
Abstract
Background
Typhoid fever, a disease caused by a gram negative bacterial species known as Salmonella typhi, constitutes a significant cause of morbidity and mortality, especially in developing nations of the world. Antibiotic therapy is the major treatment option currently but the rising incidences of resistance to existing antibiotics has necessitated the search for newer ones. The aim of this study is to apply in silico techniques to design highly potent novel imidazole-based drug candidates that strongly antagonize a cell invasion protein (SipA) of Salmonella typhi.
Methods
In this study, a set of anti-Salmonella typhi imidazole analogues were subjected to molecular docking against an important cell invasion protein of the bacterium known as SipA using PyRx graphical user interface of AutoDock Vina software. The best ligand was selected as template for designing more potent analogues. Drug-likeness, pharmacokinetic and toxicity profiles of the designed ligands were assessed through the use of Swiss ADME online tool and Osiris DataWarrior V5.5.0 chemo-informatics program. Kinetic and thermodynamic stabilities of the ligands were ascertained via Density Functional Theory’s Becke-3-parameter Lee–Yang–Parr hybrid functional and 6-31G** basis set-based quantum chemical calculations.
Results
The bioactive ligands were found to possess Gibb’s free binding energy (ΔG) values ranging from − 5.4 to − 6.7 kcal/mol against the active sites of the protease. Ligand 13 with ΔG = − 6.7 kcal/mol was used as template to design more potent analogues; B-1 and B-2 with ΔG value of − 7.8 kcal/mol and − 7.6 kcal/mol, respectively, against the protein target. When compared with ciprofloxacin used as control with ΔG value of − 6.8 kcal/mol, the designed ligands were found to be more potent. Furthermore, drug-likeness and ADMET profiling of the designed ligands revealed that they have excellent oral bioavailability and sound pharmacokinetic profiles. In addition, quantum chemical calculations revealed HOMO–LUMO energy gap of 3.58 eV and 3.45 eV; and global electrophilicity index of 4.95 eV and 4.79 eV for B-1 and B-2 ligands, respectively, indicative of their favorable kinetic and thermodynamic stabilities.
Conclusions
It is envisaged that the findings of this study would provide an excellent blueprint for developing novel antibiotics against multidrug resistant Salmonella typhi.
Similar content being viewed by others

Background
Typhoid fever is a systemic infection caused by Salmonella typhi, transmitted through the fecal–oral route (Klemm et al. 2018). The primary symptoms of this disease include prolonged high fever, headache, and malaise. If left untreated for a long time, the infection may degenerate to altered mental states, ileus, gastrointestinal bleeding, intestinal perforation, septic shock, and death (Basnyat et al. 2021; Birkhold et al. 2020). Typhoid fever is prevalent in South Asia, Southeast Asia, and sub-Saharan Africa where access to clean water, sanitation, and quality health care delivery is abysmally poor. It is a major cause of morbidity and mortality, especially among children and young adults globally with an estimated 14.3 million infections and over 135,000 deaths annually (GBD 2019; GBD 2016; Kim et al. 2019; Mogasale et al. 2014). Although, antibiotics such as chloramphenicol, ampicillin and cotrimoxazole, fluoroquinolones, third-generation cephalosporins and azithromycin have been currently recommended by the World Health Organization for the treatment of Typhoid fever infection, but the rising cases of resistance by Salmonella typhi to these drugs pose a serious threat to public health (Browne et al. 2020), necessitating the search for newer drug candidates in the antibiotic drug development pipeline.
In silico techniques such as structure-based drug design (SBDD), pharmacokinetic profiling, and quantum chemical calculations provide a safer, faster, and economical avenues in modern drug discovery. SBDD involves the use of molecular docking simulation to investigate the interaction of small molecules with the active sites of a protein target (macromolecule) whose 3D structure is known. Through the application of SBDD technique, the orientation of the therapeutic compound (pose) within the active sites of the macromolecule, its geometry conformation, and scoring, usually in form of Gibb’s free energy of binding (ΔG) are ascertained (Behl et al. 2021; Hussain et al. 2021). Also, In silico Pharmacokinetic profiling deals with the use computer aided techniques to determine the fate of a drug candidate in the biological system with respect to its absorption (A), distribution (D), metabolism (M), excretion (E), and toxicity (T) (Daina et al. 2017). This technique is used in modern pharmaceutical research to reduce attrition rates in drug development due to poor pharmacokinetics (Daoud et al. 2021). Furthermore, the safety and efficacy of a drug are grossly influenced by its ability to withstand degradation for a reasonable time within and outside biological system in order for it to optimally exert its therapeutic roles. Thus, ensuring the chemical and physical stability of therapeutic compounds is paramount to guarantee their safety and quality (Padmaja et al. 2009). The kinetic and thermodynamic stability of molecules are influenced by the energies of their frontier orbitals, which could be determined via quantum chemical calculations (Khalid et al. 2017; Ruiz-Morales, 2002; Senosiain et al. 2005).
