
Abstract
Background
Gliosarcoma is a rare variant of IDH- wild type glioblastoma with both glial and mesenchymal differentiation. It accounts for approximately 2% of glioblastomas and has a poor prognosis similar to that of classic glioblastoma. It is seen mostly between 40 and 60 years of age with a mean age over 50 years. Pediatric gliosarcoma is even rarer than gliosarcoma in adults. We describe the clinicopathological features of gliosarcoma in patients under 20 years of age and determine whether there are significant differences from gliosarcoma in adults. We also present detailed review of published literature on pediatric gliosarcoma.
Methods
Slides of gliosarcomas in patients under 20 years of age were reviewed. Clinicopathological features were noted in detail and follow up was obtained.
Results
Eleven cases of gliosarcoma were reported in patients under 20 years of age. Ages ranged from three to 19 years (mean age 13 years). Frontal, parietal and temporal lobes were the commonest locations. Mean and median tumor size was six and five cm respectively. All 11 cases demonstrated the classic biphasic pattern. In 10 cases, glial component was astrocytic and was highlighted on GFAP. Sarcomatous component in most cases resembled fibrosarcoma and was high grade in 72.7%. Glial areas were reticulin poor while sarcomatous areas were reticulin rich. In over 45% cases, bizarre tumor giant cells were seen in the sarcomatous areas. In 1 case, sarcomatous areas showed extensive bone and cartilage formation. Other histologic features included hyalinized blood vessels, hemorrhage, infarction, gemistocytic cells, rhabdoid cells etc. Follow up was available in nine patients, five received chemoradiation post resection while three received radiotherapy only. Prognosis was dismal and eight patients died within one to 14 months following resection.
Conclusions
Gliosarcomas in patients under 20 comprised 13% of all gliosarcomas reported during the study period. Frequency and mean age were higher compared to other published reports. Pathological features were similar to those described in literature. Clinicopathological features and prognosis of pediatric gliosarcomas were similar to adult gliosarcomas.
Similar content being viewed by others


Introduction
Gliosarcoma, World Health Organization (WHO) grade IV is a rare variant of Isocitrate Dehydrogenase (IDH)- wild type glioblastoma with both glial and mesenchymal differentiation and accounts for approximately 2% of glioblastomas. It has a poor prognosis similar to that of classic glioblastoma. It is seen mostly between 40 and 60 years of age with a mean age above 50 years, is more common in males and occurs mainly in the cerebral hemispheres with the temporal and frontal lobes being the commonest locations. It is characterized histologically by a biphasic pattern composed of alternating glial and mesenchymal (sarcomatous) areas. Both glial and mesenchymal components represent monoclonal proliferations. The clinical profile, imaging, spread and macroscopic appearance of this variant are similar to classic glioblastoma. It is often superficial and deceptively circumscribed. Pediatric gliosarcoma is even rarer than gliosarcoma in adults [1, 2].
Histologically, the glial component is usually astrocytic (like the classic astrocytic glioblastoma) with anaplastic features. Sarcomatous component usually manifests as a spindle cell sarcoma with nuclear atypia, mitoses and necrosis. The glial areas are reticulin poor while the sarcomatous areas are reticulin rich (highlighted on reticulin stain). On immunohistochemical (IHC) staining, glial areas express glial fibrillary acidic protein (GFAP) while the sarcomatous areas are negative [1].
Pediatric gliosarcoma, as stated above, is even rarer. To the best of our knowledge, 45 cases have been reported in literature [3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36].
Herein, we present a series of 11 cases of gliosarcoma reported in patients under 20 years of age. The aim of this study is to describe the clinicopathological features of gliosarcoma reported in patients under 20 and to determine whether there are significant differences from gliosarcomas occurring in adults. We also present a detailed review of published literature on these extremely rare tumors.
