
Abstract
Background
Epilepsy initiation involves multifactorial etiologies, including genetic susceptibility, structural anomalies, and glial cell dysregulations, particularly in astrocytes. Despite advancements in understanding various factors, the mechanisms of astrocyte dysregulation in epilepsy, critical for neural homeostasis, remain elusive, requiring comprehensive evaluation of molecular pathways and cellular interactions for future targeted interventions.
Methods
A systematic search of PubMed, ScienceDirect, and the Cochrane databases up to January 1st 2024 identified relevant studies predominantly from experimental models, forming the basis for an in-depth analysis of astrocytic contributions to epileptic pathophysiology. The aims, subjects, epilepsy induction techniques, assessment methods, and findings of each studies were presented.
Results
A total of 24 clinical trials met the inclusion criteria and were included in the systematic review. Altered potassium buffering compromises extracellular potassium regulation, fostering hyperexcitability. Aquaporin dysfunction disrupts water homeostasis, aggravating seizure susceptibility. Disturbances in glutamatergic transmission, marked by changes in glutamate transporter function, contribute to excitotoxicity, fueling epileptogenesis. Intricacies in calcium signaling and disruptions in calcium-binding proteins tip intracellular calcium balance towards hyperexcitability. Dysfunctional GABA transporters compromise inhibitory neurotransmission, upsetting excitatory–inhibitory balance. Gap junction protein dysregulation disrupts astroglial networks, impacting neuronal synchronization in epileptogenic circuitry. Compromised BBB allows entry of epileptogenic factors, exacerbating the epileptogenic milieu.
Conclusions
Collectively, these astrocytic dysregulations unveil intricate contributors to epilepsy onset and progression.
Similar content being viewed by others


Introduction
A seizure is a transient and paroxysmal event characterized by abnormal and synchronous neuronal activity in the brain, leading to various manifestations encompassing motor, autonomic, and sensory domains. Motor manifestations may include tonic, clonic, tonic–clonic, or atonic movements, presenting as convulsions or jerking motions. Autonomic manifestations may manifest as changes in heart rate, blood pressure, pupillary size, or gastrointestinal symptoms, reflecting dysregulation of the autonomic nervous system. Sensory manifestations encompass alterations in perception, such as visual disturbances, auditory hallucinations, olfactory sensations, or sensations of tingling, numbness, or abnormal sensations in the skin, reflecting aberrant sensory processing within the brain [1]. A seizure can also cause brief behavioral alterations. In seizures, a few abnormal neurons prompt changes in nearby neurons, leading to progressive synchronization and altered behavior [2]. Epileptogenesis constitutes a multifaceted process encompassing the development, progression, and maintenance of epilepsy, a chronic neurological disorder characterized by recurrent seizures. Epilepsy is increasingly recognized as a brain network disease, implicating aberrant interactions and dysregulation within neural circuits rather than isolated neuronal dysfunction [3]. Epilepsy manifests as paroxysmal, transient, and stereotyped clinical events, comprising idiopathic, symptomatic (secondary or acquired), and cryptogenic forms. Symptomatic epilepsy, the most common type, results from central nervous system lesions or abnormalities, including structural issues or disturbances in brain function [4]. The pooled lifetime prevalence of epilepsy was 7.60 per 1000 persons (95% CI 6.17–9.38), with a higher incidence observed in low/middle-income countries (LMIC) compared to high-income countries (HIC), 139.0 (95% CI 69.4–278.2) and 48.9 (95% CI 39.0–61.1), respectively [5, 6]. The prevalence of active epilepsy exhibited age-related increases, reaching peaks at 5–9 years and beyond 80 years, while global mortality rates for idiopathic epilepsy were 1.74 per 100,000 population for women and 2.09 per 100,000 population for men [7]. The complex nature of epilepsy, marked by transient electrical surges and various diagnostic criteria, underscores the significance of understanding its diverse manifestations.