Imidazole heterocycle has found immense applications over the last decades owing to its wide range of pharmacological properties making them promising drug candidates. This class of chemical compounds has been reported to possess antibacterial, antifungal, anticancer, antiviral, anti-inflammatory, and antidiabetic properties. Imidazole scaffolds are essential components of standard pharmaceuticals such as Miconazole, Econazole, Ketoconazole, Clotrimazole, Ornidazole, Azomycin, Oxiconazole, and Clonidine (Chen et al. 2018a, b; Hu et al. 2018; Rossi and Ciofalo 2020; Serdaliyeva et al. 2022; Wang et al. 2019). The antibacterial properties of nitro-imidazole scaffold is based on its ability to penetrate the cell of the organisms via passive diffusion where it experiences reduction reaction leading to the formation of nitro radical anion which eventually oxidizes the bacteria’s DNA leading to breakage of DNA strand and cell death (Edwards et al. 1993). Also, imidazoles inhibit flavohemoglobin, a protein in bacteria that metabolizes nitric oxide (NO) to nitrates and prevent NO-mediated damage, growth inhibition, and death (Helmick et al. 2005). In another study, imidazole scaffold was found to inhibit enoyl acyl carrier protein reductase (FabI), an enzyme involved in the synthesis of bacterial fatty acids (Heerding et al. 2001).
In search of novel antibiotic drug candidates that could curb the threat to public health posed by rising incidences of multidrug resistant Salmonella typhi, some in silico investigations targeting different enzymes of the bacterium has been conducted recently (Akter et al 2022; Anebi et al. 2021; Arunkumar et al. 2022; Bakthavatchalam et al. 2017; Qadir et al. 2018; Verma et al. 2020). In this study, we are aiming at the application of in silico techniques to design highly potent, non-toxic, kinetic and thermodynamically stable novel imidazole-based inhibitors of SipA protease of Salmonella typhi, an effector protein of the bacterium crucial to its virulence and pathogenicity. SipA is type-3 secretion protein that enables the bacterium to survive the harsh conditions of the extracellular milieu of the small intestine of the host, due to factors such as low pH, shear stress due to mucosal secretions or blood and host defense mechanisms and subsequently invade the intestines of its host and gains entry into the tissues where it replicates rapidly and manifest varying degree of virulence (Birhanu et al. 2021, 2018; Khan and Shamim 2022). Despite the enormous antibacterial properties of imidazole-based compounds, no in silico investigation of its antibacterial properties against Salmonella typhi has been carried out to the best of our knowledge.
Methods
Collection of data set
A set of thirty anti-Salmonella typhi imidazole derivatives was gotten from reported work (Ganguly et al. 2011). The biological activity of the ligands were expressed as Minimum Inhibitory Concentration (MIC). The 2D chemical structure and experimentally determined MIC values for each member of the data set are presented in Table 1.
Many methods abound in literature for one-pot synthesis of imidazole derivatives but are faced with drawbacks such as toxic and expensive catalysts that requires harsh reaction conditions. However, the use of green lactic acid as solvent presents an inexpensive, biodegradable, environmentally friendly approach. The green solvent (lactic acid) is easily obtainable from fermentation of carbohydrates, easy to handle, readily available, and has been reported to be used as organocatalyst in many organic reactions (Sonar et al. 2019). The reaction scheme involving the use of lactic acid as solvent for synthesis of some substituted imidazole derivative is presented in Fig. 1.

General reaction scheme for the synthesis of 2,4,5-trisubstituted imidazole
Energy minimization of the ligands
In order to obtain the most stable conformations of the ligands, their 2D chemical structures were drawn with ChemDraw Ultra 12.0 and exported unto Spartan 14 work space (www.wavefun.com) where they were subsequently optimized using Semi-empirical (pm3) method. The optimized molecules were then saved in pdb and sdf formats for further in silico analysis (Ameji et al. 2022).