Methods
The Surgical Pathology files of the Section of Histopathology, Department of Pathology and Laboratory Medicine, Aga Khan University Hospital, were searched for gliosarcomas reported between July 1, 2011 and June 30, 2019. Cases reported in patients under 20 years were identified. Slides of these cases were retrieved and were reviewed by the senior authors. The diagnosis was confirmed. Clinical and pathological features were described in detail. Follow up was obtained through verbal telephonic communication with the parents. Ethical exemption was obtained from the Aga Khan University Ethical Review Committee (ERC). All procedures performed on patient tumor samples in this study were in accordance with the ethical standards of the Institutional ERC and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Detailed review of published literature was conducted by the authors.
We systematically searched PubMed, Google Scholar and Web of Science detailed for articles on pediatric gliosarcoma with restriction of ‘original article’ and ‘case reports’. We only included articles with English abstracts. We searched using the Medical Subject Heading (MESH) terms and key words ‘pediatric gliosarcoma’. A total of 34 articles were included. Review articles were not included. We collected demographics, clinicopathological and follow up information. All selected articles were assessed for eligibility. All were found to be eligible. No duplicates were found. All 34 were included in qualitative synthesis.
Results
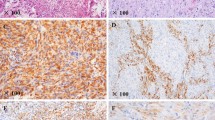
During the study period (2011–2019), 84 cases of gliosarcoma were reported. Of these, 11 (31.1%) were reported in patients younger than 20 years of age. Clinicopathological features are summarized in Table 1. Of these 11 patients, 6(54.5%) were males and 5(45.5%) were females. Ages of patients ranged from 3 to 19 years with mean and median age of 13 and 16 years respectively. Four patients (36.4%) were under 10 years of age. Of the 11 cases, 3(27.3%) were located in the frontal and 2(18.2%) in the parietal lobes; one case each was temporal, frontotemporal, temporo-parietal, parieto-occipital, occipital and sellar in location (Fig. 1a-d). All cases were received as multiple pieces of tumor tissue ranging from 1.5 cm to 12 cm in aggregate with mean and median size of 6 cm and 5 cm respectively. Gross total resection was apparently not achieved in any of the cases. On histologic examination, all 11 cases demonstrated the classic biphasic pattern (Fig. 2a). Glial component was astrocytic in 10 cases (90.9%) and oligodendroglial in 1 case (Fig. 2b). Glial component was highlighted in all 11 cases on IHC stain for GFAP (Fig. 2c). Sarcomatous component in most cases manifested as a spindle cell sarcoma resembling fibrosarcoma (Fig. 2d). Sarcomatous component was high grade in 8 (72.7%) cases and low grade in 3 (27.3%) cases. Sarcomatous areas were highlighted on IHC stain for vimentin (Fig. 3a). Glial areas were reticulin poor (Fig. 3b) while sarcomatous areas were reticulin rich (Fig. 3c). In 5 (45.5%) cases, sarcomatous areas showed considerable atypia in the form of bizarre tumor giant cells (Fig. 3d). In 1 case, the sarcomatous component showed additional lines of mesenchymal differentiation in the form of extensive bone and cartilage formation (Fig. 4a). Prominent hyalinized blood vessels (Fig. 4b) were seen in 3 (27.3%) cases while hemorrhage and infarction were noted in 2 cases. Gemistocytic (Fig. 4c) and rhabdoid (Fig. 4d) cells were seen in 1 and 2 cases respectively. Follow up was available in 9 cases. Of these 9 patients, 5 received both chemotherapy and radiotherapy post-surgery while 3 received radiotherapy only. One patient did not receive either chemotherapy or radiotherapy. Of these 9 patients, 8 died within 1 month to 14 months following surgery while the 9th patient was alive 6 months post-surgery.

