
Abstract
Background
Primary intra-axial Ewing sarcoma (EWS) is extremely rare and a highly aggressive small blue round cell tumor in the central nervous system (CNS). We reported a case of primary intra-axial CNS-EWS in a 30-year-old male and presented an extensive literature review of CNS-EWS cases.
Case presentation
A 30-year-old male had been diagnosed with a grade 2 astrocytoma in the left frontal lobe a year ago and had received chemotherapy and radiotherapy. He presented with a right-sided mass, which was revealed to be a 6 cm contrast-enhancing lesion with heterogeneous hemorrhagic areas in the right frontal lobe. The patient underwent emergent craniotomy for surgical excision. Histologically, the tumor was composed of high-grade, small, blue, round cells forming trabecular islands and rosettes which were separated by fibrovascular tissues. In immunohistochemical (IHC) examination, the tumor cells were positive for CD99 and FLI-1. Fluorescence in situ hybridization analysis revealed the presence of EWSR1 gene rearrangement. The histopathological, IHC, and molecular findings were consistent with a diagnosis of EWS. Further imaging did not show evidence of another primary site. The patient was followed up without further therapy and succumbed to the disease three months after the diagnosis.
Conclusion
EWS is very rare but an aggressive neoplasm in the CNS. Chemoradiotherapy may promote secondary cancers but it is unclear whether the development of EWS was associated with chemoradiotherapy that our case had received for astrocytoma. EWS should be considered when diagnosing a CNS tumor that shows small, blue, round cell tumor morphology. A combination of IHC and molecular tests is required for accurate diagnosis to ensure that all patients receive the most appropriate treatment to optimize outcomes.
Similar content being viewed by others


Background
Ewing sarcoma family of tumors are a group of highly aggressive and recurrent small round-cell neoplasms that include peripheral primitive neuroectodermal tumor and Ewing sarcoma (EWS) (Pisconti et al. 2020). These are different forms of the same tumor and share similar genetic changes (Campbell et al. 2018). With an estimated occurrence of 1% amongst children and adolescents, it has been reported even rarer in adults (Huguenard et al. 2021). The first case of EWS was published in 1921 by Ewing et al. (Kilpatrick et al. 2018). Since then, a limited number of central nervous system EWS (CNS-EWS) have been published (Jiahua Huang and Ghent, Robyn Levingston 2020).
A combination of histopathological appearance, radiographic findings, and clinical correlation is essential for the correct diagnosis, appropriate treatment regime, and improved survival of the patients (Saifuddin et al. 2021). Clinical presentations of EWS are diverse and depend on tumor aggressiveness. They may be nonspecific, and symptoms may be related to high intracranial pressure, focal neurological deficit, or meningism (Murphey et al. 2013). Typical symptoms include headache, nausea, vomiting, pain or fever. Symptoms are often present for more than six months before diagnosis, often correlating with advanced disease and metastases (Loarer et al. 2022; Klijanienko et al. 2012). The radiologic features of CNS-EWS seen on MRI and CT imaging usually tend to be large with a wide zone of transition/poorly defined margin. In an intra-axial location, imaging often shows a supratentorial enhancing mass that contacts the leptomeninges (VandenHeuvel et al. 2015). In an extra-axial location, high attenuation on CT and low signal intensity on T2-weighted MRI is characteristic (Li et al. 2005). Histologically, CNS-EWS consists of small, blue, round cells showing neuroepithelial differentiation defined by the presence of rosette formation. They typically show diffuse positivity for the immunohistochemical (IHC) marker CD99 in cytoplasmic membrane (Loarer et al. 2022). The detection of EWSR1 gene rearrangement by fluorescence in situ hybridization (FISH) analysis is a common diagnostic method for EWS. Additionally, reverse transcription polymerase chain reaction (RT-PCR) or RNA sequencing can be employed to identify the specific fusion partners involved in EWS.