An unprovoked seizure occurs without identifiable triggers and includes events without recognized causes, as well as those associated with stable or progressive CNS abnormalities [8]. Current evidences show that the initiation of epilepsy is multifactorial, involving genetic susceptibility, structural anomalies, traumatic brain injuries, chemical exposures, hypoxia, infections, metabolic imbalances, immunologic factors, stroke, and glial cells dysregulations among its diverse etiological factors [9]. Glial cells, especially astrocytes, enhance individual neuronal activity through gliotransmission and the tripartite synapse. While they normally play a crucial role in maintaining blood–brain barrier integrity and addressing inflammation and oxidative stress, these functions become impaired in epilepsy [10]. Despite advancements in understanding various etiological factors contributing to epilepsy, the precise mechanisms underlying astrocyte dysregulations remain elusive. One reason is that in humans, chronic temporal lobe epilepsy usually follows a seizure-free latent period that could last years, during which crucial pathophysiological changes occur. Animal models are essential to study this latent phase, vital for understanding astrocytic alterations preceding epileptic neuronal activity [11]. Studies have shown that astrocytes play pivotal roles in ion and water uptake, glucose metabolism, and communication with neurons, making these functions integral to neural homeostasis. Disruptions in these crucial astrocytic activities have been intricately linked to the pathophysiology of epilepsy [12]. Astrocytes play a crucial role in preserving the integrity of the blood–brain barrier and mitigating inflammation and oxidative stress. However, in epilepsy, these functions are compromised [13]. Epileptic conditions lead to disruptions in astrocytic communication through gap junctions, impacting ion and water balance. Activated astrocytes contribute to altered neuronal excitability by decreasing glutamate uptake and increasing adenosine metabolism. Moreover, their heightened adenosine metabolism may play a role in DNA hypermethylation and other epigenetic changes associated with epileptogenesis [10, 13]. In the context of drug-resistant epilepsy (DRE) and refractory status epilepticus (RSE), astrocytic dysfunction assumes heightened significance. Astrocytes play a crucial role in the mechanisms underlying resistance to antiseizure medications (ASMs), contributing to reduced drug efficacy and treatment failure [9]. Their involvement in pharmacoresistance encompasses various mechanisms, such as impaired drug transport across the blood–brain barrier, enhanced drug metabolism, and altered expression of drug targets or efflux transporters [14]. Moreover, astrocytic gliosis and neuroinflammation in DRE and RSE further exacerbate neuronal hyperexcitability and perpetuate seizure activity [14, 15].
As our understanding of the brain’s non-neuronal elements expands, glial cells emerge as central figures in epilepsy pathogenesis [16]. Recognizing their role as key organizers of homeostasis and contributors to inflammation and brain excitability opens avenues for innovative therapeutic approaches [17]. By evaluating the molecular pathways and cellular interactions underlying astrocyte dysregulation, this systematic review aims to provide a comprehensive overview of the current state of knowledge. Ultimately, this synthesis of evidence is crucial for guiding future research directions and developing targeted interventions that may effectively modulate astrocytic function, thereby mitigating the epileptogenic process.
Methods
Study design and inclusion criteria
In strict adherence to the PRISMA guidelines, we conducted a systematic review characterized by high methodological rigor. The study protocol underwent registration and approval in the PROSPERO database (ID: CRD496570) before the commencement of the systematic search. Inclusion criteria were defined to encompass studies addressing dysregulations in astrocytes associated with epilepsy, with no language restrictions imposed. The selected criteria included studies examining both animal and human subjects, evaluating the involvement of astrocyte components in epileptogenesis and their roles in pathological conditions predisposing to epilepsy development. Dysregulations in astrocytes were categorized, with a focus on but not limited to dysfunctions involving potassium buffering, water homeostasis, glutamatergic transmission, calcium signaling and calcium-binding proteins, gamma-aminobutyric acid (GABA) transporter, gap junction proteins, and blood–brain barrier (BBB). Studies concentrating on astrocyte roles in brain diseases unrelated to epileptogenesis or not exploring the molecular mechanisms of seizure onset were excluded.
Literature search and selection
A comprehensive literature search, following the PRISMA flowchart, was conducted on PubMed, ScienceDirect, and the Cochrane databases to identify studies investigating the impact of astrocyte dysfunctions on epileptogenesis and epilepsy-related brain diseases. The search encompassed studies published up to January 1, 2024, without backward limits, employing MeSH terms. To mitigate the potential omission of pertinent studies, the reference lists of included papers and previous reviews on similar topics were manually screened. Duplicate articles were eliminated using Microsoft Excel 16.37 (Redmond, WA, USA). The research strategy relied on the analysis of titles and abstracts. The full text of an article was retrieved if the title and abstract met the inclusion criteria. No automated tools were employed during this phase.