a T2WI Axial Image: A well-defined rounded T2WI heterogenous signal intensity mass seen in right parietal lobe with significant perilesional edema. b T2WI Sagittal Image: A well-defined rounded T2WI heterogenous signal intensity mass seen in right parietal lobe with significant perilesional edema c T1 Axial post-contrast: Avid post contrast enhancing mass with few central hypointensities identified in right posterior parietal lobe and surrounding perilesional edema. d T1 coronal post contrast image: Avid post contrast enhancing mass with few central hypointensities identified in right posterior parietal lobe and shows surrounding perilesional edema

a Classic biphasic appearance of gliosarcoma. The mesenchymal elements at top are sharply demarcated from glial component at bottom. b Oligodendroglioma as glial component was seen in one case. c. GFAP expression in the glial component. d. Mesenchymal component appeared as fibrosarcoma in most cases

a. Vimentin positivity in mesenchymal component. b While glial areas are reticulin poor, c, mesenchymal component was reticulin rich. d. Focal bizarre tumor cells in mesenchymal component

a. Trabeculae of lamellar bone in mesenchymal component. b. Prominent large vessels were noted in some cases. c & d Sheets of gemistocytes and rhabdoid cells are focally seen in few cases
Discussion
Pediatric gliosarcomas are even rarer than adult gliosarcomas. Clinical and morphological features are similar to adult gliosarcomas. However, they may mimic more common tumors on radiological and histological examination. The clinicopathological features and differential diagnostic consideration are discussed. Published literature is reviewed.
On radiological examination, the relative discreteness of these tumors may mimic meningioma [37, 38]. Similarly, on gross examination, the appearance of a firm well-circumscribed mass with attachment to dura may be mistaken with a meningioma or metastases, although such confusion is more likely in adults rather than in the pediatric age group [1]. On histological examination, the diagnosis in typical cases even in pediatric age group is straightforward when the classic biphasic pattern is well developed. However, the sarcomatous areas in both adult and pediatric gliosarcoma can resemble fibrosarcoma. Some cases may show other types of mesenchymal differentiation such as bone and cartilage formation (resembling osteo-or chondrosarcoma), smooth and striated muscle differentiation (resembling leiomyosarcoma and rhabdomyosarcoma), lipomatous differentiation (resembling liposarcoma) and primitive neural differentiation. Variable mesenchymal differentiation was seen in our series and additional mesenchymal differentiation in the form of bone and cartilage formation was seen in one case. Similarly, adenoid (epithelial) differentiation may be seen in gliosarcoma in all age groups and such areas may resemble carcinoma resulting in misdiagnosis. Squamous metaplasia may be seen and can be mistaken for squamous cell carcinoma. The presence of gemistocytic and rhabdoid cells may lead to erroneous diagnosis of gemistocytic astrocytoma and atypical teratoid/rhabdoid tumor in the pediatric age group. Gemistocytic and rhabdoid cells were seen in one and two cases respectively in our series. In pediatric patients, germ cell tumors (such as germinoma and teratoma) should also be considered in the differential diagnosis. Gliosarcoma in pediatric patients can especially be confused with teratoma if bone, cartilage or other mesenchymal components are present [22, 39,40,41,42,43]. Desmoplastic infantile astrocytoma (DIA) is another rare tumor which can be confused with gliosarcoma in the pediatric age group. However, DIA is a slow growing, WHO grade I tumor which typically occurs in infants as a large cystic mass in the supratentorial cerebral cortex and meninges and is often attached to the dura. Microscopically, it is composed of a prominent desmoplastic stroma in which streams of neoplastic astrocytes are seen. Mitotic activity and necrosis are uncommon and ki67 index is usually < 2% [1].
At the molecular level, gliosarcomas including those in the pediatric age group demonstrate Phosphatase and Tensin homolog (PTEN) and TP53 mutations and Cyclin-dependent kinase inhibitor 2A (CDKN2A) deletions. Epidermal growth factor receptor (EGFR) amplification is infrequent. Except for the last, their genetic profile is similar to that of IDH-wild type glioblastoma. Gains on chromosome 7 are seen in 75% cases while losses on chromosome 10 are seen in 35% [44]. In a study of adult gliosarcomas by Smith et al., these tumors were primarily 0–6-Methylguanine-DNA-Methyltransferase (MGMT) unmethylated (87.5%), IDH-1 preserved (100%) and EGFR wild type (100%). A 2019 study by Lowder et al. demonstrated that the most frequent alteration was copy number loss comprising 57% of total copy number changes and far exceeding the number of copy number gains (26.2%), amplifications and loss of heterozygosity events. Chromosomes 9 and 10 showed the highest number of losses while the majority of copy number gains were seen on chromosome 7 [45, 46]. Recently, Graham et al. reported a gliosarcoma in an eleven-year-old girl and a twelve-year-old boy. The latter had neurofibromatosis type 1 (NF1), the first reported case of pediatric gliosarcoma in a child with NF1. Whole-exome sequencing showed higher mutational burden in the patient without NF. NF1 patient survived without progression while patient without NF1 died of disease [36].