Molecular Docking evaluation
Molecular Docking simulation is an in silico technique that allows accurate prediction of the strength of association between a ligand and a target macromolecule. The optimized ligands were prepared using the AutoDock Vina interface and saved as pdbqt files. The 3D crystal structure of SipA protein was retrieved from protein data bank at www. rcsb. org/pdb (PDB Code: 1Q5Z). The protein was firstly prepared on Discovery Studio software interface where attached water molecules and heteroatoms were removed. It was further exported unto the AutoDock Vina platform where missing atoms were checked, repaired, and polar hydrogen and Kollman charges were added to the macromolecule. The prepared protein was then saved as pdbqt file formats. As a way of ascertaining the binding affinity values of the investigated imidazole analogues against SipA enzyme of Salmonella typhi, the bioactive ligands were docked against the active sites of the protease using the PyRx graphical user interface of AutoDock Vina software (Ameji et al. 2022).
Design of novel derivatives
In line with a major objective of this study, which is the design of highly potent imidazole-based drug candidates, the ligand with the best binding affinity (highest negative ΔG value) was selected as template molecule and subsequently subjected to pharmacophoric modifications giving rise to some new derivatives. The new derivatives were then docked against the SipA target and the analogues with better ΔG values than the template molecule were selected as the novel drug candidates to be subjected to more analysis. As a quality control measure, an approved antibiotic, ciprofloxacin was docked against SipA macromolecule and the computed binding affinity value was compared with those of the novel ligands (Ameji et al. 2022).
Drug-likeness assessment
Drug-likeness is primarily concerned with the evaluation of the oral bioavailability of therapeutic compounds. This important parameter was evaluated for the designed ligands using the Lipinski’s rule of five and the Veber’s rule. The former rule states that a drug-like compound must not violate any of the following indices; molecular weight (Mw) ≤ 500, number of hydrogen bond donors (HBD) ≤ 5, octanol/ water partition coefficient (Log P) ≤ 5 and number of hydrogen bond acceptors (HBA) ≤ 10. The latter rule on the other hand, states that a drug-like molecule must have number of rotatable bonds (NRB) and topological polar surface area (TPSA) that is not greater than 10 and 140 Å2, respectively (Ameji et al. 2022). The drug-likeness of the novel ligands were evaluated with the aid of SwissADME online tool at www.swissadme.ch/.
Pharmacokinetic profiling
Pharmacokinetics describes the time course of drugs and their effects in the body. It include absorption (A), distribution (D), metabolism (M), excretion (E), and toxicity (T) of drugs. The SwissADME (www.swissadme.ch/) online resource and Osiris DataWarrior V5.5.0 chemo-informatics program were used to assess the ADMET profiles of the designed ligands.
Assessment of kinetic and thermodynamic stabilities
Thermodynamic stability is the stability of the lowest energy state of a system while kinetic stability is the stability of the highest energy state of a system. Ensuring the chemical and physical stability of therapeutic compounds is paramount to guarantee their safety and quality. The knowledge of the energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) give idea on stabilities of molecules (Khalid et al. 2017). The LUMO serves as electron acceptor with an associated energy expressed as electron affinity (EA), while the HOMO plays the role of electron donors and its energy is linked to ionization potential (IP). The charge transfer interaction within a molecule is explained by the HOMO–LUMO energy gap defined in Eq. 1. High HOMO–LUMO energy gap in a molecule connote low chemical reactivity and high kinetic stability. This is because, it is not energetically feasible to add electron to the high-lying LUMO by removing electrons from the low-lying HOMO (Ruiz-Morales, 2002; Senosiain et al. 2005). Molecules with HOMO–LUMO energy gap > 1.30 are generally considered to be kinetically stable (Aihara 1999).
where ELUMO and EHOMO denote energy of LUMO and energy of HOMO, respectively.
An important quantum chemical descriptor of thermodynamic stability of a molecule is the global electrophilicity index (ω). ω is defined as the energy decrease that accompanies the flow of electron from HOMO to LUMO in molecules. ω is defined in Eq. 2.
where η is the global chemical hardness and µ the electronic chemical potential which describes the charge transfer within a system in the ground state. Equations 3 and 4, defines η and µ, respectively.
The higher the value of ω in a molecule, the higher its reactivity and vice versa (Senosiain et al. 2005).
The Spartan’14 software (www.wavefun.com) was used to perform quantum chemical calculations on the designed ligands and the standard antibiotics. Geometry optimization and frequency calculations were carried out using DFT alongside the B3LYP standard principle in conjunction with the split-valence 6-31G** basis function. The choice of this method is anchored on its computational efficiency and high accuracy in obtaining geometries, zero-point energy and frequencies (Halls et al. 2001; Miar et al. 2021).