The EWSR1 gene (22q12) is combined with one of the individual members of the ETS group of transcription factors, such as the FLI1 gene (11q24) in 85% of cases or the ERG gene (21q22) in 5–10% of cases, resulting in specific chromosomal translocations. Less frequently, FEV (2q36), ETV1 (7p21), or ETV4 (also known as E1AF; 17q21) are coupled with EWSR1 (Delattre et al. 1994; Kaneko et al. 1997; Peter et al. 1997; Jeon et al. 1995; Urano et al. 1998; Shing et al. 2003). There are other unusual variants, such as a t(16;21) rearrangement in the FUS (16p11) gene compared to the ERG (21q22) gene (Shing et al. 2003; Berg et al. 2009). Additionally, it has been shown that the t(2;16)(q36;p11) rearrangement is the cause of the FUS-FEV combination gene (Ng et al. 2007). Two novel translocations have recently been discovered: t(4;22), involving EWSR1 and SMARCA5 (4q31), and t(20;22), involving EWSR1 and NFATC2 (20q13) (Szuhai et al. 2009; Sumegi et al. 2011).
Here, we reported a rare case of a primary intracranial CNS-EWS in a 30-year-old male and discussed the case with its clinical, radiological, histopathological, and molecular features. We also presented an extensive literature review including all intracranial and extracranial CNS-EWS cases.
Case presentation
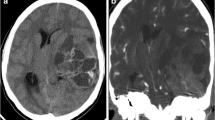
A 30-year-old male, who had been diagnosed with a grade 2 astrocytoma in the left frontal lobe at another hospital a year ago and had received three cycles of chemotherapy and radiotherapy, presented with a right-sided mass. A computed tomography (CT) scan revealed a 6 cm of contrast-enhancing mass with heterogeneous hemorrhagic areas in the right frontal lobe. Enhanced scanning showed that the frontal horns of the right lateral ventricle were obliterated and subfalcine herniation was present due to the mass effect. The patient underwent emergent craniotomy for surgical excision of the lesion. As shown in Fig. 1, histologically, the tumor was well demarcated from the surrounding brain tissue. It consisted of uniform, small-blue-round cells with scant cytoplasm and indistinct cytoplasmic membranes. The tumor cells formed trabecular islands and rosettes that were separated by fibrovascular tissues. Immunohistochemically, the tumor cells were positive for CD99 (membranous), FLI-1, NKX2.2, vimentin, S100, CD56, NSE, p53; and negative for cytokeratin AE1/AE3, cytokeratin 7, cytokeratin 20, synaptophysin, chromogranin, TTF-1, desmin, EMA, GFAP, Olig-2, CD3, CD20, CD138, SALL-4, PLAP, and neurofilament. The Ki-67 labelling index of the tumor was 70%. Fluorescence in situ hybridization (FISH) analysis revealed EWSR1 gene rearrangement (EWSR1 Dual Color Break Apart Probe, REF Z-2096-200, LOT P144-PF1, Zytolight). The histopathological, IHC, and molecular findings were consistent with a diagnosis of EWS. A subsequent CT scan did not show evidence of another primary site. Due to the poor overall condition of our patient, he was followed up without further therapy and succumbed to the disease three months after the diagnosis.

Diagnostic features of Ewing sarcoma. a The tumor was well demarcated from the surrounding brain tissue. The tumor cells formed trabecular islands and rosettes that were separated by fibrovascular tissues. b The tumor consisted of uniform, small-blue-round cells with scant cytoplasm and indistinct cytoplasmic membranes. Rosette formation is evident. c Immunohistochemically, CD99 showed diffuse membranous positivity. d- Fluorescence in situ hybridization showed EWSR1 gene rearrangement
Discussion and conclusion
Primary CNS-EWS is a very rare entity and only 94 cases have been reported in spinal cord and cranium in the English literature, as summarized in Table S1 in the supplementary file. Primary intracranial CNS-EWS is even rarer that only 31 adult cases reported as of October 2022 (Hu et al. 2022). Our case was a 30-year-old male patient who had been diagnosed with a grade 2 astrocytoma and treated with chemoradiotherapy a year prior to being diagnosed with primary intra-axial CNS-EWS. The histological, IHC, and molecular features of the tumor were typical of those reported in the literature. Histologically, the tumor was composed of high-grade, small, blue, round cells forming trabecular islands and rosettes which were separated by fibrovascular tissues. Mitotic activity was high and focal areas of necrosis were present. Immunohistochemically, the tumor cells were positive for CD99 (membranous), FLI-1, CD56, NSE; and FISH analysis revealed EWSR1 gene rearrangement. The histopathological, IHC, and molecular findings were consistent with a diagnosis of CNS-EWS. Table 1 summarizes cases reported in the literature of patients with intra-axial primary CNS-EWS.