Occasional case reports documenting pediatric gliosarcomas in locations other than the cerebral hemispheres have been published. Neelima et al. reported a case occurring in the thalamus [21]. A case of pediatric gliosarcoma associated with NF1 was recently reported by Dogan et al. [33]. Granados et al. reported a pineal gliosarcoma in a five-year-old girl, the first reported case in this unusual location [29].
Various studies have emphasized the importance of gross total resection in achieving relatively better prognosis [23, 32]. Studies have shown that subtotal resection is the most important variable in the dismal prognosis associated with pediatric gliosarcomas in most cases [31]. However, a number of studies have shown that prognosis is dismal even in cases where apparent gross total resection was achieved and in spite of aggressive chemo and radiotherapy post resection [15, 20, 26], median overall survival and event free survival have been only a few months, mostly under a year [15, 20, 37]. Few studies, however, have reported better prognosis and long-term survival with aggressive treatment (gross total resection, chemotherapy and radiotherapy [18, 24, 47]. However, overall, pediatric gliosarcomas share a dismal prognosis with adult gliosarcoma. Thus, although a longer survival has been reported in a few cases, the majority of patients demonstrate an extremely poor prognosis with early recurrence and death within a few months after surgery even after apparent gross total resection and aggressive post-surgical chemo and radiotherapy [26, 31]. This was true for our cases except for two patients who survived for 14 months and 2 years respectively post resection. Both these patients received chemo and radiotherapy. Findings of comprehensive literature review are summarized in Table 2.
Mallick et al. published a series of five cases of pediatric gliosarcoma and investigated the value of concurrent and adjuvant temozolamide in the treatment of these tumors. They showed that temozolamide is well tolerated by pediatric patients and survival data with temozolamide therapy was encouraging. The two-year progression free and overall survival rates were 44.2 and 62.9%, respectively [27].
Limitations
-
1)
Follow up was available in only 9 out of 11 cases
-
2)
Molecular workup was not performed.
Conclusions
Pediatric gliosarcomas are extremely rare. Clinicopathological features of pediatric gliosarcoma are similar to adult gliosarcoma. However, pediatric gliosarcomas may mimic more common tumors on radiological and histological examination. On histological examination, gliosarcomas may sometimes mimic sarcoma and carcinoma if specific mesenchymal and glandular differentiation is present. In pediatric age group, osteosarcoma, fibrosarcoma, teratoma, and atypical teratoid/rhabdoid tumor should be excluded. Like their adult counterparts, pediatric gliosarcomas have a dismal prognosis in spite of aggressive chemoradiation. Slightly better survival times have been demonstrated in some studies with gross total resection although other studies have shown extremely poor survival even with apparent gross total resection.
Availability of data and materials
Data and materials of this work are available from the corresponding author on reasonable request.
Abbreviations
- WHO:
-
World Health Organization
- IHC:
-
Immunohistochemical
- GFAP:
-
Glial fibrillary acidic protein
- ERC:
-
Ethical Review Committee
- OS:
-
Overall survival
- PFS:
-
Progression free survival
- DIA:
-
Desmoplastic infantile astrocytoma
- PTEN:
-
Phosphatase and Tensin homolog
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- EGFR:
-
Epidermal growth factor receptor
- IDH:
-
Isocitrate Dehydrogenase
- MGMT:
-
Methylguanine-DNA-Methyltransferase
- NF1:
-
Neurofibromatosis type 1
- MESH:
-
Medical Subject Heading
References
Burger PC, Giangaspero F, Ohgaki H, Bieruat W. Gliosarcoma. In: Louis DN, Ohgaki H, WK WODC, Ellison DW, Figarella–Branger D, Perry A, et al., editors. WHO Classification of Tumours of the Central Nervous System. Revised 4th Edition. International Agency for Research on Cancer (IARC) Lyon; 2016. p. 48–9.