Results
Identification of template molecule
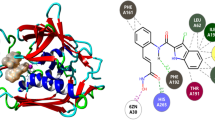
Antibiotics such as quinolones and beta lactams function by binding to a target enzyme of the bacterium, disrupting its enzymatic functions leading to inactivation or death of the microbe. The result of the molecular docking investigation of the studied bioactive imidazole-based ligands and Ciprofloxacin (R) against SipA protease of Salmonella typhi is presented in Table 2. Potency of the ligands varies directly with the negative value of Binding affinity (ΔG). Thus, ligand 13 with ΔG value of − 6.7 kcal/mol was selected as the template molecule to be used to design more potent analogues. The 3D and 2D diagrams of interaction of the ligand within the binding pocket of SipA protease are shown in Fig. 2.

3D and 2D diagram of interaction of the template molecule with SipA
Newly designed ligands
Potency of a bioactive molecule against any macromolecular target is a function of certain features within its moiety called pharmacophores. In order to obtain more potent derivatives of the template, it was subjected to pharmacophoric modifications leading to the design of ligand B-1 and B-2 with ΔG value of − 7.8 kcal/mol and − 7.6 kcal/mol, respectively. Figure 3 gives the 2D chemical structures of B-1, B-2 and R, while their 2D diagram of interactions with the amino acid residues of the target are presented in Fig. 4.

2D chemical structures of the novel ligands and R. a: 3-((2-(2-benzyl-5-nitro-1H-imidazol-1-yl)-1-chloroethyl)amino)phenol. b: 3-((1-chloro-2-(2-(4-hydroxybenzyl)-5-nitro-1H-imidazol-1-yl)ethyl)amino)phenol. c: 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylic acid

2D diagram of interaction of the designed ligands and R with amino acid residues of SipA target
Drug-likeness and ADMET properties of B-1, B-2 and R
Drug-likeness and ADMET properties of a bioactive ligand are functions of certain physicochemical features of the molecule. The computed values of molecular descriptors of drug-likeness and ADMET properties of the novel ligands and R are presented in Tables 3 and 4, respectively.
Quantum chemical descriptors of B-1, B-2 and R
Electronic features of B-1, B-2 and R derived from quantum mechanical calculations are presented in Table 5. The shapes and energies of the LUMO and HOMO for the ligands are shown in Fig. 5

The shapes and energy of the Frontier orbitals of the designed ligands and R
Discussion
SipA protein in Salmonella typhi plays unique roles in pathogenicity and virulence in the organism. It aids the survival of the pathogen in the intestine of the host and enhances its penetration into relevant tissues where it create a niche for itself and unleash varying degree of virulence. Binding of bioactive ligands to this macromolecular target inhibits its enzymatic roles and consequently predisposes the bacterial species to the harsh conditions of the extracellular milieu of the small intestine of the host leading to its inactivation or death. The result of molecular docking simulation study on the bioactive imidazole analogues against the SipA target of Salmonella typhi is presented in Table 2. The binding affinity (ΔG) values of the ligands range from − 5.4 to − 6.7 kcal/mol. However, ligand 13 with the best ΔG value of − 6.7 kcal/mol was selected as template molecule for designing more potent analogues. Ligand 13 whose 2D and 3D diagram of interactions with the active sites of SipA protease is shown in Fig. 2, was found to interact with the amino acid residues of the target via pi-alkyl interactions with ARG542; a pi-sigma interaction with LEU 634; a pi-anion and a Van der waals interaction with ASP 540; and five conventional hydrogen bonds with LEU 631, LEU 640, GLY 630, GLU 543, and ALA 541 amino acid residues.
Two novel ligands; B-1 and B-2 (Fig. 3) with ΔG value of − 7.8 kcal/mol and − 7.6 kcal/mol, respectively, against the SipA target, were designed through structural modification of the template ligand. The novel ligands were found to more potent when compared with the standard ligand, ciprofloxacin (R) with ΔG value of − 6.8 kcal/mol against the target. Ligands B-1 and B-2 were found to interact with the active sites of the SipA target via similar mechanism. They both bind to the target via; pi-alkyl interactions with LYS 518, PRO 613 and PRO 513 amino acid side chains, a pi-sigma interaction with VAL 608, and two conventional hydrogen bonds with LEU 517 and PHE 514 (Fig. 4a,b). Also, the standard ligand (R) binds to the target protease (Fig. 3c) through; a pi-alkyl interaction with LYS 551; a pi-sigma with THR 544; and a conventional hydrogen bond with SER 526.
Drug-likeness defines the oral bioavailability of bioactive compounds. This parameter plays a significant role in drug discovery and development because substantial number of drugs are administered through the oral route. The physicochemical properties of B-1 and B-2 ligands that could be used to describe their drug-likeness are presented in Table 3. The result shows that the ligands, like R obey both the Lipinski’s and Veber’s rules, an indication of their excellent oral bioavailability.