A total of 583 papers were identified, of which 47 underwent full-text screening due to indications in their titles or abstracts suggesting a discussion of astrocyte dysregulations in the pathogenesis of epilepsy. Furthermore, only primary research articles were included, while several reviews were consulted for general information. Among the 23 papers, the majority did not specifically address astrocyte dysregulations but rather focused on therapeutic approaches related to astrocyte regulation or epilepsy in general. Ultimately, only papers explicitly discussing the dysfunctions of astrocyte components in epilepsy or a model thereof were included, amounting to a total of 24 papers (Fig. 1).

PRISMA flowchart
Qualitative data extraction
A comprehensive and systematic data extraction process was diligently executed to acquire comprehensive demographic, baseline clinical, and outcome-related data from the selected studies. This methodological rigor facilitated a nuanced evaluation of the effects of dysregulations in each astrocyte component on the pathogenesis of epilepsy. The categorization of mechanisms of action in astrocyte dysregulation afforded a comprehensive understanding of epilepsy pathophysiology, thereby enhancing the precision and depth of the research findings.
In accordance with the aforementioned criteria, all articles underwent screening and identification by two reviewers. Disagreements were resolved through discussion and consensus, and when discussion failed to lead to consensus, a third researcher mediated. Extracted qualitative data included authorship, publication year, study objectives, and principal findings.
Results
Study selection
From the literature search conducted with the abovementioned queries, 583 articles were identified, which were subsequently reduced to 171 after the removal of duplicates. Following exclusion based on title and abstract, 47 papers were obtained for eligibility assessment. Subsequently, these were screened for relevance, culminating in the inclusion of 24 in accordance with our inclusion criteria and the overarching objective of the review. The full text was accessible for all 24 included studies [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41], all of which were incorporated into our qualitative analysis (Fig. 1).
Findings of included studies
The synthesized evidence from diverse studies presents a comprehensive landscape of astrocytic dysregulations implicated in epileptogenesis (Table 1). These findings collectively showcase the intricate interplay between astrocyte dysfunction and key molecular components, shedding light on potential mechanisms underpinning the development and progression of epilepsy. Three studies [18,19,20] demonstrated that increased potassium levels lead to frequency-dependent synaptic facilitation, contributing to neuronal hyperexcitability. In addition, the downregulation of astrocytic inward rectifier potassium channels 4.1 (Kir4.1) channel in the Leucine-Rich Glioma-Inactivated 1 (Lgi1)-related seizure model suggests a functional impairment, potentially heightening seizure susceptibility by compromising astrocytes’ ability to maintain extracellular homeostasis. In addition, four studies [21,22,23,24] found that downregulation of Aquaporin 4 (AQP4) disrupts astrocytic homeostasis, potentially leading to powerful epileptogenic and cognitive effects. AQP4 plays a protective role in post-traumatic seizures by promoting astrogliosis and preventing microgliosis, with dysregulation involving specific upregulation and mislocalization in post-traumatic epilepsy. Three studies [25,26,27] showed that Glutamate Transporter 1 (GLT-1) dysfunction, characterized by impaired clearance of synaptic glutamate in astrocytes, fosters epileptogenesis by elevating extracellular glutamate levels. This disruption leads to heightened neuronal excitability, synchronization, and increased susceptibility to seizures. Furthermore, four studies [28,29,30,31] suggest that dysregulation in Ca2+ signaling contributes to epilepsy by disrupting the delicate balance of intracellular calcium concentrations, leading to abnormal neuronal excitability and synaptic transmission. The involvement of calcium-binding proteins such as S-100B further exacerbates epileptogenesis by influencing processes, such as neuroinflammation, oxidative stress, and neuronal plasticity. Three studies [32, 34, 35] showed that dysregulation of the GABA transporter 1 (GAT-1) in epilepsy leads to impaired GABAergic synaptic transmission, resulting in reduced inhibitory control and heightened neuronal excitability. Four studies [36,37,38,39] found that dysregulation of gap junction protein Connexin 43 (Cx43) in astrocytes induces a breakdown in intercellular communication, leading to impaired astrocytic network coordination. This disruption in astrocyte coupling hinders the efficient spread of potassium ions and other signaling molecules, contributing to an imbalance in extracellular homeostasis and increased neuronal hyperexcitability, ultimately promoting epileptogenesis. Finally, three studies [33, 40, 41] showed that impaired BBB integrity allows the infiltration of pro-inflammatory molecules and immune cells into the brain parenchyma, fostering a neuroinflammatory environment. Concurrently, dysregulated transforming growth factor β receptor (TGF-βR) signaling disrupts the astrocytic response to inflammation, compromising their neuroprotective functions and exacerbating neuronal hyperexcitability, thereby contributing to the onset and progression of epilepsy. Each facet of astrocytic dysfunction contributes synergistically to the heightened neuronal excitability characteristic of epileptogenesis. These studies underscore the integral role of astrocytes in the pathophysiology of epilepsy.
