hereditary chorea: What’s new? Movement Disorders Clinical Practice, 3(4), 342–354. https://doi.org/10.1002/mdc3.12312
Shah, P. A., & Kuchhai, F. A. (2009). Galactosemia with chorea—An unusual presentation. Indian Journal of Pediatrics, 76(1), 97–98. https://doi.org/10.1007/s12098-009-0037-x
Shimohata, T., Hara, K., Sanpei, K., Nunomura, J., Maeda, T., Kawachi, I., & Honma, Y. (2007). Novel locus for benign hereditary chorea with adult onset maps to chromosome 8q21.3 q23.3. Brain, 130(Pt 9), 2302–2309. https://doi.org/10.1093/brain/awm036
Singhi, P., Saini, A. G., Sankhyan, N., Gupta, P., & Vyas, S. (2015). Blindness, dancing extremities, and corpus callosum and brain stem involvement: An unusual presentation of fulminant subacute sclerosing panencephalitis. Journal of Child Neurology, 30(1), 87–90. https://doi.org/10.1177/0883073813520498
Spitz, M. A., Lenaers, G., Charif, M., Wirth, T., Chelly, J., Abi-Warde, M. T., & Roubertie, A. (2021). Paroxysmal dyskinesias revealing 3-hydroxy-isobutyryl-CoA hydrolase (HIBCH) deficiency. Neuropediatrics, 52(5), 410–414. https://doi.org/10.1055/s-0040-1722678
Sweney, M. T., Newcomb, T. M., & Swoboda, K. J. (2015). The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, CAPOS and beyond. Pediatric Neurology, 52(1), 56–64. https://doi.org/10.1016/j.pediatrneurol.2014.09.015
Synofzik, M., Schule, R., Schulte, C., Kruger, R., Lindig, T., Schols, L., & Asmus, F. (2010). Complex hyperkinetic movement disorders associated with POLG mutations. Movement Disorders, 25(14), 2472–2475. https://doi.org/10.1002/mds.23307
Taurin, G., Golfier, V., Pinel, J. F., Deburghgraeve, V., Poirier, J. Y., Edan, G., & Verin, M. (2002). Choreic syndrome due to Hashimoto’s encephalopathy. Movement Disorders, 17(5), 1091–1092. https://doi.org/10.1002/mds.10230
Tonekaboni, S. H., & Mollamohammadi, M. (2014). Neurodegeneration with brain iron accumulation: An overview. Iranian Journal of Child Neurology, 8(4), 1–8.
Tranchant, C., Bhatia, K. P., & Marsden, C. D. (1995). Movement disorders in multiple sclerosis. Movement Disorders, 10(4), 418–423. https://doi.org/10.1002/mds.870100403
Traschutz, A., Cortese, A., Reich, S., Dominik, N., Faber, J., Jacobi, H., & Group, R. F. C. S. (2021). Natural history, phenotypic spectrum, and discriminative features of multisystemic RFC1 disease. Neurology, 96(9), e1369–e1382. https://doi.org/10.1212/WNL.0000000000011528
Tubing, J., Bohnenpoll, J., Spiegler, J., Gillessen-Kaesbach, G., Baumer, T., Max, C., & Munchau, A. (2018). Methylphenidate can improve chorea in NKX2.1 and ADCY5 mutation-positive patients-a report of two children. Mov Disorders Clinical Practice, 5(3), 343–345. https://doi.org/10.1002/mdc3.12608
van der Weijden, M. C. M., Rodriguez-Contreras, D., Delnooz, C. C. S., Robinson, B. G., Condon, A. F., Kielhold, M. L., & Verbeek, D. S. (2021). A gain-of-function variant in dopamine D2 receptor and progressive chorea and dystonia phenotype. Movement Disorders, 36(3), 729–739. https://doi.org/10.1002/mds.28385
Vaswani, P. A., Kimchi, E. Y., & Hung, A. Y. (2020). CRMP-5-IgG associated paraneoplastic chorea. Movement Disorders Clinical Practice, 7(6), 713–715. https://doi.org/10.1002/mdc3.13019
Venkatesan, E. P., Ramadoss, K., Balakrishnan, R., & Prakash, B. (2014). Essential thrombocythemia: Rare cause of chorea. Annals of Indian Academy of Neurology, 17(1), 106–107. https://doi.org/10.4103/0972-2327.128569
Vynogradova, I., Savitski, V., & Heckmann, J. G. (2014). Hemichorea associated with CASPR2 antibody. Tremor Other Hyperkinet Movements (N Y), 4, 239. https://doi.org/10.7916/D8VM49C5
Walker, F. O. (2007). Huntington’s disease. Lancet, 369(9557), 218–228.