Pharmacokinetic and toxicity profiling of therapeutic molecules with reference to their absorption, distribution, metabolism, excretion, and toxicity (ADMET) at early stage of drug discovery is a crucial component of modern drug design owing to the facts that these properties play pivotal roles in the success or failure rates of drug candidates. Pharmacokinetic profiles of B-1 and B-2 ligands presented in Table 4 show that they possess good gastrointestinal absorption just like the standard ligand, R.
Also, aqueous solubility deals with the maximum amount of a substance that dissolves completely in water at a given temperature and pressure. It is an essential parameter for consideration in modern drug design since drugs must be reasonably soluble in order to exert significant pharmacological response (Birch et al. 2019; Oja et al. 2022). Poor aqueous solubility has been pointed out to be responsible for slow absorption of drugs, leading to poor bioavailability and gastrointestinal mucosal toxicity (Shah et al. 2017). Ligands R, B-1, B-2, with estimated solubility (ESOL) value of 15.7 mg/ml, 0.006 mg/ml, and 0.009 mg/ml, respectively, were found to exhibit moderate to high solubility (Table 4). Likewise, Blood Brain Barrier (BBB) is an endothelial cell layer of the brain that restrict entry of toxins and xenobiotic into the brain (Alsenan et al. 2021; Tong et al. 2022; Wang et al. 2019). The result in Table 4 shows that all the ligands possess no BBB permeating potentials.
Furthermore, P-glycoprotein (P-gp) are membrane transporters that protects the body from harmful substances by extruding via efflux action, substrate xenobiotic absorbed in the intestines back to the lumen, removing drugs from the kidneys and liver into the urine and bile, respectively, and maintaining integrity of BBB by limiting cellular uptake of its substrates from blood circulation into the brain (Chen et al. 2018a, b; Miyake et al. 2021). The designed ligands, unlike R were found to be non-substrate of P-gp and are such could be unperturbed by the efflux action of P-gp.
Additionally, the ligands were screened against cytochrome P450 (CYP45) which performs the role of metabolism and biotransformation of drugs in the biological system. The result (Table 4) revealed that ligands B-1 and B-2, unlike R are inhibitors of all the five isoforms of CYP45 namely; CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 and as such may not be easily metabolized by these proteins. Toxicity evaluation of the ligands revealed that B-1 and B-2 are neither tumorigenic nor possess irritating effect. However, the two ligands could be mutagenic and pose threat to the reproductive system.
Stability of therapeutic compounds is necessary in order to guarantee their safety and efficacy. Drugs must exhibit reasonable level of stability for it to withstand unwanted physical and chemical degradation that affects its safety and pharmacological response. The stability of molecules is governed by the energies of their frontier orbitals (HOMO and LUMO energies). Thermodynamic stability refers to the stability of the lowest energy state of a system while kinetic stability is the stability of the highest energy state of a system. The former is governed by the quantum chemical descriptor known as global electrophilicity index (ω) while the latter is governed by the HOMO/LUMO energy gap. Figure 5 presents the HOMO and LUMO regions of the designed ligands and R. The energy gap value of 3.58 eV, 3.45 eV, and 4.38 eV computed for B-1, B-2, and R, respectively (Table 5) are > 1.30 eV. Thus, the novel ligands, like R are kinetically stable. Likewise, the ω value of 4.95 eV, 4.75 eV, and 2.70 eV were computed for ligands B-1, B-2, and R, respectively (Table 5). The low values of this descriptor is an indication of high thermodynamic stability of the ligands.
Summarily, this study is an in silico research and as such the aforementioned research findings will require further in vitro and in vivo validations.
Conclusions
The rising cases of drug resistant strains of Salmonella typhi has necessitated the search for newer drug candidates in the drug development pipeline. In search for novel antibiotic drugs against this bacterium, a set of bioactive imidazole analogues were subjected to molecular docking-based virtual screening against a cell invasion protein, SipA of the organism using the PyRx graphical user interface of AutoDock Vina software. The ligands were found to bind to the target sites of the macromolecular target with binding affinity (ΔG) values that ranges from − 5.4 to − 6.7 kcal/mol. Ligand 13 with the best dock score of − 6.7 kcal/mol was used as template to design more potent analogues; B-1 and B-2 with ΔG value of − 7.8 and − 7.6 kcal/mol, respectively. When compared with a standard drug, ciprofloxacin (ΔG value of − 6.8 kcal/mol) used as control, the designed ligand were found to be more potent. Ligands B-1 and B-2 were found to obey both the Lipinski’s rule of five and Veber’s rule, an indication of their positive drug-likeness. Also, ADMET profiling of the designed ligands reveals their excellent pharmacokinetic and toxicity profiles. Furthermore, DFT-based quantum chemical calculation revealed an appreciable kinetic and thermodynamic stability of the ligands. Further in vivo and in vitro investigations of these newly designed ligands may lead to discovery of novel antibiotics that could curb the rising cases of drug resistance in Salmonella typhi.