Discussion
The following discussion section systematically organizes clinical studies focused on various dysregulations within astrocytes, covering a spectrum of dysfunctions. This categorization prioritizes critical aspects, including potassium buffering, water homeostasis, glutamatergic transmission, calcium signaling, calcium-binding proteins, GABA transporter, gap junction proteins, and BBB integrity. This structured approach aims to provide a thorough analysis of the intricate involvement of astrocytes in epileptogenesis. Each category serves as a discrete analytical framework, elucidating the impact of astrocytic dysregulation on epilepsy, elucidating potential mechanisms, pathways, and therapeutic targets associated with each facet (Fig. 2).

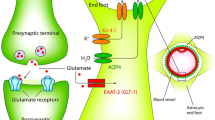
Astrocytes fulfill diverse functions crucial for neuronal homeostasis. A In the context of potassium buffering, astrocytes employ Kir4.1 channels to facilitate the entry of K+ released during neuronal activity, promoting its distribution into capillaries. B Addressing water homeostasis, the astrocytic water channel Aquaporin-4 (AQP4) orchestrates water flow between the extracellular space and the blood, maintaining osmotic balance. C In glutamatergic transmission, astrocytes utilize glutamate transporters (GLT-1 or GLAST) to uptake glutamate, subsequently converting it to glutamine, a process critical for neurotransmission. D In calcium signaling, voltage-gated channels and Ca2+ waves stimulate gliotransmitter release from astrocytes, influencing neuronal excitability. E Gamma-aminobutyric acid (GABA) transporter GAT-1 facilitates the removal of GABA from the synaptic cleft, impacting synaptic transmission. F Astrocytic gap junction proteins, such as Cx43, enable intercellular communication, contributing to spatial buffering of ions. G Astrocytes play a role in the blood–brain barrier (BBB) integrity, where disruptions, as seen in neuroinflammatory conditions, activate microglia and lead to neuroinflammation
Potassium buffering
Potassium buffering, a crucial homeostatic function performed by astrocytes, is integral to the regulation of extracellular potassium concentrations within the central nervous system. Under normal physiological conditions, neurons generate action potentials, leading to potassium efflux into the extracellular space. Astrocytes actively participate in maintaining the ionic balance by swiftly taking up excess potassium ions through various mechanisms [42]. This intricate process involves the activity of Kir channels, particularly Kir4.1, abundantly expressed on astrocytic endfeet. Kir4.1 channels enable astrocytes to effectively clear extracellular potassium, preventing its accumulation and ensuring a stable neuronal environment [43].
These channels play a crucial role in regulating the brain's extracellular potassium ([K+]o) and water fluxes. During physiological activation and repetitive activation, nonhomogeneous increases in [K+]o occur, particularly in specific cortical layers, such as the pyramidal layer of Ammon's horn in the hippocampus or deep layers in the neocortex. The localized increase in [K+]o depolarizes astrocytes, leading to spatial buffering of [K+]o [44, 45]. Dysregulation in astrocytic Kir4.1 channels has been implicated in epileptogenesis. Studies suggest that downregulation or functional impairment of Kir4.1 channels results in decreased potassium buffering capacity, leading to elevated extracellular potassium levels [46]. This heightened potassium concentration in the extracellular milieu has profound effects on neuronal excitability, potentially contributing to the generation and propagation of epileptic events. Elevated extracellular K+ concentration ([K+]o), resulting from defective K+ regulation, is strongly associated with seizure initiation during hypersynchronous neuronal activities when [K+]o reaches peaks of 10–12 mM [47].