Walker, R. H. (2011). Further evidence for celiac disease-associated chorea. Tremor Other Hyperkinet Movements (N Y). https://doi.org/10.7916/D82806BC
Wang, Z. B., Liu, J. Y., Xu, X. J., Mao, X. Y., Zhang, W., Zhou, H. H., & Liu, Z. Q. (2019). Neurodegeneration with brain iron accumulation: Insights into the mitochondria dysregulation. Biomedicine & Pharmacotherapy, 118, 109068. https://doi.org/10.1016/j.biopha.2019.109068
Weiner, S. M., Otte, A., Schumacher, M., Brink, I., Juengling, F. D., Sobanksi, T., & Peter, H. H. (2000). Alterations of cerebral glucose metabolism indicate progress to severe morphological brain lesions in neuropsychiatric systemic lupus erythematosus. Lupus, 9(5), 386–389. https://doi.org/10.1191/096120300678828370
Wolff, M., Brunklaus, A., & Zuberi, S. M. (2019). Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia, 60(Suppl 3), S59–S67. https://doi.org/10.1111/epi.14935
Wright, R. A., Pollock, M., & Donaldson, I. M. (1992). Chorea and tuberous sclerosis. Movement Disorders, 7(1), 87–89. https://doi.org/10.1002/mds.870070119
Yamagishi, T., Inoue, K., Ouchi, H., Shibano, K., & Hara, K. (2020). A case of anti-SRY-related HMG-box gene 1 (SOX1) antibody-positive chorea. Rinsho Shinkeigaku, 60(12), 852–856. https://doi.org/10.5692/clinicalneurol.cn-001454
Yim, S. H., Choi, Y. H., Heo, K., & Cho, K. H. (2019). A case of dyskinesia after levetiracetam administration. BMC Neurology, 19(1), 292. https://doi.org/10.1186/s12883-019-1519-8
Yokoyama, Y., Hosokawa, N., Kudo, T., Goda, H., Ito, K., Suzuki, M., & Funakoshi, R. (2020). Chorea-like symptoms and high blood concentration of ceftriaxone in a patient undergoing hemodialysis: A case report. Journal of Infection and Chemotherapy, 26(3), 285–288. https://doi.org/10.1016/j.jiac.2019.10.005
Zech, M., Bardakjian, T. M., Stoklosa, M., Ploski, R., Jech, R., Gonzalez-Alegre, P., & Winkelmann, J. (2021). A neurodevelopmental disorder with dystonia and chorea resulting from clustering CAMK4 variants. Movement Disorders, 36(2), 520–521. https://doi.org/10.1002/mds.28398
Acknowledgements
We thank Michaela Winkelmann (Deutsche Huntington-Hilfe e. V.) for her helpful comments.
AWMF registry No. Is 030/028; level of guideline: S2k; last update: November 24th, 2022; Valid until: November 23th, 2027; Date September 20th, 2023.
Contributing scientific societies
German Neurological Society (DGN). German Association for Psychiatry, Psychotherapy, and Psychosomatics e.V. (DGPPN). German Society of Human Genetics e.V. (GfH). Swiss Neurological Society (SNG). Austrian Society of Neurology (ÖGN). German HD Association (Deutsche Huntington-Hilfe e.V.).
Methodological approach
The project was managed by the coordinator Carsten Saft. The topics were worked on by all other authors based on the current data situation and then coordinated in two Delphi rounds by the guideline group. This S2k level guideline (AWMF-registry number 030/028) is based on a systematic pubmed search. The core statements were evaluated according to the guidelines of the Oxford Centre for Evidence-based Medicine—Levels of Evidence and from this a strength of recommendation was derived. A strong recommendation corresponds in the formulation to a “should”, a recommendation to a “should” and an open recommendation to a “can”. In a second Delphi round, all recommendations were finally agreed upon by the neurological guidelines group. Based on this expert consensus, the formulation of the core statements was evaluated as strong agreement in the case of > 95% of all experts, as agreement in the case of 75–95%, as majority agreement in the case of > 50–75%, and as no agreement in the case of < 50%. In this abbreviated guideline we only refer to agreements of 90–100%. The final recommendations of this guideline were established through a Delphi process (strength of consensus > 75–95% for all recommendations), achieved over two rounds of voting. All statements for which consensus strength is not specified separately were met with a consensus of > 95%. The guideline was reviewed by the Guideline Committee of the German Neurological Society and approved by the German Neurological Society. Interdisciplinarity was established. As a Patient organization the German HD Association (Deutsche Huntington-Hilfe e.V.) was involved.
Funding
Open Access funding enabled and organized by Projekt DEAL. All funding was provided by the Deutsche Gesellschaft für Neurology DGN (German Neurological Society). This guideline has been produced without any influence or support from industry and is provided by the authors free of charge.
Author information
Authors and Affiliations
Contributions
CS: leading author of the S2k-Guideline of the German Neurological Society (DNG) for German-Speaking Countries, conception of guideline development process, literature research, interpretation of literature, discussion, approval of recommendations, preparation of the manuscript. GBL: discussion, approval of recommendation, interpretation of literature, editing of manuscript. JMB, MD, HHJ, RK, JP, HPN, KR, RR, KS: interpretation of literature, discussion, approval of recommendations, editing of manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Given by all authors and the German Neurological society.
Competing interests
All participants in the guideline have submitted their declarations of interest (AWMF form for the declaration of interests in the context of guideline projects) to the coordinator or the Editorial Office for Guidelines of the DGN in time and completely filled out. The evaluation of the declarations of interest with regard to thematic relevance to the guideline was carried out by Carsten Saft. The external evaluation of the interests in the overall view was also carried out by NN AWMF. No conflicts of interest were found, so no consequences, e.g. abstentions, were taken. For reasons of transparency, the interests of the participants and the consequences drawn from them are listed on the respective AWMF guideline website and are also shown in the appendix of the short version. No competing interests with regard to the contents (see attached CoI-satement A detailed listing is available at https://dgn.org/leitlinien/.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saft, C., Burgunder, JM., Dose, M. et al. Differential diagnosis of chorea (guidelines of the German Neurological Society). Neurol. Res. Pract. 5, 63 (2023). https://doi.org/10.1186/s42466-023-00292-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42466-023-00292-2