
Abstract
Background
Actinobacteria is as a group of advanced filamentous bacteria. Rare Actinobacteria are of special interest as they are rarely isolated from the environments. They are a major source of important bioactive compounds. Determining the proper strategy for the identification of Actinobacteria harboring biosynthetic gene clusters and producing bioactive molecules is a challenging platform.
Methodology
In this review, we discuss a consequence of microbiological and molecular methods for the identification of rare Actinobacteria. In addition to that, we shed light on rare Actinobacteria’s significance in antibiotic production. We also clarified molecular approaches for the manipulation of novel biosynthetic gene clusters via PCR screening, fosmid libraries, and Illumina whole-genome sequencing in combination with bioinformatics analysis.
Conclusion
Perceptions of the conventional and molecular identification of Actinobacteria were conducted. This will open the door for the genetic manipulation of novel antibiotic gene clusters in heterologous hosts. Also, these conclusions will lead to constructing new bioactive molecules via genetically engineering biosynthetic pathways.
Similar content being viewed by others

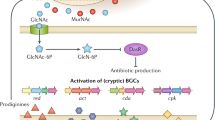
Introduction
Actinobacteria is a group of aerobic, spore-forming Gram-positive bacteria; they belong to the order Actinomycetales. It harbors GC content of DNA ranges from just under 50% (e.g., Hoyosella and Tropheryma) to over 70% (e.g., Frankia and Streptomyces), which are recorded from 16S ribosomal cataloging and DNA-rRNA pairing studies (Korn-Wendisch & Schneider, 1992). Now, it signifies one of the largest taxonomic groups recognized within the domain Bacteria (Ventura et al., 2007). The name Actinobacteria originated from Greek “atkis” (a ray) and “mykes” (fungus), having criteria of both Bacteria and Fungi (Das & Khosla, 2009), but it has sufficient different features to restrict it into kingdom Bacteria.
Actinobacteria are generally aerobic; however, some genera are facultative or obligatory anaerobic. They are chemoheterotrophic using diverse energy sources and complex polymers. Most of the Actinobacteria are free-living in a wider range of habitats in nature such as water (Ma et al., 2009), soil (Elbendary et al., 2018), the greatest depth of the ocean (Pathom-Aree et al., 2006), Antarctica (Rego et al., 2019), and desert soil (Busarakam et al., 2016). It has been reported that Actinobacteria are isolated from all layers of soil but gradually decreases with increasing the depth (Takahashi & Omura, 2003). In addition, some are pathogens for humans (Könönen & Wade, 2015), animals (Ertaş et al., 2005), and plants (Lerat, 2009). Actinobacteria have different morphological features that ranged from relatively simple rods and cocci to complex mycelial organization similar to eukaryotes. They were considered as an intermediate group between bacteria and fungi but now are recognized as prokaryotic organisms. Actinobacteria are considered as Gram stain-positive or Gram stain variable. The cell wall of Actinobacteria is rigid to keep the shape of the cell and avoid bursting at high osmotic pressure (Colquhoun et al., 1998). It is formed of different complex compounds such as peptidoglycan, teichoic, teichuronic acid, and polysaccharides (Davenport et al., 2000). The trend of Actinobacteria identification has been and still augmented, as it is a challenging signal for further exploration of new natural compounds. In this review, we highlight the procedures of rare Actinobacteria identification and manipulation of their biosynthetic gene cluster (Fig. 1).

Identification of bioactive rare Actinobacteria. The following flow chart demonstrates steps for the identification of Actinobacteria on the genus level and their biosynthetic genes. Additionally, it gives different options to manipulate these genes and hence monitor the biosynthetic capability of the Actinobacteria
Taxonomy of Actinobacteria
According to the first edition of Bergey’s Manual of Systematic Bacteriology, Actinobacteria belonged to the order Actinomycetales and was subdivided into 4 families Streptomycetaceae, Actinomycetaceae, Actinoplanaceae, and Mycobacteriaceae. The taxonomy of Actinobacteria has evolved considerably over time with the buildup of information. In the second edition of Bergey’s Manual of Systematic Bacteriology, Actinobacteria were included separately in the fifth volume. Phylum Actinobacteria is separated into 6 classes: Actinobacteria, Acidimicrobiia, Coriobacteriia, Nitriliruptoria, Rubrobacteria, and Thermoleophilia. Class Actinobacteria is subdivided into 16 orders: Actinomycetales, Actinopolysporales, Bifidobacteriales, Catenulisporales, Corynebacteriales, Frankiales, Glycomycetales, Jiangellales, Kineosporiales, Micrococcales, Micromonosporales, Propionibacteriales, Pseudonocardiales, Streptomycetales, Streptosporangiales, and Incertae sedis.
Bergey’s Manual of Systematics of Archaea and Bacteria showed that phylum Actinobacteria includes 5 classes, 19 orders, 50 families, and 221 genera. However, many novel taxa continue to be discovered, so this listing is certainly unfinished. The class Actinobacteria and fundamental taxonomic ranks above the genus level were proposed exclusively on the basis of 16S rRNA gene sequence-based groups and taxon-specific 16S rRNA gene sequences. This classification represented an obvious change in the classification of Actinobacteria above the genus level as it showed that previous classifications based on the form and function did not reflect natural relationships. Actinobacteria have been assigned the rank of a phylum as the phylogenetic depth signified by the lineage resembles that of existing phyla on the basis of its branching position in 16S rRNA gene trees (Barka et al., 2016).
Basics of Actinobacteria identification
Originally, classical approaches for the identification of Actinobacteria were based on morphological observations, chemotaxonomy, and physiological criteria. Morphological observations were performed to identify an unknown strain to the genus level including identification of aerial mycelium presence, color of substrate mycelium, color of aerial mycelium, ornamentation of spores, and production of soluble pigments (Barka et al., 2016; Shirling & Gottlieb, 1966; Messaoudi et al., 2015; Amin et al., 2017a). Chemotaxonomic criteria such as the detection of diaminopimelic acid (DAP) isomers are one of the most important cell wall properties of Actinobacteria. Determination of the DAP isomers LL (Levo) form or the meso is usually sufficient for the characterization of the Actinobacteria groups (Messaoudi et al., 2015; Amin et al., 2017a; Hasegawa et al., 1983).
A wide range of physiological characteristics has been evaluated, including carbohydrate utilization profile, nitrogen source utilization profile, degradation or hydrolysis of numerous substrates, and sensitivity to various inhibitors (Shirling & Gottlieb, 1966; Messaoudi et al., 2015).
The identification of Actinobacteria via traditional methods such as phenotypic characteristics is not as accurate as genotypic methods. 16S rRNA gene sequence analysis has been recognized as a powerful tool for the identification of poorly described, rarely isolated, or phenotypically aberrant strains and can lead to a unique phylogenetic analysis of newly isolated strains (Heuer et al., 1997; Monciardini et al., 2002; Busti et al., 2006). However, it is not yet possible to complete a comprehensive suprageneric classification of Actinobacteria from the results of partial sequencing of the 16S rRNA and could not distinguish between closely related species or even genera (Colquhoun et al., 1998). An updated taxonomy of the phylum Actinobacteria based on 16S rRNA trees was recently described (Korn-Wendisch & Schneider, 1992). That bring up to date classification removed the taxonomic ranks of subclasses and suborders, moving the prior subclasses and suborders to the ranks of classes and orders, respectively (Barka et al., 2016). The phylum is consequently distributed into six separate classes including Actinobacteria, Acidimicrobiia, Coriobacteriia, Nitriliruptoria, Rubrobacteria, and Thermoleophilia. The class Actinobacteria contains 16 orders, comprising previously suggested orders, Actinomycetales and Bifidobacteriales (Hasegawa et al., 1983). The order Actinomycetales is now delimited only to the members of the family Actinomycetaceae (Barka et al., 2016). Several genera were identified by 16S rRNA gene sequence analysis such as Streptomyces, Micromonospora, Kribbella, Actinomadura, and Saccharopolyspora (Patel et al., 2004).