Secondary cancers are distinct from primary tumors and may arise months or years after the initial treatment. Alkylating agents such as cisplatin and topoisomerase-II inhibitors such as etoposide have been implicated in the etiology of secondary cancers. Moreover, these agents have a synergistic effect on secondary leukemia when combined in chemotherapy (Shimatani et al. 2019). Ewing sarcomas are rare secondary malignant neoplasms that have been reported in a few cases following chemotherapy for a primary tumor. A study of 3844 patients treated in the last three international EWS trials revealed that EWS occurred after a variety of malignancies, mainly acute lymphoblastic leukemia’s and lymphomas (Kaiser et al. 2022). Zengin et al. reported a case of cutaneous EWS after two cycles of BEP chemotherapy for testis tumor (Tanik et al. 2014). Wolpert et al. (2016). Although chemotherapy is associated with secondary cancer in the long term, it is unclear whether the development of EWS is associated with the chemotherapy in our patient who was diagnosed with astrocytoma a year prior to being diagnosed with EWS.
Most EWSs are diagnosed in the second decade and usually occur in the bone and soft tissues. When these tumors are detected in different age groups and locations, they may not be suspected clinically and radiologically. However, when histopathological examination reveals a small round blue cell tumor (SBRCT), EWS should be included in the differential diagnosis. The diagnosis of EWS can be supported by a few IHC stains. CD99 is a sensitive IHC marker and is positive in around 95% of EWS cases, however, it lacks specificity. FLI1 is a relatively specific marker for EWS, but it may also be expressed in other SBRCTs such as lymphoblastic leukaemia’s, lymphomas, and several soft-tissue sarcomas. Additionally, about 15% of EWS cases have variant translocations that do not involve FLI1. Molecular testing is essential to confirm the diagnosis. Detection of EWSR1 gene rearrangement by FISH and/or FET-ETS gene fusions by RT-PCR may suffice for the diagnosis of EWS, when typical histopatological EWS features are present. However, some other CNS tumors may show EWSR gene rearrangement, namely intracranial mesenchymal tumor with FET::CREB fusion.
The current treatment of choice for patients with EWS is radical surgical resection followed by chemo-radiotherapy, radiotherapy or a combination thereof. However, these tumors have a highly malignant and aggressive phenotype and approximately 25% of patients present with distant metastasis at the time of diagnosis, making complete resection difficult (Jin et al. 2016). In our case, the patient had been diagnosed with grade 2 astrocytoma in the left frontal lobe and treated with chemo-radiotherapy. A year later, he developed EWS and died three months after tumor resection. No additional treatment could be given because our patient’s overall condition was poor.
In conclusion, EWS is very rare but an aggressive neoplasm in the CNS. It should be considered in the differential diagnosis of a CNS tumor that shows SBRCT morphology. A combination of IHC and molecular tests is required for accurate diagnosis to ensure that all patients receive the most appropriate treatment to optimize outcomes.
Data Availability
All data generated or analysed during this study are included in this published article.
Abbreviations
- CNS:
-
Central nervous system
- FISH:
-
Fluorescence in situ hybridization
- EWS:
-
Ewing sarcoma
- CNS-EWS:
-
Central nervous system-Ewing sarcoma
- IHC:
-
Immunohistochemical
- RT-PCR:
-
reverse transcription polymerase chain reaction
- CT:
-
Computed tomography
- SBRCT:
-
Small blue round cell tumor
- S:
-
Surgery
- R:
-
Radiotherapy
- C:
-
Chemotherapy
- DOD:
-
Died of disease
- AWD:
-
Alive with disease
- AWND:
-
Alive with no disease
References
Berg T, Kalsaas A-H, Buechner J, Busund L-T. Ewing sarcoma–peripheral neuroectodermal tumor of the kidney with a FUS–ERG fusion transcript. Cancer Genet Cytogenet. 2009;194(1):53–7.