Astrocytic Kir4.1 channel dysfunction, induced by KCNJ10 gene mutations or downregulation, elevates extracellular potassium and glutamate levels, triggering neuronal hyperexcitation in epileptogenesis [48]. Research involving glial-specific conditional Kir4.1 KO mice confirmed disrupted K+ homeostasis, offering additional mechanistic evidence supporting the role of abnormal Kir4.1 expression and function in the epileptic phenotype [49, 50]. Linkage analysis conducted in patients presenting with seizures, ataxia, sensorineural deafness, mental retardation, and electrolytic imbalance (SeSAME or EAST syndrome) implicated KCNJ10. Sequencing of the affected KCNJ10 gene in individuals with SeSAME or EAST syndrome revealed loss-of-function mutations within the channel, and subsequent heterologous expression experiments confirmed the functional impact of these mutations on Kir4.1, leading to depolarization [51, 52]. The compromised potassium buffering capacity of astrocytes may induce frequency-dependent synaptic facilitation. The aberrant accumulation of potassium, particularly in the context of epileptic events characterized by increased neuronal activity, may enhance synaptic transmission [53]. This heightened synaptic facilitation, dependent on N-methyl-d-aspartate (NMDA) receptor activity, further contributes to neuronal hyperexcitability and network synchronization. Consequently, the dysregulation of potassium buffering in astrocytes emerges as a pivotal factor in the pathophysiological cascade leading to epileptogenesis [53, 54].
Water hemostasis
Dysregulation of water homeostasis, specifically involving AQP4, constitutes a pivotal aspect of astrocyte dysfunction in the epileptogenic process. AQP4, a water channel protein predominantly expressed in astrocytes, plays a fundamental role in maintaining water balance within the central nervous system [55]. In normal physiological conditions, AQP4 facilitates the movement of water across astrocytic membranes, ensuring the precise regulation of extracellular fluid volume. This process is particularly critical in the context of ion and water homeostasis within the brain, where astrocytes actively participate in the intricate maintenance of the microenvironment [56]. During epileptogenesis, dysregulation of AQP4 emerges as a significant contributor to pathological alterations in water homeostasis. Experimental studies have demonstrated that changes in AQP4 expression and function are associated with epileptic activity [21,22,23,24]. The sensitivity of neuronal excitability to osmolarity and changes in the extracellular space is manifested through reductions in the extracellular space, resulting in increased concentrations of extracellular ions and neurotransmitters, thereby intensifying ephaptic interactions among closely interacting neurons and fostering heightened synchronous firing and bursting activity, contributing significantly to the complexities of epileptogenesis [57, 58].
Roles in the etiology of water homeostasis dysfunction in astrocytes encompass AQP4 misexpression, AQP4 mislocalization, dysregulation of AQP4 isoforms, loss of AQP4 polarity, and inadequacy in phosphorylation of AQP4 [59]. Following systemic kainic acid-induced status epilepticus in rats, hippocampal AQP4 expression was found to be mislocalized, with reduced density in the adluminal endfeet of astrocytes during the latent phase before chronic epileptic seizures [60]. In a mouse model of post-traumatic epilepsy, AQP4 subcellular redistribution was observed [24]. Furthermore, the pivotal protein in anchoring AQP4 to perivascular end feet astrocytes is dystrophin, a component of the dystrophin-associated protein complex (DAPC) [61]. This complex interacts with AQP4 via α-syntrophin. The anchoring system is vital for AQP4’s physiological role in fluid circulation and ion homeostasis between blood and brain tissue [62]. Through the deletion of α-syntrophin, a considerable and consistently ranging proportion (79–94%) of the perivascular AQP4 pool is eliminated and could precedes the occurrence of chronic seizures [59, 63]. In addition, dysregulation of AQP4 phosphorylation, triggered by CaM activation and PKA-mediated phosphorylation at S276, plays a crucial role in epileptogenesis. The resulting AQP4 translocation to the plasma membrane contributes to seizure initiation, presenting a promising avenue for therapeutic intervention in epilepsy [64].
Glutamatergic transmission
Astrocytes play a pivotal role in maintaining glutamatergic homeostasis within the central nervous system, primarily through the glutamate transporters GLAST (Glutamate Aspartate Transporter) and GLT-1. GLAST and GLT-1, localized on astrocytic processes ensheathing synapses, function to efficiently clear the neurotransmitter glutamate from the extracellular space, preventing excitotoxicity [65]. Dysregulation of these transporters can lead to aberrant glutamate levels, thereby influencing neuronal excitability and contributing to the pathogenesis of epilepsy. In the intrahippocampal kainic acid model of temporal lobe epilepsy, an early upregulation of astrocyte glutamate transporters GLT-1 and GLAST occurs, implying their potential involvement in epilepsy development [66]. Downregulation or impaired function of these transporters results in compromised glutamate clearance, leading to an accumulation of glutamate. Elevated synaptic glutamate concentrations induce overstimulation of postsynaptic glutamate receptors, precipitating hyperexcitability and even excitotoxic neuronal death [67]. Thus, alterations in GLAST and GLT-1 expression, function, or localization can disrupt the delicate balance of glutamatergic neurotransmission.