Definition of rare Actinobacteria
Rare Actinobacteria are defined as certain types of Actinobacteria that are difficult to isolate. Molecular tools indicate that the so-called rare Actinobacteria are relatively abundant in various habitats, and they can be retrieved in large numbers if a suitable isolation method is available. We believe that exploring rare Actinobacteria, which is difficult to isolate, will result in chemical diverse active compounds (Donadio et al., 2002). Some genera belonged to this group such as Actinomadura, Actinoplanes, Amycolatopsis, Actinokineospora, Acrocarpospora, Actinosynnema, Catenuloplanes, Cryptosporangium, Dactylosporangium, Kibdelosporangium, Kineosporia, Kutzneria, Microbiospora, Microtetraspora, Nocardia, Nonomuraea, Planomonospora, Planobispora, Pseudonocardia, Saccharomonospora, Saccharopolyspora, Saccharothrix, Streptosporangium, Spirilliplanes, Thermomonospora, Thermobifida, Virgosporangium, Micromonospora, and some uncommon species of Streptomyces (Lazzarini et al., 2000; Mazza et al., 2003).
Rare Actinobacteria habitats
Although soil is the main habitat of rare Actinobacteria, they also can be isolated from different ecological niches. The population and types of rare actinomycetes in each ecosystem are affected by various factors such as soil type, pH, humus content, and humus type (Tiwari & Gupta, 2013). An Egyptian research group selectively isolated rare Actinobacteria genera including Actinomadura, Actinoplanes, and Micromonospora from soil samples in Egypt (Abd-allah et al., 2012). Another study declared the isolation of rare Actinobacteria from shallow water sediments of the Trondheim Fjord (Norway) including Micromonospora, Actinocorallia, Actinomadura, Knoellia, Glycomyces, Nocardia, Nocardiopsis, Nonomuraea, Pseudonocardia, Rhodococcus, and Streptosporangium genera (Bredholdt et al., 2007). Biodiversity of rare Actinobacteria in water was also reported from Lake Baikal revealing isolates belong to the genus Micromonospora (Terkina et al., 2002). Additionally, rare Actinobacteria inhabit extremophile ecological niches such as caves with low temperatures, high relative humidity, low amount of organic nutrients, and high mineral concentrations. Isolates belonging to the Nocardia and Micromonospora genera were isolated from El Gola cave, Sinai, Egypt (Mansour, 2003). Moreover, Nocardia altamirensis was isolated from the Altamira Cave, Cantabria, Spain (Jurado et al., 2008). The extreme desiccation condition of hyper-arid deserts is often accompanied by high temperature, low water activity, and intense radiation (Bull, 2011). Amin et al. declared the isolation of Micromonospora and Kribbella genera from the Sinai Desert, Egypt (Tolba et al., 2013).
Isolation of rare Actinobacteria
Isolation of rare Actinobacteria by conventional dilution plate methods is difficult. They require a complicated procedure for isolation, preservation, and cultivation because they are usually masked by fast growers such as bacteria, fungi, and common streptomyces (Lazzarini et al., 2000). So, new isolation methodologies were developed focusing on physical and chemical pretreatments for isolation samples’ previous dilution plate methods. Many pretreatments are used such as dry heat, phenol treatments (Hayakawa et al., 2004), sucrose gradient centrifugation (Yamamura et al., 2003), and sodium dodecyl sulfate treatment. These treatments eliminate non-filamentous bacteria from the samples and suppress fungal growth that in turn promotes the growth of slow-growing rare Actinobacteria (Stanek et al., 2011). Appropriate selective media containing macromolecules like casein, chitin, and humic acid are important for promoting the growth of rare Actinobacteria and suppressing bacterial and fungal contaminants. It has been confirmed that the addition of antibacterial and antifungal antibiotics such as anisomycin, cycloheximide, gentamicin, kanamycin, nalidixic acid, novobiocin, nystatin, penicillin, primaricin, polymyxin, rifampicin, streptomycin, tunicamycin, and vancomycin to the isolation media promotes the selection of rare Actinobacteria (Hong et al., 2009).
Role of rare Actinobacteria in antibiotic production
In 2018, the World Health Organization declared the occurrence of antimicrobial resistance everywhere in the world as a great challenge to public health (Roca et al., 2015; Tillotson, 2018). The emergence and spread of multi-resistant pathogens became near to all known antibiotics (Yong et al., 2009), which cause the urgent need for searching for new antibiotics. The emergence of multidrug resistance among bacteria including Staphylococcus aureus, members of the ESKAPE pathogens, and latterly extreme drug-resistant Mycobacterium tuberculosis is a major worldwide public health threat (Pfaller et al., 1998; Vajs et al., 2017). It has been reported that numerous bioactive compounds were isolated from Actinobacteria and inhibited multidrug-resistant pathogens such as vancomycin-resistant Enterococci, methicillin-resistant Staphylococcus aureus, Shigella dysenteriae, Klebsiella sp., Escherichia coli, and Pseudomonas aeruginosa (Selvameenal et al., 2009; Severin et al., 2014). Nowadays, it is highly important to explore new antibiotics in order to combat multidrug-resistant pathogens. We believe that exploring the biosynthetic potential of rare Actinobacteria will reveal novel structures with useful biological activities (Koehn & Carter, 2005; Baltz, 2006; Pelaez, 2006; Bull & Stach, 2007; Dancer, 2004).
Rare Actinobacteria isolates are a rich source of antibacterial agents, anti-parasitics, antifungal agents, herbicides, pesticides, anticancer, and immunosuppressive agents and enzymes (Takahashi & Omura, 2003; Magarvey et al., 2004; Singh & Barrett, 2006; Hacene et al., 2000). Different genera of Actinobacteria produce valuable bioactive molecules such as rifamycins from Amycolatopsis mediterranei (Solanki et al., 2008), erythromycin from Saccharopolyspora erythraea (Oliynyk et al., 2007), teicoplanin from Actinoplanes teichomyceticus (Somma et al., 1984), vancomycin from Amycolatopsis orientalis (Lazzarini et al., 2000), lupinacidins from Micromonospora lupini (Igarashi et al., 2007), neorustmicin from M. carbonacea (Yun-yang et al., 2007), rifamycin S from M. rifamycinica (Huang et al., 2009), erythromycin from Actinopolyspora sp. (Huang et al., 2009), and roseoflavin from Streptomyces davawensis (Grill et al., 2008).
The antibiotics produced by Actinobacteria are divided into numerous classes based upon their structure such as aminoglycosides (e.g., streptomycin and kanamycin), ansamycins (e.g., rifampin) (Floss & Yu, 1999), anthracyclines (e.g., doxorubicin) (Kremer et al., 2001), β-lactam (cephalosporins) (Kollef, 2009), macrolides (e.g., erythromycin), and tetracycline. Antibiotics produced by rare Actinobacteria have various mechanisms of action such as inhibition of cell wall synthesis (vancomycin), cell membrane damage (polyene), inhibition of DNA and RNA synthesis (quinolones and fluoroquinolones), protein synthesis inhibition (aminoglycosides, macrolides, and tetracyclines), and inhibition of essential enzymes required for folate metabolism (trimethoprim and sulfonamide).