Availability of data and materials
Not applicable.
Abbreviations
- SipA:
-
Salmonella invasion protein A
- HOMO:
-
Highest occupied molecular orbital
- LUMO:
-
Lowest unoccupied molecular orbital
- ADMET:
-
Absorption, distribution, metabolism, excretion, toxicity
- SBDD:
-
Structure-based drug design
- HBA:
-
Hydrogen bond acceptor
- HBD:
-
Hydrogen bond donor
- Mw:
-
Molecular weight
- cLogP:
-
Consensus octanol water partition coefficient
- NRB:
-
Number of rotatable bond
- TPSA:
-
Topological polar surface area
- ESOL:
-
Estimated solubility
- GIA:
-
Gastrointestinal absorption
- BBB:
-
Blood brain barrier penetration
- P-gp+ :
-
P-glycoprotein substrate
References
Aihara J (1999) Reduced HOMO−LUMO gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A 103(37):7487–7495
Akter T, Chakma M, Tanzina AY, Rumi MH, Shimu MSS et al (2022) Curcumin analogues as a potential drug against antibiotic resistant protein, b-lactamases and L, D-transpeptidases involved in toxin secretion in Salmonella typhi: a computational approach. Biomedinformatics 2:77–100. https://doi.org/10.3390/biomedinformatics2010005
Alsenan S, Al-Turaiki I, Hafez A (2021) A deep learning approach to predict blood-brain barrier permeability. PeerJ Comput Sci. 7:e515. https://doi.org/10.7717/peerj-cs.515
Ameji JP, Uzairu U, Shallangwa GA, Uba S (2022) Virtual screening of novel pyridine derivatives as effective inhibitors of DNA gyrase (GyrA) of salmonella typhi. Curr Chem Lett 12:1–16
Anebi E, Ibrahim MT, Shallangwa GA, Isyaku S, Abdulsalam S, Danmallam AM (2021) Molecular docking study, drug-likeness and pharmacokinetic properties (ADMET) prediction of some novel thiophene derivatives as Salmonella typhi inhibitors. Bayero J Pure Appl Sci 14(2):235–244. https://doi.org/10.4314/bajopas.v14i2.29
Arunkumar M, Mahalakshmi M, Ashokkumar V, Aravind MK, Gunaseelan S, Mohankumar V et al (2022) Evaluation of seaweed sulfated polysaccharides as natural antagonists targeting Salmonella typhi OmpF: molecular docking and pharmacokinetic profiling. Beni-Suef Univ J Basic Appl Sci 11:8. https://doi.org/10.1186/s43088-021-00192-x
Bakthavatchalam YD, Kumar TD, Ayubi IA, Shankar BA, Babu P, Munusamy E, Thukkaram B, Ravi R, Doss CGP, Veeraraghavan B (2017) In vitro efficacy and in silico analysis of cefixime–ofloxacin combination for Salmonella typhi from bloodstream infection. J Appl Microbiol 123:615–624
Basnyat B, Qamar FN, Rupali P, Ahmed T, Parry CM (2021) Clinical update: enteric fever. BMJ 372:n437. https://doi.org/10.1136/BMJ.N437
Behl T, Kaur I, Sehgal A, Singh S, Bhatia S, Al-Harrasi A et al (2021) Bioinformatics accelerates the major tetrad: a real boost for the pharmaceutical industry. IJMS 22:1–28
Birch H, Redman AD, Letinski DJ, Lyon DY, Mayer P (2019) Determining the water solubility of difficult-to-test substances: a tutorial review. Anal Chim Acta 1086:16–28
Birhanu BT, Park N, Lee S, Hossain MA, Park S (2018) Inhibition of Salmonella Typhimurium adhesion, invasion, and intracellular survival via treatment with methyl gallate alone and in combination with marbofloxacin. Vet Res 49:101. https://doi.org/10.1186/s13567-018-0597-8
Birhanu BT, Lee EB, Lee SJ, Park SC (2021) Targeting Salmonella Typhimurium invasion and intracellular survival using pyrogallol. Front Microbiol 12:631426. https://doi.org/10.3389/fmicb.2021.631426
Birkhold M, Coulibaly Y, Coulibaly O, Dembélé P, Kim DS, Sow S et al (2020) Morbidity and mortality of typhoid intestinal perforation among children in Sub-Saharan Africa 1995–2019: a scoping review. World J Surg 44(9):2892–2902. https://doi.org/10.1007/s00268-020-05567-2
Browne AJ, Hamadani BHK, Kumaran EAP, Rao P, Longbottom J et al (2020) Drug-resistant enteric fever worldwide, 1990 to 2018: a systematic review and meta-analysis. BMC Med 18:1–22. https://doi.org/10.1186/s12916-019-1443-1