GLT-1, an Na+-dependent transmembrane symporter, serves as the predominant astrocytic glutamate transporter in the adult human brain, responsible for more than 90% of synaptic glutamate clearance and exhibiting expression levels surpassing GLAST by four to six times in astrocytes [68, 69]. It plays a pivotal role in maintaining the synaptic glutamate gradient, and its dysregulation has been associated with excitotoxicity, neuronal death, and neurological disorders, as evidenced by studies on GLT-1 knockout mice experiencing lethal spontaneous seizures and significant neuronal loss, while functional GLT-1 prevented post-traumatic seizures in a rat traumatic brain injury (TBI) model [70,71,72]. Various factors, such as genetic predisposition, trauma, or inflammation, may instigate changes in glutamate transporter activity. Jen and colleagues demonstrated that a heterozygous mutation in GLT-1 resulted in reduced glutamate uptake, contributing to neuronal hyperexcitability and leading to manifestations, such as seizures, hemiplegia, and episodic ataxia [73].
Moreover, dysfunction in astrocytic glutamate transporters can impact synaptic plasticity and long-term potentiation, processes crucial for normal neuronal function [74]. Dysregulated glutamate transport may contribute to hyperexcitability and the generation of abnormal neuronal circuits, fostering a conducive environment for the initiation and propagation of seizures [75]. Glutamate transporters tightly control synaptic transmission, influencing long-term plasticity by regulating the spatiotemporal profile of glutamate transients and potentially determining the sensitivity of synapses to various plasticity paradigms [76]. In addition, the altered expression or function of GLAST and GLT-1 in epilepsy may not only affect synaptic transmission but also influence the surrounding microenvironment. The intricate interplay between astrocytic glutamate transporters and neuronal activity underscores their significance in epileptogenesis.
Ceftriaxone, functioning as a transcriptional activator, has demonstrated the capacity to enhance GLT-1 expression during the initial stages of epileptogenesis. This effect may potentially ameliorate cognitive impairments associated with epilepsy by addressing the deficit in glutamate uptake [27, 77]. Although, administration of ceftriaxone has demonstrated negative effects on hippocampal synaptic plasticity and memory recognition [78]. A study conducted by Sha and colleagues found that inhibition of Hsp90 increases GLT-1 levels by disrupting the association between Hsp90β and GLT-1, preventing GLT-1 degradation and suggesting a potential therapeutic target for epilepsy and excitotoxicity through up-regulation of GLT-1 in reactive astrocytes [79].
Ca2+ signaling and calcium binding protein
The dysregulation of Ca2+ signaling and calcium-binding proteins represents a pivotal aspect of astrocyte dysfunction in the pathogenesis of epileptogenesis. Ca2+ serve as crucial intracellular messengers, participating in diverse cellular processes, including neurotransmitter release, gene expression, and modulation of astrocytic function [80, 81]. In the context of astrocyte dysfunction in epileptogenesis, aberrations in Ca2+ signaling are integral to the intricate interplay of cellular events leading to the development and perpetuation of epilepsy [82]. Astrocytes actively engage in bidirectional communication with neurons, and alterations in Ca2+ dynamics profoundly influence this intercellular signaling [83]. Calcium waves within astrocytic networks contribute to the regulation of neuronal activity, with disturbances in these waves being implicated in epileptic pathophysiology [84]. Intracellular Ca2+ oscillations in astrocytes are tightly regulated by various mechanisms, including purinergic signaling and release from intracellular stores, such as the endoplasmic reticulum [85]. In various neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and epilepsy, calcium ion (Ca2+) serves as a crucial second messenger, influencing neuronal excitability. The interplay of voltage-dependent calcium channels (VDCCs), intracellular calcium-binding proteins, and calcium channels within intracellular stores contributes to epileptogenesis [86].