Factors affecting antibiotic production
There are many natural products to be discovered from rare Actinobacteria (Bérdy, 2012). Searching for valuable antibiotics begins with screening unusual rare Actinobacteria to detect the best source of novel bioactive metabolites, followed by optimization of culture conditions for maximum antimicrobial compound production, antibiotic assay, chemical characterization, and identification of antibiotic substances. The nature of antibiotics produced by Actinobacteria depends upon the species, strain, and culturing conditions such as cell density, pH, incubation period, carbon sources, and nitrogen sources. The ability of Actinobacteria cultures to form antibiotics is not a fixed property but can be greatly increased or completely lost under different conditions of nutrition and cultivation. The cell density is an important factor in attaining the highest antimicrobial yield. It has been reported in some studies that the optimum pH range for antimicrobial metabolite production by Actinobacteria is 6–7 (Hamid et al., 2015; Amin et al., 2018; Amin et al., 2017b; Pharm, 2010; Ahmad et al., 2017), depending upon each strain. The incubation period of Actinobacteria greatly affects the yield of antibiotics. It was shown that the highest level of antimicrobial agent production was recorded after 6 to 8 days of incubation (Liang et al., 2008). Carbon sources are essential components in the culture media. Several reports showed that the optimum antimicrobial agent production depends upon the type and concentration of carbon sources used in culture media such as (starch, glucose, maltose, fructose, glycerol, and molasses) (Amin et al., 2017a; Amin et al., 2017b; Pharm, 2010; Taurino et al., 2011; Wang et al., 2017; Abdelwahed et al., 2012). Also, the type and amount of nitrogen sources such as ammonium sulfate, ammonium nitrate, ammonium chloride, peptone, soya bean meal, and yeast extract greatly influence the antimicrobial production by Actinobacteria (Amin et al., 2017a; Amin et al., 2017b; Ahmad et al., 2017; Taurino et al., 2011; Wang et al., 2017; Abdelwahed et al., 2012).
Antibiotic crude substances were extracted after the optimized fermentation step. Purification of the crude substance is performed using paper or thin-layer chromatography and high-performance liquid chromatography analysis which separates the metabolites according to the retention factor value. The further physiochemical analysis is important for the identification of antimicrobial agents such as infrared analysis, mass spectroscopy (Tiwari & Gupta, 2012), and nuclear magnetic resonance spectroscopy (Bérdy, 2012).
It has been shown that the manipulation of genes that encode the enzymes involved in the biosynthetic pathways is considered as a hopeful alternative approach for redesigning antibiotic structures to create new activities and overwhelmed microbial resistance to current drugs. Studying the functional analysis of biosynthetic genes is crucial for such approaches. In addition to that, the development of genetic manipulation methodologies and heterologous hosts is more genetically agreeable for antibiotic expression of biosynthetic genes (Sánchez et al., 2002).
Polymerase chain reaction screening for genes that encode the enzymes responsible for antibiotic production and studying their phylogeny and biotechnological manipulation of these genes are valuable tools for drug discovery (Amos et al., 2015; Okami & Hotta, 1988; Walsh, 2002). Bioinformatics tools are crucial for analyzing huge genomic and proteomic data that will help in the field of drug discovery and detecting novel antibiotics (Bérdy, 2012; Amos et al., 2015).
Molecular approaches for manipulating antibiotic biosynthetic clusters
Screening of non-ribosomal peptide synthetase and polyketide synthase genes
Non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) as non-ribosomal peptides (NRPs) and polyketides (PKs) are the two major classes of secondary metabolites with diverse chemical structures and a valuable source of pharmaceutically important molecules. Recent advances in genomics and genome sequencing have shown that the potential of Actinobacteria to produce molecules of pharmacological interest has been greatly under-evaluated. Full genome sequencing showed that there are three biosynthetic pathways: peptides manufactured by the conventional ribosomal assembly, NRPS metabolites, and polyketides. Non-ribosomal peptide synthetases and polyketide synthases are a group of enzymes, coded by genes responsible for the production of important antibiotic groups. The presence of NRPS and PKS genes in an Actinobacteria strain is highly related to their biosynthetic potential. The NRPS mechanism was first described in 1971 during a research on gramicidin S and tyrocidin biosynthesis. NRPSs are modularly organized with each module responsible for the incorporation of a specific amino acid. The modules consist of at least three core domains catalyzing a specific reaction in the incorporation of a monomer (Lipmann et al., 1971). Firstly, the adenylation (A) domain selects the cognate amino acid which it activates by transforming it into an aminoacyl adenylate. The thiolation or peptidyl carrier protein (PCP) domain covalently binds the activated monomer to the synthetase by a phosphopantetheinyl arm. The condensation (C) domain catalysis the formation of a peptide bond between the amino acids linked to two adjacent modules. (A) dedicated loading module, carrying only the (A), and PCP domains is the first module of the NRPS, whereas a termination module containing a thioesterase (TE) domain, which releases the peptide from the synthetase, concludes the assembly line (Lipmann et al., 1971).
Many NRPSs feature secondary specialized domains within modules that allow residue modifications. Epimerization (E) domains lead to D-isomer forms of amino acids; methylation (M), oxidation (Ox), reduction (R), formylation (F), and heterocyclization (Cy) domains enable NRPSs to biosynthesize an impressive number of diversified peptides with broad biological activities that cannot be produced by the classical ribosomal machinery (Felnagle et al., 2008). PKS consists of modules of at least three core domains: an acyltransferase (AT) domain which selects the suitable extender unit and transmits it to the acyl carrier protein (ACP) domain, wherever a thioester bond is made fixing the growing polyketide to the synthase, and a ketosynthase (KS) domain. The KS domain is responsible for the condensation between the extender unit present on the acyl carrier protein (ACP) domain of the same module and the polyketide intermediate bound to the (ACP) domain of the preceding module. Additional secondary domains such as ketoreductase (KR), oxidation (Ox), dehydratase (DH), methyltransferase (MT), enoylreductase (ER), and methylation (M) domains modify the growing polyketide molecule. Type II PKSs often feature a cyclase (Cy) domain leading to the formation of aromatic structures. The last module possesses a thioesterase (TE) domain catalyzing the release of the final product from the enzyme (Meurer et al., 1997; Moore & Hertweck, 2002).
Important antibiotics are produced by NRPS gene clusters such as amphomycin produced by Streptomyces canus (Yang et al., 2014), cephamycin from Streptomyces clavuligerus (Alexander & Jensen, 1998), daptomycin from Streptomyces roseosporus (Miao et al., 2006), and teicoplanin from Actinoplanes teichomyceticus (Somma et al., 1984). Antibiotics produced by PKS gene clusters are rifamycin from Amycolatopsis mediterranei (Stratmann et al., 1999), tetracycline from Streptomyces rimosus (Petković et al., 2006), and actinorhodin from Streptomyces coelicolor. Some antibiotics are hybrid from both NRPS and PKS gene clusters such as pristinamycin IIA from Streptomyces pristinaespiralis (Voelker & Altaba, 2001) and virginiamycin from Streptomyces virginiae (Pulsawat et al., 2007).
Several PCR assays using diverse primers were used to target PKS and NRPS genes, which is useful in selecting potent strains with a diverse biosynthetic potential due to different associations of modules (Amos et al., 2015). Recently, a research group studied the biosynthetic capability of Micromonospora sp. Rc5 isolated from Egyptian soils via NRPS and PKS PCR assays. They used eight pairs of primers demonstrating low similarity PKS gene clusters in Micromonospora sp. Rc5 compared to related PKS sequences to a database. The results revealed that these distinct clusters would probably be responsible for the production of different bioactive molecules (Amin et al., 2017c). Another study explored European soil sample biosynthetic potential using newly designed primers in NRPS and PKS PCR assays. The results revealed a surprising number of phylogenetically divergent NRPS and PKS sequences to some rare Actinobacteria such as Actinospica, Catenulispora, and Nonomuraea. They also suggested that these NRPS and PKS sequences may encode for novel bioactive compounds (Amos et al., 2015). They assumed that NRPS and PKS PCR assay limitations are the specific primer sequence designs in underexplored taxa and a great sequencing effort needed to discover all the gene clusters in these soils (Amos et al., 2015).