Galanis E, Buckner JC, Dinapoli RP, Scheithauer BW, Jenkins RB, Wang CH, et al. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: north central Cancer treatment group results. J Neurosurg. 1998;89:425–30.
Goldstein SJ, Young B, Markesberry WR. Congenital malignant gliosarcoma. AJNR Am J Neuroradiol. 1981;2:475–6.
McKeever PE, Wichman A, Chronwall B, Thomas C, Howard R. Sarcoma arising from a gliosarcoma. South Med J. 1984;77:1027–32.
Cerame MA, Guthikonda M, Kohli CM. Extraneural metastases in gliosarcoma: a case report and review of the literature. Neurosurgery. 1985;17:413–8.
Lee YY, Castillo M, Nauert C, Moser RP. Computed tomography of gliosarcoma. AJNR Am J Neuroradiol. 1985;6:527–31.
Takaue Y, Sullivan MP, Ramirez I, Cleary KR, van Eys J. Second malignant neoplasm in treated Hodgkin’s disease. Report of a patient and scope of the problem. Am J Dis Child. 1986;140:49–51.
Chadduck WM, Gollin SM, Gray BA, Norris JS, Araoz CA, Tryka AF. Gliosarcoma with chromosome abnormalities in a neonate exposed to heptachlor. Neurosurgery. 1987;21:557–9.
Radkowski MA, Naidich TP, Tomita T, Byrd SE, McLone DG. Neonatal brain tumors: CT and MR findings. J Comput Assist Tomogr. 1988;12(1):10–20.
Ono N, Nakamura M, Inoue HK, Tamura M, Murata M. Congenital gliosarcoma; so-called sarcoglioma. Childs Nerv Syst. 1990;6:416–20.
Kaschten B, Flandroy P, Reznik M, Hainaut H, Stevenaert A. Radiation-induced gliosarcoma. Case report and review of the literature. J Neurosurg. 1995;83:154–62.
Lach M, Wallace CJ, Krcek J, Curry B. Radiation-associated gliosarcoma. Can Assoc Radiol J. 1996;47:209–12.
Kepes JJ, Bastian FO, Weber ED. Gliosarcoma developing from an irradiated ependymoma. Acta Neuropathol (Berl). 1996;92:515–9.
Rizk T, Nabbout R, Koussa S, Akatcherian C. Congenital brain tumor in a neonate conceived by in vitro fertilization. Childs Nerv Syst. 2000;16:501–2.
Okami N, Kawamata T, Kubo O, Yamane F, Kawamura H, Hori T. Infantile gliosarcoma: a case and a review of the literature. Childs Nerv Syst. 2002;18:351–5.
Malde R, Jalali R, Muzumdar D, Shet T, Kurkure P. Gliosarcoma occurring 8 years after treatment for a medulloblastoma. Childs Nerv Syst. 2004;20:243–6.
Deb P, Sharma MC, Chander B, Mahapatra AK, Sarkar C. Giant cell glioblastoma multiforme: report of a case with prolonged survival and transformation to gliosarcoma. Childs Nerv Syst. 2006;22:314–9.
Salvati M, Lenzi J, Brogna C, Frati A, Piccirilli M, Giangaspero F, Raco A. Childhood’s gliosarcomas: pathological and therapeutical considerations on three cases and critical review of the literature. Childs Nerv Syst. 2006;22:1301–6.
Hocwald O, McFadden D, Osiovich H, Dunham C. Congenital gliosarcoma: detailed clinicopathologic documentation of a rare neoplasm. Pediatr Dev Pathol. 2009;12:398–403.