Chen L, Zhao B, Fan ZJ, Liu XM, Wu QF, Li HP, Wang HX (2018a) Synthesis of novel 3,4-chloroisothiazole-based imidazoles as fungicides and evaluation of their mode of action. J Agric Food Chem 66:7319–7327
Chen C, Lee M, Weng C, Leong MK (2018b) Theoretical prediction of the complex P-glycoprotein substrate efflux based on the novel hierarchical support vector regression scheme. Molecules 23:1820. https://doi.org/10.3390/molecules23071820
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717. https://doi.org/10.1038/srep42717
Daoud NEK, Deb PK, Alzweiri M, Borah P, Venugopala KN, Hourani W et al (2021) ADMET profiling in drug discovery and development: perspectives of in silico, in vitro and integrated approaches. Curr Drug Metab. https://doi.org/10.2174/1389200222666210705122913
Edwards DI (1993) Review Nitroimidazole drugs—action and resistance mechanisms I. Mechanisms of Action. J Antimicrob Chemother 31:9–20
Ganguly S, Vithlani VV, Kesharwani AK, Kuhu R, Baskar L, Mitramazumder P, Sharon A, Dev A (2011) Synthesis, antibacterial and potential anti-HIV activity of some novel imidazole analogs. Acta Pharm 61:187–201. https://doi.org/10.2478/v10007-011-0018-2
GBD (2016) Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017; 390: 1211–59.
GBD (2019) Typhoid and Paratyphoid Collaborators. The global burden of typhoid and paratyphoid fevers: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect Dis. 19(4):369–81.
Halls MD, Velkovski J, Schlegel HB (2001) Harmonic frequency scaling factors for Hartree-Fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set. Theor Chem Acc 105(6):413–421
Heerding DA, Chan G, DeWolf WE, Fosberry AP, Janson JA, Jaworski DD, McManus E, Miller WH, Moore TD, Payne DJ, Qiu X, Rittenhouse SF, Slater-Radosti C, Smith W, Takata DT, Vaidya KS, Yuan CCK, Huffman WF (2001) 1,4-Disubstituted imidazoles are potential antibacterial agents functioning as inhibitors of enoyl acyl carrier protein reductase (FabI). Bioorg Med Chem Lett 1:2061–2065
Helmick RA, Fletcher AE, Gardner AM, Gessner CR, Hvitved AN, Gustin MC, Gardner PR (2005) Imidazole antibiotics inhibit the nitric oxide dioxygenase function of microbial flavohemoglobin. Antimicrob Agents Chemother 49(5):1837–1843
Hu Y, Shen YF, Wu XH, Tu X, Wang GX (2018) Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur J Med Chem 143:958–969
Hussain W, Rasool N, Khan YD (2021) Insights into machine learning-based approaches for virtual screening in drug discovery: existing strategies and streamlining through FP-CADD. Curr Drug Discov Technol 18:463–472
Khalid M, Ali M, Aslam M, Sumrra SH, Khan MU, Raza N, Kumar N, Imran M (2017) Frontier molecular, natural bond orbital, UV-VIS spectral study, solvent influence on geometric parameters, vibrational frequencies and solvation energies of 8-hydroxyquinoline. IJPSR 8(2):457–469. https://doi.org/10.13040/IJPSR.0975-8232.8(2).457-69
Khan M, Shamim S (2022) Understanding the mechanism of antimicrobial resistance and pathogenesis of Salmonella enterica Serovar Typhi. Microorganisms 10:2006. https://doi.org/10.3390/microorganisms10102006
Kim J, Im J, Parajulee P, Holm M, Espinoza LMC, Poudyal N, Mogeni OD, Marks F (2019) A systematic review of typhoid fever occurrence in Africa. Clin Infect Dis 69(6):492–498. https://doi.org/10.1093/cid/ciz525
Klemm JE, Shakoor S, Page JA, Qamar NF, Judge K, Saeed KD et al (2018) Emergence of an extensively drug-resistant Salmonella enterica serovar Typhi clone harboring a promiscuous plasmid encoding resistance to fluoroquinolones and third-generation cephalosporins. Mbio 9:e00105-e118. https://doi.org/10.1128/mBio.00105-18
Miar M, Shiroudi A, Pourshamsian K, Oliaey RA, Hatamjafari F (2021) Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo[d] thiazole-2(3H)-imine and its para-substituted derivatives: Solvent and substituent effects. J Chem Res 2021:147–158