Actinobacteria genomic libraries
A genomic library is a collection of the total genomic DNA from a single organism and digested with a restriction enzyme to cut the DNA into fragments of a specific size. The fragments are then inserted into the vector. Genomic libraries are commonly used for sequencing applications. Fosmid vectors are cosmids which are a type of hybrid plasmid that contains a lambda phage cos sequence, which use the F-plasmid origin of replication and partitioning mechanisms to allow cloning of large DNA fragments. Fosmids can hold DNA inserts of up to 40 kb in size. Fosmids contain several main functional elements such as OriV (origin of replication): the sequence starting with which the plasmid DNA will be replicated in the recipient cell; tra-region (transfer genes): genes coding the F-pilus and DNA transfer process, antibiotic resistance genes as a selectable marker and lambda phage cos site sequence for packaging insert DNA integrated within fosmid into phages (Hall, 2004).
A fosmid library is prepared by extracting the genomic DNA from the target organism. DNA must be sheared into fragments of approximately 35 kb in size and cloned into the fosmid vector (Hall, 2004). The ligation mix is then packaged into phage particles, and the DNA is transfected into the bacterial host such as Escherichia coli and bacterial cells were grown on Luria-Bertani media containing an antibiotic marker. Bacterial clones with a fosmid library can only grow on the media due to antibiotic resistance genes carried on the fosmid. Fosmids may be convenient to construct libraries from complex genomes. Fosmids have high structural stability and have been found to maintain human DNA effectively even after 100 generations of bacterial growth (Shizuya et al., 1992).
Fosmid libraries have been constructed for a variety of organisms, including bacteria (Fitz-Gibbon et al., 1997), fungi (Magrini et al., 2004), plants (Meyer et al., 2008), animals (Zhang et al., 2007), and humans. Fosmid libraries can be successfully used to capture and express many functional genes such as those associated with antibiotic resistance (Udikovic-Kolic et al., 2014; Amos et al., 2014). In addition to that, fosmid libraries are used to evaluate the diversity of biosynthetic gene clusters and the discovery of several new bioactive compounds (Feng et al., 2011; Kallifidas et al., 2012). Similar studies showed the construction of Actinobacteria fosmid libraries with distinct clones with PKS genes (Parsley et al., 2011). Moreover, it was reported the screening of distinct NRPS and PKS genes in fosmid libraries from environmental DNA from soil (Amos et al., 2015).
It was demonstrated that only 40% of the genes from the genomes of 32 prokaryotes could be detected when expressed in E. coli (Gabor et al., 2004). The study also revealed significant differences in the predicted expression modes between distinct taxonomic groups of organisms. Another study showed that E. coli, Pseudomonas putida, and Streptomyces lividans differed in their abilities to express heterologous gene clusters (Martinez et al., 2004). Our previous work on the biosynthetic NRPS and PKS genes using a fosmid library indicated that it was a successful way to capture the genes. However, no gene expression was recorded. We concluded that the biosynthetic gene cluster integration in the fosmids was incomplete. We recommend the use of bacterial vectors with large DNA intake capacity for complete gene expression.
Whole-genome sequencing
Metagenomics is a novel strategy for the identification of bacteria diversity, and the next-generation DNA sequencing technologies increase scientific interests in understanding the microbial diversity inhabiting different environments. Several new methods for DNA sequencing were developed in the mid to late 1990s and were implemented in commercial DNA sequencers by the year 2000. These were called the “next-generation” sequencing methods such as reversible dye terminators (Illumina sequencing) (Bentley et al., 2008), massively parallel signature sequencing (Brenner et al., 2000), 454 pyrosequencing (Margulies et al., 2005), polony sequencing (Shendure et al., 2005), sequencing by oligonucleotide ligation detection (Mardis, 2008), ion torrent sequencing by synthesis (Rusk, 2010), single-molecule real-time sequencing by synthesis (Eid et al., 2009), and DNA nanoball sequencing (Drmanac et al., 2010). An important application of next-generation sequencing is whole-genome sequencing. Whole-genome sequencing is a powerful tool for genomics research and the most comprehensive method for analyzing the genome such as determining the sequence of individual genes, clusters of genes or operons, full chromosomes, or entire genomes of any organism. It reduces sequencing costs and produces large volumes of data (Bentley, 2006). Whole-genome sequencing is commonly associated with sequencing human genomes, livestock (Eck et al., 2009), plants (Goff et al., 2002), and microbes (Qiao et al., 2012). Whole-genome sequencing provides a high-resolution, base-by-base view of the genome; identifies potential causative variants for further follow-on studies of gene expression and regulation mechanism; and delivers large volumes of data in a short amount of time to support the assembly of novel genomes.
Illumina genome sequencing
Illumina (Bennett, 2004) is an American company founded by Shankar Balasubramanian and David Klenerman in 1998 and developed a sequencing method based on reversible dye terminator technology and engineered polymerases. Illumina gains the massively parallel sequencing technology invented in 1997 by Pascal Mayer and Laurent Farinelli, which is now implemented in Illumina’s Hi-Seq genome sequencers.
High-throughput Hi-Seq genome sequencers are used for whole-genome sequencing of numerous microbes and plant (Li et al., 2012), human, and animal genomes (Eck et al., 2009). The company provides a line of products and services that serve the sequencing, genotyping, and gene expression markets. Its tools allow the researchers to make genetic tests and provide medical information based on genomics and proteomics. Illumina genome sequencing technologies allow researchers to sequence DNA and RNA much more quickly to obtain the sequence of multiple strands at once and cheaply than the previously used Sanger sequencing; thus, it revolutionized the study of genomics and molecular biology (Pettersson et al., 2009).
In Illumina sequencing (Bentley, 2006; Bennett, 2004; Bennett et al., 2005), the process started with the fragmentation of purified DNA into 100–150-bp reads by enzymatic digestion or temperature. The small DNA fragments are linked to adapters which are a kind of molecular modifications and act as reference points during amplification, sequencing, and analysis. In this process, one end of a single DNA molecule is attached to a flow cell surface (it had a complementary sequence with adapters at certain regions). DNA fragments subsequently bend over and hybridize to complementary adapters creating a “bridge,” thereby forming the template for the synthesis of their complementary strands by DNA polymerases.
After the amplification step, a flow cell with more than 40 million clusters is produced, wherein each cluster is composed of approximately 1000 clonal copies of a single template molecule. The templates are sequenced in a massively parallel fashion using a DNA sequencing-by-synthesis approach that employs reversible terminators with removable fluorescent moieties and special DNA polymerases that can incorporate these terminators into growing oligonucleotide chains. The terminators are labeled with fluorescence of 4 different colors to distinguish among the different bases at the given sequence position, and a computer determines each base was added by the wavelength of the fluorescent tag and records every spot on the chip (Bentley, 2006; Bennett, 2004; Bennett et al., 2005).
Bioinformatics analysis and genome assembly
The production of raw sequence data is only the beginning of its detailed bioinformatics analysis (Severin et al., 2014). Many new methods for sequencing and correcting sequencing errors were developed.
Occasionally, raw reads provided by the sequencer are accurate and precise only in a part of their length. The use of the entire read may lead to artifacts in the downstream analyses such as genome assembly, single nucleotide polymorphism calling, or gene expression assessment. Two classes of trimming programs have been introduced, based on the window-based or the running-sum classes of algorithms (Del Fabbro et al., 2013). Trimming programs such as Trimmomatic to trim raw reads and remove adapters was widely used (Bolger et al., 2014). Samtools (Li et al., 2009) and bwa-mem software (Li & Durbin, 2009) were used for qualitative filtering the reads and assembling the genome.