Karremann R, Rausche U, Fleischhack G, Nathrath M, Pietsch T, Kramm CM, Wolff JE. Clinical and epidemiological characteristics of pediatric gliosarcomas. J Neuro-Oncol. 2010;97:257–65.
Neelima R, Abraham M, Kapilamoorthy TR, Hingwala DR, Radhakrishnan VV. Pediatric gliosarcoma of thalamus. Neurol India. 2012;60:674–6.
Ravisankar S, Chander RV, Devadoss PK. Pediatric gliosarcoma with fibrosarcomatous differentiation: report of a rare case. Indian J Pathol Microbiol. 2012;55:521–4.
Moscote – Salazara LR, Alcala – Cerra G, Gutierez – Paternina JJ, Penagos Gonzelezc PJ, Zubieta Vega C, Chater – Cure G, Alberto Meneses C, Saenz M (2014) Pediatric Gliosarcoma: case report and literature review. Bol Asoc Med PR 106: 43–47.
Martin J, Devadoss P, Kannan K, Kumar Sundarraj S. Malignant pediatric gliosarcoma defies general survival data. Case Rep Med. 2014, 2014:175679. https://doi.org/10.1155/2014/175679.
Burzynski SR, Janicki TJ, Burzynski GS, Marszalek A. Long term survival (> 13 years) in a child with recurrent pontine gliosarcoma: a case report. J Pediatr Hematol Oncol. 2014;36:433–9.
Savant HV, Balasubramaniam S, Mahajan V. Giant parietal lobe infantile gliosarcoma in a 5-year-old child. J Pediatr Neurosci. 2015;10:159–61.
Mallick S, Gandhi AK, Sharma DN, Gupta S, Haresh KP, Rath GK, Julka PK. Pediatric gliosarcoma treated with adjuvant radiotherapy and temozolamide. Childs Nerv Syst. 2015;31:2341–4.
Meena US, Sharma S, Chopra S, Jain SK. Gliosarcoma: a rare variant of glioblastoma multiforme in pediatric patient: case report and review of literature. World J Clin Cases. 2016;4:302–5.
Granados AM, Ospina C, Paredes S. Pineal gliosarcoma in a 5-year-old girl. Radiol Case Rep. 2017;13:244–7.
Yao K, Duan Z, Wang Y, Zhao X, Fan T, Qi X. Spinal cord gliosarcoma with rhabdomyoblastic differentiation: a case report. Int J Clin Exp Pathol. 2017;10:9779–85.
Dutta G, Gupta R, Garg M, Singh D, Singh H, Srivastava AK, Jagetia A. Giant parieto-occipital lobe pediatric gliosarcoma: report of a rare entity and review of literature. Surg Neurol Int. 2018;9:111. https://doi.org/10.4103/sni.sni_31_18.
Bouali S, Bahri K, Zehani A, Haj AB, Said IB, Kallel J. Complete surgical resection of a congenital gliosarcoma with long time survival: case report and review of the literature. Human Pathology: Case Reports. 2020;21:200375.
Dogan GM, Sigirci A, Cengiz A, Erbay MF, Gokce H. A case of gliosarcoma in a child with neurofibromatosis type 1. Ann Med Res. 2020;27:2214–7.
Jeng F, Reynolds A. Retrobulbar chlorpromazine injection in a child with gliosarcoma invasion into the orbits. BMJ Case Rep. 2020;13:e233394. https://doi.org/10.1136/bcr-2019-233394.
Bukhari SS, Junaid M, Afzal A, Kulsoom A. Primary pediatric cerebellar gliosarcoma. Surg Neurol Int. 2020;11:96.
Graham RT, Bell EH, Webb A, Zhao Y, Timmers C, Fleming JL, Sells BE, Robison NJ, Palmer JD, Finlay JL, Chakravarti A. Pediatric Gliosarcoma with and without Neurofibromatosis type 1: a whole-exome comparison of 2 patients. J Pediatr Hematol Oncol. 2020;00:000–0.