Miyake T, Tsutsui H, Haraya K, Tachibana T, Morimoto K, Takehara S, Ayabe M, Kobayashi K, Kazuki Y (2021) Quantitative prediction of P-glycoprotein-mediated drug–drug interactions and intestinal absorption using humanized mice. Br J Pharmacol 178:4335–4351. https://doi.org/10.1111/bph.15612
Mogasale V, Maskery B, Ochiai RL et al (2014) Burden of typhoid fever in low-income and middle-income countries: a systematic, literature-based update with risk-factor adjustment. Lancet Glob Health 2:e570–e580
Oja M, Sild S, Piir G, Maran U (2022) Intrinsic aqueous solubility: mechanistically transparent data-driven modeling of drug substances. Pharmaceutics 14:2248. https://doi.org/10.3390/pharmaceutics14102248
Padmaja L, Ravikumar C, Sajan D, Joe HI et al (2009) Density functional study on the structural conformations and intramolecular charge transfer from the vibrational spectra of the anticancer drug combretastatin-A2. J Raman Spectrosc 40:419–428
Qadir IM, Mushtaq H, Mobeen T (2018) In-silico study of potential carboxylic acid derivatives as D-glutamate ligase inhibitors in Salmonella typhi. Kuwait J Sci 45(1):100–107
Rossi R, Ciofalo M (2020) An updated review on the synthesis and antibacterial activity of molecular hybrids and conjugates bearing imidazole moiety. Molecules 25:5133
Ruiz-Morales Y (2002) HOMO−LUMO gap as an index of molecular size and structure for polycyclic aromatic hydrocarbons (PAHs) and asphaltenes: a theoretical study I. J Phys Chem A 106:11283–11308
Senosiain JP, Klippenstein SJ, Miller JA (2005) The reaction of acetylene with hydroxyl radicals. J Phys Chem A 109:6045–6055
Serdaliyeva D, Nurgozhin T, Satbayeva E, Khayitova M, Seitaliyeva A, Ananyeva L (2022) Review of pharmacological effects of imidazole derivatives. J Clin Med Kazakhstan 19(3):11–15. https://doi.org/10.23950/jcmk/12117
Shah P, Goodyear B, Michniak-Kohn BB (2017) A review: enhancement of solubility and oral bioavailability of poorly soluble drugs. Adv Pharm J 2(5):161–173
Sonar J, Pardeshi S, Dokhe S, Pawar R, Kharat K, Zine A, Matsagar B, Wu K, Thore S (2019) An efficient method for the synthesis of 2,4,5-trisubstituted imidazoles using lactic acid as promoter. SN Appl Sci 1:1045. https://doi.org/10.1007/s42452-019-0935-0
Tong X, Wang D, Ding X, Tan X, Ren Q, Chen G et al (2022) Blood–brain barrier penetration prediction enhanced by uncertainty estimation. J Cheminformatics 14:44. https://doi.org/10.1186/s13321-022-00619-2
Verma AK, Danyaya AI, Kumar A, Shuaibu BS et al (2020) Virtual screening, molecular docking, and ADME/T analysis of natural product library against cell invasion protein SipB from Salmonella enterica serotype typhi: In silico analysis. Acta Sci Pharm Sci 4(8):20–30
Wang PY, Wang MW, Zeng D, Xiang M, Rao JR, Liu QQ, Liu LW, Wu ZB, Li Z, Song BA et al (2019) Rational optimization and action mechanism of novel imidazole (or Imidazolium)-labeled 1,3,4 oxadiazole thioethers as promising antibacterial agents against plant bacterial diseases. J Agric Food Chem 67:3535–3545
Acknowledgements
We are grateful to staff members in the Physical Chemistry Unit of Chemistry Department, Ahmadu Bello University, Zaria-Nigeria, for providing the essential facilities to carry out this research work.
Funding
No fund was received for this work.
Author information
Authors and Affiliations
Contributions
AU outlined and designed the research work. GAS and SU analyzed and supervised the study. APJ handled the computational chemistry software and drafted the manuscript. In addition, all authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ameji, J.P., Uzairu, A., Shallangwa, G.A. et al. Design, pharmacokinetic profiling, and assessment of kinetic and thermodynamic stability of novel anti-Salmonella typhi imidazole analogues. Bull Natl Res Cent 47, 6 (2023). https://doi.org/10.1186/s42269-023-00983-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-023-00983-5