Genome assembly refers to the process of taking a large number of short DNA sequences and putting them back together to create a representation of the original chromosomes from which the DNA originated. Automated sequencing machines produce millions of small DNA fragments “read” which can read up to 1000 nucleotides. A genome assembly algorithm works by taking all the pieces and aligning them to one another and detecting overlapping regions of the reads. These overlapping reads can be merged and the process continues (Paszkiewicz & Studholme, 2010).
Genome assembly is a very difficult computational problem, made more difficult because many genomes contain large numbers of identical sequences, known as repeats. These repeats may be thousands of nucleotides long, and some occur in dissimilar positions, particularly in the large genomes of plants and animals. The resulting genome is called a draft genome sequence; it is produced by combining the information sequenced contigs together in the correct orientation and order and then link them to create scaffolds (Paszkiewicz & Studholme, 2010).
Scaffolds are larger DNA fragments positioned along the physical map of the chromosome. Also, contigues obtained from the sequencing genomes can be assembled on the basis of the most similar reference genome on the database to fill the gaps (Darling et al., 2011). Several tools are used in this process such as mauve Aligner 2.4. software (Rissman et al., 2009) and CAP3 assembly program for contigue assembly (Huang & Madan, 1999).
Genome annotation
Genome annotation is the process of identifying the elements of the genome and attaching biological information to these elements (Stein, 2001). After genome assembly is performed, gene annotation is required to determine the structural and functional identity of those genes (Kisand & Lettieri, 2013). Automatic annotation tools try to perform all this by computer analysis, rather than manual annotation which involves human expertise. The popular online automated annotation systems are Rapid Annotation using Subsystem Technology (RAST) which is an automated service for annotating complete or approximately complete bacterial genomes. It also provides high-quality genome annotations for these genomes in phylogenetic tree analysis (Aziz et al., 2008), IMG (Integrated Microbial Genomes based on BLAST p) system which serves as a community resource for comparative analysis of publicly accessible genomes in a wide-ranging integrated context. IMG contains both draft and complete microbial genomes (Markowitz et al., 2009) and Prokaryotic Genomes Automatic Annotation Pipeline (PGAAP) developed at the National Center for Biotechnology Information (NCBI) that is based on gene prediction algorithms with homology-based methods. PGAAP annotates both complete genomes and draft genomes encompassing multiple contigs (Tatusova et al., 2016).
In 2018, a research group declared the annotation of two rare Actinobacteria genomes derived from Illumina whole-genome sequencing using mainly PGAAP system. They assume that this annotation opens the door for highlighting significant biosynthetic gene clusters in rare Actinobacteria (Amin et al., 2019).
Identification of biosynthetic gene clusters by antiSMASH
The genes encoding the biosynthetic pathways that are responsible for the production of secondary metabolites are usually clustered together on the chromosome in biosynthetic gene clusters. Recently, genome mining of such biosynthetic gene clusters (BGCs) is considered as an important method to identify new molecules, leading to the detection of lots of novel compounds. A variety of computational tools have been developed to analyze specific classes of secondary metabolites (Weber et al., 2015).
In 2011, a tool called the antibiotics and Secondary Metabolite Analysis SHell (antiSMASH) was introduced as a Web server for genomic identification and analysis of BGCs of any type, thus facilitating rapid genome annotation of a wide range of bacterial and fungal strains (Blin et al., 2013). Although antiSMASH is capable of annotating extensive chemical structures of secondary metabolites, it is still limited to annotate peptides and polyketides coded by modular assembly lines only. Annotation of chemical compounds coded by cyclization and tailoring reactions is still limited. A multiple possible end product compound strategy can be applied to overcome this limitation. This is important to prevent the replication of existing compounds for effective drug discovery and comparative analysis of unknown and known gene clusters (Weber et al., 2015).
Bioinformatics analysis using antiSMASH 3.0 predicts secondary metabolite gene clusters in rare Actinobacteria. Amin et al. mined the whole genomic sequence of Micromonospora sp. Rc5 isolated from the Egyptian desert using antiSMASH server. This study demonstrated out reads of 33 potential secondary metabolite gene clusters including PKS, NRPS, hybrid polyketide synthases, terpenes, lantipeptides, saccharides, siderophore, bacteriocin, arylpolyene, and unidentified clusters (Amin et al., 2019). Another study reporting the annotation of the draft genome sequence of Micromonospora sp. DSW705 using antiSMASH analysis predicts 3 PKS gene clusters, 1 NRPS gene clusters, and 3 hybrid PKS/NRPS gene clusters responsible for antitumor rakicidin synthesis (Komaki et al., 2016).
Conclusion
Rare Actinobacteria is a great potential source of antibiotic production against multidrug-resistant pathogens. Conventional and molecular identification of rare Actinobacteria is a valuable tool. However, further investigations including DNA-DNA hybridization and additional chemotaxonomic and biochemical tests are required to identify their species level. Molecular approaches for the identification of biosynthetic gene cluster are useful in detecting the antimicrobial potential of rare Actinobacteria with uncommon biochemical pathways. This will help in the development of novel bioactive metabolites. The current review provides information that helps to control antimicrobial drug resistance problems and will enhance health care in Egypt and worldwide. In addition, it introduces promising methodologies to support the research of drug discovery in Egypt.
Availability of data and materials
Not applicable.
Abbreviations
- antiSMASH:
-
Antibiotics and Secondary Metabolite Analysis SHell
- BGCs:
-
Biosynthetic gene clusters
- DAP:
-
Diaminopimelic acid
- NRPS:
-
Non-ribosomal peptide synthetase
- PKS:
-
Polyketide synthase
References
Abd-allah N, Tolba S, Hatem D (2012) Selective isolation of rare actinomycetes from different types of Egyptian soil. Egypt J Exp Biol 8(2):175–182
Abdelwahed N, Abdallah NA, El-Ghawas DE, El-Din SMB, El-Diwany AL (2012) Isolation, identification and optimization of antimicrobial metabolites produced by soil derived actinomycetes. Egypt J Exp Biol (Bot). 8(2):205–217
Ahmad MS, El-Gendy AO, Ahmed RR, Hassan HM, El-Kabbany HM, Merdash AG. Exploring the antimicrobial and antitumor potentials of Streptomyces sp. AGM12-1 isolated from Egyptian soil. Frontiers in Microbiology. 2017 2017-March-13;8(438). English.
Alexander DC, Jensen SE (1998) Investigation of the Streptomyces clavuligerus cephamycin C gene cluster and its regulation by the CcaR protein. J Bacteriol 180(16):4068–4079
Amin DH, Abolmaaty A, Borsetto C, Tolba S, Abdallah NA, Wellington EMH (2019) In silico genomic mining reveals unexplored bioactive potential of rare actinobacteria isolated from Egyptian soil. Bull Nat Res Cent 43(1):78
Amin DH, Abolmaaty A, Tolba S, Abdallah NA, Wellington EM (2017a) Phylogenic characteristics of a unique antagonistic Micromonospora sp. Rc5 to S. aureus isolated from Sinai Desert of Egypt. Curr Res Microbiol Biotechnol 5(6):1295–1306
Amin DH, Abolmaaty A, Tolba S, Abdallah NA, Wellington EM (2018) Phylogenic characteristics of a unique antagonistic Micromonospora sp. Rc5 to S. aureus isolated from Sinai Desert of Egypt. Ann Res Rev Biol 9:1–15
Amin DH, Borsetto C, Tolba S, Abolmaaty A, Abdallah NA, Wellington EM. Phylogenic Analysis of NRPS and PKS Genes associated with antagonistic Micromonospora Rc5 and Streptomyces Ru87 isolates. J Adv Biol Biotechnol 2017c:1-22.