Campbell K, Shulman D, Janeway KA, DuBois SG. Comparison of epidemiology, clinical features, and outcomes of patients with reported Ewing sarcoma and PNET over 40 years justifies current WHO classification and treatment approaches. Sarcoma. 2018;2018.
Delattre O, Zucman J, Melot T, Garau XS, Zucker J-M, Lenoir GM, et al. The ewing family of tumors–a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med. 1994;331(5):294–9.
Huguenard AL, Li YD, Sharifai N, Perkins SM, Dahiya S, Chicoine MR. Multifocal primary central nervous system ewing sarcoma presenting with intracranial hemorrhage and leptomeningeal dissemination: illustrative case. J Neurosurg Case Lessons. 2021;1(10):1–9.
Hu X, Huang Q, Wang J, Li D, Wang P, Cai J. Case report: primary intracranial EWs/PNET in adults: clinical experience and literature review. Front Oncol. 2022;12.
Jay V, Zielenska M, Lorenzana A, Drake J. An unusual cerebellar primitive neuroectodermal tumor with t(11; 22) translocation: pathological and molecular analysis. Pediatr Pathol Lab Med. 1996;16(1):119–28.
Jeon I-S, Davis JN, Braun BS, Sublett JE, Roussel MF, Denny CT, et al. A variant ewing’s sarcoma translocation (7; 22) fuses the EWS gene to the ETS gene ETV1. Oncogene. 1995;10(6):1229–34.
Jiahua Huang F, Ghent, Robyn Levingston MSD. Intracranial ewing sarcoma – a case report Jiahua. Surg Neurol Int. 2020;1–4.
Jin SG, Jiang XP, Zhong L. Congenital ewing’s Sarcoma/Peripheral primitive neuroectodermal tumor: a Case Report and Review of the literature. Pediatr Neonatol. 2016;57(5):436–9.
Kaiser I, Kauertz K, Zöllner SK, Hartmann W, Langer T, Jürgens H et al. Secondary Malignancies after Ewing Sarcoma—Epidemiological and Clinical Analysis of an International Trial Registry. Cancers (Basel). 2022;14(23).
Kaneko Y, Kobayashi H, Handa M, Satake N, Maseki N. EWS-ERG fusion transcript produced by chromosomal insertion in a Ewing sarcoma. Genes, Chromosom Cancer. 1997;18(3):228–31.
Ke C, Duan Q, Yang H, Zhu F, Yan M, Xu SP, Zhou S, Wan F, Shu K, Lei T, Xia LM. Meningeal Ewing sarcoma/peripheral PNET: clinicopathological, immunohistochemical and FISH study of four cases. Neuropathology. 2017;37(1):35–44.
Kilpatrick SE, Reith JD, Rubin B. Ewing sarcoma and the history of similar and possibly related small round cell tumors: from whence have we come and where are we going? Adv Anat Pathol. 2018;25(5):314–26.
Klijanienko J, Couturier J, Bourdeaut F, Fréneaux P, Ballet S, Brisse H, et al. Fine-needle aspiration as a diagnostic technique in 50 cases of primary ewing sarcoma/peripheral neuroectodermal tumor. Institut Curie’s experience. Diagn Cytopathol. 2012;40(1):19–25.
Le Loarer F, Baud J, Azmani R, Michot A, Karanian M, Pissaloux D. Advances in the classification of round cell sarcomas. Histopathology. 2022;80(1):33–53.
Li W-Y, Brock P, Saunders DE. Imaging characteristics of primary cranial ewing sarcoma. Pediatr Radiol. 2005;35:612–8.
Murphey MD, Senchak LT, Mambalam PK, Logie CI, Klassen-Fischer MK, Kransdorf MJ. From the radiologic pathology archives: ewing sarcoma family of tumors: radiologic-pathologic correlation. Radiographics. 2013;33(3):803–31.
Ng TL, O’Sullivan MJ, Pallen CJ, Hayes M, Clarkson PW, Winstanley M, et al. Ewing sarcoma with novel translocation t (2; 16) producing an in-frame fusion of FUS and FEV. J Mol Diagnostics. 2007;9(4):459–63.