Swaidan MY, Hussaini M, Sultan I, Mansour A. Radiological findings in gliosarcoma. A single institution experience. Neuroradiol J. 2012;25:173–80.
Romero-Rojas AE, Diaz-Perez JA, Ariza-Serrano LM, Amaro D, Lozano-Castillo A. Primary gliosarcoma of the brain: radiologic and histopathologic features. Neuroradiol J. 2013;26:639–48.
Kepes JJ, Fulling KH, Garcia JH. The clinical significance of "adenoid" formations of neoplastic astrocytes, imitating metastatic carcinoma, in gliosarcomas. A review of five cases. Clin Neuropathol. 1982;1:139–50.
Tada T, Katsuyama T, Aoki T, Kobayashi S, Shigematsu H. Mixed glioblastoma and sarcoma with osteoid-chondral tissue. Clin Neuropathol. 1987;6:160–3.
Mørk SJ, Rubinstein LJ, Kepes JJ, Perentes E, Uphoff DF. Patterns of epithelial metaplasia in malignant gliomas. II Squamous differentiation of epithelial-like formations in gliosarcomas and glioblastomas. J Neuropathol Exp Neurol. 1988;47:101–18.
Hayashi K, Ohara N, Jeon HJ, Akagi S, Takahashi K, Akagi T, Namba S. Gliosarcoma with features of chondroblastic osteosarcoma. Cancer. 1993;72:850–5.
Fukuda T, Yasumichi K, Suzuki T. Immunohistochemistry of gliosarcoma with liposarcomatous differentiation. Pathol Int. 2008;58:396–401.
Actor B, Cobbers JM, Büschges R, Wolter M, Knobbe CB, Lichter P, et al. Comprehensive analysis of genomic alterations in gliosarcoma and its two tissue components. Genes Chromosomes Cancer. 2002;34(4):416–27. https://doi.org/10.1002/gcc.10087.
Smith DR, Wu CC, Saadatmand HJ, Isaacson SR, Cheng SK, Sisti MB, et al. Clinical and molecular characteristics of gliosarcoma and modern prognostic significance relative to conventional glioblastoma. J Neuro-Oncol. 2018 Apr;137(2):303–11. https://doi.org/10.1007/s11060-017-2718-z Epub 2017 Dec 20.
Lowder L, Hauenstein J, Woods A, Chen H-R, Rupji M, Kowalski J, et al. Gliosarcoma: distinct molecular pathways and genomic alterations identified by DNA copy number/SNP microarray analysis. J Neuro-Oncol. 2019;143(3):381–92. https://doi.org/10.1007/s11060-019-03184-1 Epub 2019 May 9.
Castelli J, Feuvret L, Haoming QC, Biau J, Jouglar E, Berger A, et al. Prognostic and therapeutic factors of gliosarcoma from a multi-institutional series. J Neuro-Oncol. 2016;129:85–92.
Acknowledgements
Not applicable.
Funding
No financial support was provided for this study.
Author information
Authors and Affiliations
Contributions
NU and ZA performed the histological and IHC evaluation. HI and SR was involved in literature review and drafted the manuscript; JA-G participated with the corresponding, reviewing, editing the drafted manuscript as per journal policy, and submission of the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed on patient tumor samples in this study were in accordance with the ethical standards of the Institutional Ethics Committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Since this was a retrospective observational study and did not involve actual patients or patient’s images, ethical exemption was obtained from the Aga Khan University Ethical Review Committee (2020–3340-8831). Informed consent was obtained from parents and/or legal guardians for participants who are under age 18.
Consent for publication
Written informed consent was obtained from parents and/or legal guardians for participants who are under age 18.
Competing interests
It is declared that all authors have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Din, N.U., Ishtiaq, H., Rahim, S. et al. Gliosarcoma in patients under 20 years of age. A clinicopathologic study of 11 cases and detailed review of the literature. BMC Pediatr 21, 101 (2021). https://doi.org/10.1186/s12887-021-02556-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-021-02556-9