Amin DH, Tolba S, Abolmaaty A, Abdallah NA, Wellington EM (2017b) Phylogenetic and antimicrobial characteristics of a novel Streptomyces sp. Ru87 isolated from Egyptian soil. Int J Curr Microbiol App Sci. 6(8):2524–2541
Amos GC, Borsetto C, Laskaris P, Krsek M, Berry AE, Newsham KK et al (2015) Designing and implementing an assay for the detection of rare and divergent NRPS and PKS clones in European, Antarctic Cuban soils. PloS One 10(9):e0138327
Amos GC, Zhang L, Hawkey P, Gaze WH, Wellington E (2014) Functional metagenomic analysis reveals rivers are a reservoir for diverse antibiotic resistance genes. Vet Microbiol 171(3):441–447
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA et al (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9(1):75
Baltz RH (2006) Molecular engineering approaches to peptide, polyketide and other antibiotics. Nat Biotechnol 24(12):1533–1540
Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Klenk H-P et al (2016) Taxonomy, physiology, and natural products of Actinobacteria. Microbiol Mol Biol Rev 80(1):1–43
Bennett S (2004) Solexa ltd. Pharmacogenomics. 5(4):433–438
Bennett ST, Barnes C, Cox A, Davies L, Brown C (2005) Toward the $1000 human genome. Pharmacogenomics 6(4):373–382
Bentley DR (2006) Whole-genome re-sequencing. Curr Opin Genet Dev 16(6):545–552
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG et al (2008) Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456(7218):53–59
Bérdy J (2012) Thoughts and facts about antibiotics: where we are now and where we are heading. J Antibiot 65(8):385–395
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E et al (2013) antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41(W1):W204–WW12
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Bredholdt H, Galatenko OA, Engelhardt K, Fjærvik E, Terekhova LP, Zotchev SB (2007) Rare actinomycete bacteria from the shallow water sediments of the Trondheim Fjord, Norway: isolation, diversity and biological activity. Environ Microbiol 9(11):2756–2764
Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH, Johnson D et al (2000) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature biotechnology. 18(6):630
Bull A (2011) Extremophiles handbook
Bull AT, Stach JE (2007) Marine actinobacteria: new opportunities for natural product search and discovery. Trends Microbiol 15(11):491–499
Busarakam K, Bull AT, Trujillo ME, Riesco R, Sangal V, van Wezel GP et al (2016) Modestobacter caceresii sp. nov., novel actinobacteria with an insight into their adaptive mechanisms for survival in extreme hyper-arid Atacama Desert soils. Syst Appl Microbiol 39(4):243–251
Busti E, Monciardini P, Cavaletti L, Bamonte R, Lazzarini A, Sosio M et al (2006) Antibiotic-producing ability by representatives of a newly discovered lineage of actinomycetes. Microbiol 152(3):675–683
Colquhoun JA, Mexson J, Goodfellow M, Ward AC, Horikoshi K, Bull AT (1998) Novel rhodococci and other mycolate actinomycetes from the deep sea. Antonie van Leeuwenhoek 74(1-3):27–40
Dancer SJ (2004) How antibiotics can make us sick: the less obvious adverse effects of antimicrobial chemotherapy. Lancet Infect Dis 4(10):611–619
Darling AE, Tritt A, Eisen JA, Facciotti MT (2011) Mauve assembly metrics. Bioinformatics 27(19):2756–2757
Das A, Khosla C (2009) Biosynthesis of aromatic polyketides in bacteria. Acc Chem Res 42(5):631–639
Davenport RJ, Curtis TP, Goodfellow M, Stainsby FM, Bingley M (2000) Quantitative use of fluorescent in situ hybridization to examine relationships between mycolic acid-containing actinomycetes and foaming in activated sludge plants. Appl Environ Microbiol 66(3):1158–1166
Del Fabbro C, Scalabrin S, Morgante M, Giorgi FM (2013) An extensive evaluation of read trimming effects on Illumina NGS data analysis. PloS One 8(12):e85024
Donadio S, Carrano L, Brandi L, Serina S, Soffientini A, Raimondi E et al (2002) Targets and assays for discovering novel antibacterial agents. J Biotechnol 99(3):175–185
Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG et al (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327(5961):78–81
Eck SH, Benet-Pagès A, Flisikowski K, Meitinger T, Fries R, Strom TM (2009) Whole genome sequencing of a single Bos taurus animal for single nucleotide polymorphism discovery. Genome Biol 10(8):R82
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G et al (2009) Real-time DNA sequencing from single polymerase molecules. Science 323(5910):133–138
Elbendary AA, Hessain AM, El-Hariri MD, Seida AA, Moussa IM, Mubarak AS et al (2018) Isolation of antimicrobial producing actinobacteria from soil samples. Saudi J Biol Sci 25(1):44–46
Ertaş HB, Kiliç A, Özbey G, Muz A (2005) Isolation of Arcanobacterium (Actinomyces) pyogenes from abscessed cattle kidney and identification by PCR. Turk J Vet Anim Sci 29(2):455–459
Felnagle EA, Jackson EE, Chan YA, Podevels AM, Berti AD, McMahon MD et al (2008) Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol Pharm 5(2):191–211
Feng Z, Kallifidas D, Brady SF (2011) Functional analysis of environmental DNA-derived type II polyketide synthases reveals structurally diverse secondary metabolites. Proc Nat Acad Sci 108(31):12629–12634
Fitz-Gibbon S, Choi AJ, Miller JH, Stetter KO, Simon MI, Swanson R et al (1997) A fosmid-based genomic map and identification of 474 genes of the hyperthermophilic archaeonPyrobaculum aerophilum. Extremophiles 1(1):36–52
Floss HG, Yu T-W (1999) Lessons from the rifamycin biosynthetic gene cluster. Curr Opin Chem Biol 3(5):592–597
Gabor EM, Alkema WB, Janssen DB (2004) Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol 6(9):879–886
Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296(5565):92–100
Grill S, Busenbender S, Pfeiffer M, Köhler U, Mack M (2008) The bifunctional flavokinase/flavin adenine dinucleotide synthetase from Streptomyces davawensis produces inactive flavin cofactors and is not involved in resistance to the antibiotic roseoflavin. J Bacteriol 190(5):1546–1553
Hacene H, Daoudi-Hamdad F, Bhatnagar T, Baratti J, Lefebvre G (2000) H107, a new aminoglycoside anti-Pseudomonas antibiotic produced by a new strain of Spirillospora. Microbios 102(402):69–77
Hall BG (2004) Predicting the evolution of antibiotic resistance genes. Nature Reviews Microbiology. 2(5):430–435
Hamid AA, Ariffin S, Mohamad SAS (2015) Identification and optimal growth conditions of Actinomycetes isolated from mangrove environment. Malaysian J Anal Sci 19(4):904–910
Hasegawa T, Takizawa M, Tanida S (1983) A rapid analysis for chemical grouping of aerobic actinomycetes. J Gen Appl Microbiol 29(4):319–322
Hayakawa M, Yoshida Y, Iimura Y (2004) Selective isolation of bioactive soil actinomycetes belonging to the Streptomyces violaceusniger phenotypic cluster. J Appl Microbiol 96(5):973–981
Heuer H, Krsek M, Baker P, Smalla K, Wellington E (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63(8):3233–3241
Hong K, Gao A-H, Xie Q-Y, Gao HG, Zhuang L, Lin H-P et al (2009) Actinomycetes for marine drug discovery isolated from mangrove soils and plants in China. Marine Drugs 7(1):24–44
Huang H, Wu X, Yi S, Zhou Z, Zhu J, Fang Z et al (2009) Rifamycin S and its geometric isomer produced by a newly found actinomycete, Micromonospora rifamycinica. Antonie van Leeuwenhoek 95(2):143
Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res 1999;9(9):868-877.