Peter M, Couturier J, Pacquement H, Michon J, Thomas G, Magdelenat H, et al. A new member of the ETS family fused to EWS in ewing tumors. Oncogene. 1997;14(10):1159–64.
Pisconti S, Della Vittoria Scarpati G, Buonerba C, Messinese S, Carella R, Di Marzo M et al. Management of ewing sarcoma family of tumors: a short description of a rare primitive uterine pPNET and literature review. Onco Targets Ther. 2020;1179–84.
Saifuddin MSAH, Ng CY, Abdullah MS. Skull base primary ewing sarcoma: a radiological experience of a rare disease in an atypical location. Am J Case Rep. 2021;22(1):1–7.
Shimatani A, Aono M, Hoshi M, Oebisu N, Iwai T, Takada N, et al. Secondary osteosarcoma in patients previously treated for childhood cancer: three case reports. Mol Clin Oncol. 2019;10(1):153–8.
Shing DC, McMullan DJ, Roberts P, Smith K, Chin S-F, Nicholson J, et al. FUS/ERG gene fusions in Ewing’s tumors. Cancer Res. 2003;63(15):4568–76.
Sumegi J, Nishio J, Nelson M, Frayer RW, Perry D, Bridge JA. A novel t (4; 22)(q31; q12) produces an EWSR1–SMARCA5 fusion in extraskeletal ewing sarcoma/primitive neuroectodermal tumor. Mod Pathol. 2011;24(3):333–42.
Szuhai K, IJszenga M, de Jong D, Karseladze A, Tanke HJ, Hogendoorn PCW. The NFATc2 gene is involved in a novel cloned translocation in a ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res. 2009;15(7):2259–68.
Tanik S, Zengin K, Albayrak S, Eryilmaz R, Yilmaz D, Pirinçci N. Cutaneous ewing’s sarcoma secondary to chemotherapy given for testis tumor: Case report. Int J Surg Case Rep. 2014;5(12):972–4.
Urano F, Umezawa A, Yabe H, Hong W, Yoshida K, Fujinaga K, et al. Molecular analysis of Ewing’s sarcoma: another fusion gene, EWS-E1AF, available for diagnosis. Japanese J cancer Res. 1998;89(7):703–11.
VandenHeuvel KA, Al-Rohil RN, Stevenson ME, Qian J, Gross NL, McNall-Knapp R, et al. Primary intracranial ewing’s sarcoma with unusual features. Int J Clin Exp Pathol. 2015;8(1):260–74.
Weil RJ, Zhuang Z, Pack S, Kumar S, Helman L, Fuller BG, Mackall CL, Oldfield EH. Intramedullary Ewing sarcoma of the spinal cord: consequences of molecular diagnostics: case report. J Neurosurg Spine. 2001;95(2):270–5.
Wolpert F, Grotzer MA, Niggli F, Zimmermann D, Rushing E, Bode-Lesniewska B. Ewing’s Sarcoma as a Second Malignancy in Long-Term Survivors of Childhood Hematologic Malignancies. Sarcoma. 2016;2016.
Yang MJ, Whelan R, Madden J, Mulcahy Levy JM, Kleinschmidt-DeMasters BK, Hankinson TC, Foreman NK, Handler MH. Intracranial Ewing sarcoma: four pediatric examples. Child’s Nervous Syst. 2018;34:441–8.
Acknowledgements
Not applicable.
Funding
The authors did not receive financial support for the research, authorship and/or publication.
Author information
Authors and Affiliations
Contributions
FD conceptualised, analysed and edited the manuscript. MTGM contributed in a thorough literature survey, writing and editing the manuscript. AK analysed and interpreted the patient data. All three authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient has given consent to have this manuscript published in an academic journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dirilenoğlu, F., George Moyo, M.T. & Kahraman, A. Primary intra-axial Ewing sarcoma of the central nervous system: report of a rare case with literature review. Surg Exp Pathol 6, 12 (2023). https://doi.org/10.1186/s42047-023-00137-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42047-023-00137-x