Igarashi Y, Trujillo ME, Martínez-Molina E, Yanase S, Miyanaga S, Obata T et al (2007) Antitumor anthraquinones from an endophytic actinomycete Micromonospora lupini sp. nov. Bioorganic Medicinal Chem Lett 17(13):3702–3705
Jurado V, Boiron P, Kroppenstedt RM, Laurent F, Couble A, Laiz L et al (2008) Nocardia altamirensis sp. nov., isolated from Altamira cave, Cantabria, Spain. Int J Syst Evol Microbiol 58(9):2210–2214
Kallifidas D, Kang H-S, Brady SF (2012) Tetarimycin A, an MRSA-active antibiotic identified through induced expression of environmental DNA gene clusters. J Am Chem Soc 134(48):19552–19555
Kisand V, Lettieri T (2013) Genome sequencing of bacteria: sequencing, de novo assembly and rapid analysis using open source tools. BMC Genomics 14(1):211
Koehn FE, Carter GT (2005) The evolving role of natural products in drug discovery. Nat Rev Drug Discov 4(3):206–220
Kollef MH (2009) New Antimicrobial agents for methicillin-resistant ‘Staphylococcus aureus’. Crit Care Resusc 11(4):282
Komaki H, Ichikawa N, Hosoyama A, Hamada M, Harunari E, Ishikawa A et al (2016) Draft genome sequence of Micromonospora sp. DSW705 and distribution of biosynthetic gene clusters for depsipeptides bearing 4-amino-2, 4-pentadienoate in actinomycetes. Stand Genomic Sci 11(1):84
Könönen E, Wade WG (2015) Actinomyces and related organisms in human infections. Clin Microbiol Rev 28(2):419–442
Korn-Wendisch F, Schneider J (1992) Phage typing—a useful tool in actinomycete systematics. Gene 115(1):243–247
Kremer L, Van Dalen E, Offringa M, Ottenkamp J, Voute P (2001) Anthracycline-induced clinical heart failure in a cohort of 607 children: long-term follow-up study. J Clin Oncol 19(1):191–196
Lazzarini A, Cavaletti L, Toppo G, Marinelli F (2000) Rare genera of actinomycetes as potential producers of new antibiotics. Antonie van Leeuwenhoek 78(3):399–405
Lerat S (2009) simao-Beaunoir AM, Beaulieu C. Genetic and physiological determinants of Streptomyces scabies pathogenicity. Mol Plant Pathol 10(5):579–585
Li D, Deng Z, Qin B, Liu X, Men Z (2012) De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genomics 13(1):192
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079
Liang J, Xu Z, Liu T, Lin J, Cen P (2008) Effects of cultivation conditions on the production of natamycin with Streptomyces gilvosporeus LK-196. Enzyme Microbial Technol 42(2):145–150
Lipmann F, Gevers W, Kleinkauf H, Roskoski R (1971) Polypeptide synthesis on protein templates: the enzymatic synthesis of gramicidin S and tyrocidine. Adv Enzymol Relat Areas Mol Biol 35:1–34
Ma G, Xia Z, Wu S (2009) Inhibiting effect of seven marine actinomycete strains against vegetable pathogenic microorganisms. Crops 5:3–9
Magarvey NA, Keller JM, Bernan V, Dworkin M, Sherman DH (2004) Isolation and characterization of novel marine-derived actinomycete taxa rich in bioactive metabolites. Appl Environ Microbiol 70(12):7520–7529
Magrini V, Warren WC, Wallis J, Goldman WE, Xu J, Mardis ER et al (2004) Fosmid-based physical mapping of the Histoplasma capsulatum genome. Genome Res 14(8):1603–1609
Mansour SR (2003) The occurrence and distribution of soil actinomycetes in Saint Catherine area, South Sinai, Egypt. Pak Biol Sci 6(7):721–728
Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genetics 24(3):133–141
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437(7057):376
Markowitz VM, Mavromatis K, Ivanova NN, Chen I-MA, Chu K, Kyrpides NC (2009) IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics 25(17):2271–2278
Martinez A, Kolvek SJ, Yip CLT, Hopke J, Brown KA, MacNeil IA et al (2004) Genetically modified bacterial strains and novel bacterial artificial chromosome shuttle vectors for constructing environmental libraries and detecting heterologous natural products in multiple expression hosts. Appl Environ Microbiol 70(4):2452–2463
Mazza P, Monciardini P, Cavaletti L, Sosio M, Donadio S (2003) Diversity of Actinoplanes and related genera isolated from an Italian soil. Microbial Ecol 45(4):362–372
Messaoudi O, Bendahou M, Benamar I, Abdelwouhid D-E (2015) Identification and preliminary characterization of non-polyene antibiotics secreted by new strain of actinomycete isolated from sebkha of Kenadsa, Algeria. Asian Pacific J Trop Biomed 5(6):438–445
Meurer G, Gerlitz M, Wendt-Pienkowski E, Vining LC, Rohr J, Hutchinson CR (1997) Iterative type II polyketide synthases, cyclases and ketoreductases exhibit context-dependent behavior in the biosynthesis of linear and angular decapolyketides. Chem Biol 4(6):433–443
Meyer J, Deleu W, Garcia-Mas J, Havey M (2008) Construction of a fosmid library of cucumber (Cucumis sativus) and comparative analyses of the eIF4E and eIF (iso) 4E regions from cucumber and melon (Cucumis melo). Mol Gen Genomics 279(5):473–480
Miao V, Coëffet-Le Gal M-F, Nguyen K, Brian P, Penn J, Whiting A et al (2006) Genetic engineering in Streptomyces roseosporus to produce hybrid lipopeptide antibiotics. Chem Biol 13(3):269–276
Monciardini P, Sosio M, Cavaletti L, Chiocchini C, Donadio S (2002) New PCR primers for the selective amplification of 16S rDNA from different groups of actinomycetes. FEMS Microbiol Ecol 42(3):419–429
Moore BS, Hertweck C (2002) Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat Prod Rep 19(1):70–99
Okami Y, Hotta K. editors, Goodfellow M., Williams ST and Mordarski M. Academic Press Inc, New York; 1988.
Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S et al (2007) Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol 25(4):447
Parsley LC, Linneman J, Goode AM, Becklund K, George I, Goodman RM et al (2011) Polyketide synthase pathways identified from a metagenomic library are derived from soil Acidobacteria. FEMS Microbiol Ecol 78(1):176–187
Paszkiewicz K, Studholme DJ (2010) De novo assembly of short sequence reads. Brief Bioinformatics 11(5):457–472
Patel JB, Wallace RJ, Brown-Elliott BA, Taylor T, Imperatrice C, Leonard DG et al (2004) Sequence-based identification of aerobic actinomycetes. J Clin Microbiol 42(6):2530–2540
Pathom-Aree W, Stach JE, Ward AC, Horikoshi K, Bull AT, Goodfellow M (2006) Diversity of actinomycetes isolated from Challenger Deep sediment (10,898 m) from the Mariana Trench. Extremophiles 10(3):181–189
Pelaez F (2006) The historical delivery of antibiotics from microbial natural products—can history repeat? Biochem Pharmacol 71(7):981–990
Petković H, Cullum J, Hranueli D, Hunter IS, Perić-Concha N, Pigac J et al (2006) Genetics of Streptomyces rimosus, the oxytetracycline producer. Microbiol Mol Biol Rev 70(3):704–728
Pettersson E, Lundeberg J, Ahmadian A (2009) Generations of sequencing technologies. Genomics 93(2):105–111
Pfaller MA, Jones RN, Doern GV, Kugler K, Group SP (1998) Bacterial pathogens isolated from patients with bloodstream infection: frequencies of occurrence and antimicrobial susceptibility patterns from the SENTRY antimicrobial surveillance program (United States and Canada, 1997). Antimicrob Agents Chemother 42(7):1762–1770
Pharm M (2010) Thesis submitted in partial fulfillment for the award of degree of: Vinayaka Missions University
Pulsawat N, Kitani S, Nihira T (2007) Characterization of biosynthetic gene cluster for the production of virginiamycin M, a streptogramin type A antibiotic, in Streptomyces virginiae. Gene 393(1):31–42
Qiao J, Chen L, Li Y, Wang J, Zhang W, Chen S (2012) Whole-genome sequence of Nocardiopsis alba strain ATCC BAA-2165, associated with honeybees. J Bacteriol 194(22):6358–6359
Rego A, Raio F, Martins TP, Ribeiro H, Sousa AG, Séneca J et al (2019) Actinobacteria and Cyanobacteria diversity in terrestrial Antarctic microenvironments evaluated by culture-dependent and independent methods. Front Microbiol 10
Rissman AI, Mau B, Biehl BS, Darling AE, Glasner JD, Perna NT (2009) Reordering contigs of draft genomes using the Mauve aligner. Bioinformatics 25(16):2071–2073
Roca I, Akova M, Baquero F, Carlet J, Cavaleri M, Coenen S et al (2015) The global threat of antimicrobial resistance: science for intervention. New Microbes New Infect 6:22–29
Rusk N (2010) Torrents of sequence. Nat Methods 8(1):44
Sánchez M, Prim N, Rández Gil F, Pastor F, Diaz P (2002) Engineering of baker’s yeasts, E. coli and Bacillus hosts for the production of Bacillus subtilis lipase A. Biotechnol Bioeng 78(3):339–345
Selvameenal L, Radhakrishnan M, Balagurunathan R (2009) Antibiotic pigment from desert soil actinomycetes; biological activity, purification and chemical screening. Indian J Pharm Sci 71(5):499
Severin J, Lizio M, Harshbarger J, Kawaji H, Daub CO, Hayashizaki Y et al (2014) Interactive visualization and analysis of large-scale sequencing datasets using ZENBU. Nat Biotechnol 32(3):217–219
Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM et al (2005) Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309(5741):1728–1732
Shirling E, Gottlieb D (1966) Methods for characterization of Streptomyces species1. Int J Syst Evol Microbiol 16(3):313–340
Shizuya H, Birren B, Kim U-J, Mancino V, Slepak T, Tachiiri Y et al (1992) Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc Nat Acad Sci 89(18):8794–8797
Singh SB, Barrett JF (2006) Empirical antibacterial drug discovery—foundation in natural products. Biochem Pharmacol 71(7):1006–1015
Solanki R, Khanna M, Lal R (2008) Bioactive compounds from marine actinomycetes. Indian J Microbiol 48(4):410–431
Somma S, Gastaldo L, Corti A (1984) Teicoplanin, a new antibiotic from Actinoplanes teichomyceticus nov. sp. Antimicrobial Agents Chemother 26(6):917–923
Stanek RJ, Maher MB, Norton NB, Mufson MA (2011) Emergence of a unique penicillin-resistant Streptococcus pneumoniae serogroup 35 Strain. J Clin Microbiol 49(1):400–404
Stein L (2001) Genome annotation: from sequence to biology. Nature Rev Genet 2(7):493–503
Stratmann A, Toupet C, Schilling W, Traber R, Oberer L, Schupp T (1999) Intermediates of rifamycin polyketide synthase produced by an Amycolatopsis mediterranei mutant with inactivated rifF gene. Microbiol 145(12):3365–3375
Takahashi Y, Omura S (2003) Isolation of new actinomycete strains for the screening of new bioactive compounds. J Gen Appl Microbiol 49(3):141–154
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L et al (2016) NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44(14):6614–6624
Taurino C, Frattini L, Marcone GL, Gastaldo L, Marinelli F (2011) Actinoplanes teichomyceticus ATCC 31121 as a cell factory for producing teicoplanin. Microbial Cell Factor 10(1):82
Terkina I, Drukker V, Parfenova V, Kostornova TY (2002) The biodiversity of actinomycetes in Lake Baikal. Microbiol 71(3):346–349
Tillotson G (2018) A crucial list of pathogens. Lancet Infect Dis 18(3):234–236
Tiwari K, Gupta RK (2012) Rare actinomycetes: a potential storehouse for novel antibiotics. Crit Rev Biotechnol 32(2):108–132
Tiwari K, Gupta RK (2013) Diversity and isolation of rare actinomycetes: an overview. Crit Rev Microbiol 39(3):256–294
Tolba ST, Nagwa AAA, Hatem D (2013) Molecular characterization of rare actinomycetes using 16S rRNA-RFLP. African J Biol Sci 9:185–197
Udikovic-Kolic N, Wichmann F, Broderick NA, Handelsman J (2014) Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization. Proc Nat Acad Sci 111(42):15202–15207
Vajs J, Proud C, Brozovic A, Gazvoda M, Lloyd A, Roper DI et al (2017) Diaryltriazenes as antibacterial agents against methicillin resistant Staphylococcus aureus (MRSA) and Mycobacterium smegmatis. Eur J Med Chem 127:223–234
Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald GF, Chater KF et al (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71(3):495–548
Voelker F, Altaba S (2001) Nitrogen source governs the patterns of growth and pristinamycin production in ‘Streptomyces pristinaespiralis’. Microbiol 147(9):2447–2459
Walsh CT (2002) Combinatorial biosynthesis of antibiotics: challenges and opportunities. ChemBioChem. 3(2-3):124–134
Wang L, Zhang M, Li Y, Cui Y, Zhang Y, Wang Z et al (2017) Application of response surface methodology to optimize the production of antimicrobial metabolites by Micromonospora Y15. Biotechnol Biotechnol Equip 31(5):1016–1025
Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R et al (2015) antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43(W1):W237–WW43
Yamamura H, Hayakawa M, Iimura Y (2003) Application of sucrose-gradient centrifugation for selective isolation of Nocardia spp. from soil. J Appl Microbiol 95(4):677–685
Yang H-J, Huang X-Z, Zhang Z-L, Wang C-X, Zhou J, Huang K et al (2014) Two novel amphomycin analogues from Streptomyces canus strain FIM-0916. Nat Prod Res 28(12):861–867
Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K et al (2009) Characterization of a new metallo-β-lactamase gene, blaNDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 53(12):5046–5054
Yun-yang L, Yang X, Tian-en W, Zhen-tai C, Yuan-rong C (2007) Neorustmicin produced by M. carbonacea FIM 99-663. Chin J Antibiot 32(11):657
Zhang L, Bao Z, Cheng J, Li H, Huang X, Wang S et al (2007) Fosmid library construction and initial analysis of end sequences in Zhikong scallop (Chlamys farreri). Marine Biotechnol 9(5):606–612
Acknowledgements
We would like to thank the Microbial Resources Center (Cairo MIRCEN) and Ain Shams Specialized Hospital for providing the strains of foodborne and bloodborne pathogens strains.
Funding
We are very grateful for the scholarship provided by Egyptian missions and the British Council in Egypt (Newton-Mosharafa program 2016–2017) to complete and conduct the molecular studies at the School of Life Sciences, Lab C123, University of Warwick, UK.
Author information
Authors and Affiliations
Contributions
This work was carried out in collaboration between all authors. NAA, ST, AA, and EMHW designed the study and wrote the protocol. DHA and CB managed the lab work of the study. AA managed the paper organization. DHA and AA wrote the first draft of the manuscript. DHA and AA managed the literature searches. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Amin, D.H., Abdallah, N.A., Abolmaaty, A. et al. Microbiological and molecular insights on rare Actinobacteria harboring bioactive prospective. Bull Natl Res Cent 44, 5 (2020). https://doi.org/10.1186/s42269-019-0266-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-019-0266-8