Furthermore, calcium-binding proteins play a pivotal role in shaping astrocytic responses to changes in Ca2+ levels. These proteins, such as calmodulin and S100B, exert modulatory effects on intracellular signaling cascades [87]. S100B exerts its effects on astrocyte function through interactions with various cellular targets. S100B, featuring a helix–loop–helix structure and a calcium-binding domain, plays a crucial role in neurological disorders by activating the MAPK pathway, inducing increased NF-kB expression, and influencing cellular processes, such as survival, proliferation, and gene up-regulation [88]. Notably, S100B modulates intracellular calcium levels, acting as both a sensor and effector in calcium-mediated signaling pathways within astrocytes [89]. Dysregulation of calcium-binding proteins in astrocytes has been associated with altered synaptic transmission, compromised neuronal homeostasis, and heightened susceptibility to seizures [31]. Moreover, S100B is intricately linked to neuroinflammatory processes, further exacerbating astrocyte dysfunction in epilepsy [90]. The release of S100B from astrocytes into the extracellular space can activate microglia and perpetuate a proinflammatory milieu. This inflammatory cascade, coupled with disruptions in astrocytic calcium homeostasis mediated by S100B, creates a conducive environment for epileptogenic changes within the neural circuitry [91, 92]. In the epileptogenic milieu, the dysregulation of S100B extends beyond its role in calcium signaling and inflammation. S100B has been implicated in gliosis, contributing to the reactive astrocytic phenotype observed in epilepsy [93, 94]. The sustained activation of astrocytes, characterized by altered morphology and function, is a hallmark of epileptogenic processes, and S100B appears to play a modulatory role in this regard.
GABA transporter
GABA, the primary inhibitory neurotransmitter, modulates excitatory neurotransmission upon release from interneurons. Astrocytic glutamine is transported to GABAergic neurons, where it undergoes conversion to glutamate and then promptly to GABA via glutamate decarboxylase (GAD) [95]. GABA, crucially reliant on the tricarboxylic acid (TCA) cycle intermediaries, faces deficits in transmitter production during cellular energy metabolism inefficiencies, often observed after compromised tissue perfusion and heightened neuronal metabolic demand [96]. Presynaptically stored GABA is released onto various postsynaptic terminals, and the majority is reclaimed by its transporter, GAT-1, into the presynaptic neuron for recycling into vesicles. Under normal conditions, GAT-1 is responsible for the majority of GABA reuptake in astrocytes, preventing excessive accumulation in the synaptic cleft and maintaining inhibitory tone. However, in epileptogenesis, alterations in the expression and function of GAT-1 lead to impaired GABAergic neurotransmission [97]. This dysfunction may manifest as reduced GABA uptake, resulting in prolonged GABAergic signaling and increased susceptibility to seizures [98, 99]. GABA transporter dysregulation, encompassing various isoforms, such as GAT-1, plays a pivotal role in the complex process of astrocyte dysfunction during epileptogenesis [99]. GABA, a major inhibitory neurotransmitter in the central nervous system, is actively reuptaken by astrocytes through GABA transporters, ensuring precise regulation of its extracellular concentrations [100]. The dysregulation of GABA transporters, notably GAT-1, disrupts this delicate balance, contributing to aberrant inhibitory signaling within neuronal networks [101]. Moreover, dysregulated GABA transporters influence the availability of GABA for extrasynaptic signaling, impacting tonic inhibition. The heightened tonic inhibition stems from compromised GABA uptake by the GABA transporter GAT-1 in the tested genetic models, playing a crucial role in seizure genesis [102]. The altered GABAergic tone can contribute to hyperexcitability in neuronal circuits, fostering a pro-epileptic environment. The intricate interplay between GABA transporters and the homeostatic control of inhibitory neurotransmission underscores their significance in the epileptogenic process.
Gap junction protein
Gap junction protein dysregulation, particularly involving Cx43, stands as a pivotal factor in the intricate process of astrocyte dysfunction contributing to epileptogenesis [103]. Astrocytes, integral to maintaining neuronal homeostasis, express Cx43, forming gap junctions that facilitate direct intercellular communication [103]. In epileptic conditions, alterations in Cx43 expression and function disrupt normal signaling cascades between astrocytes, compromising their regulatory roles [104]. Astrocytic Cx channels have been implicated in epilepsy, particularly in the sclerotic hippocampus of temporal lobe epilepsy (TLE) patients, where astrocytic gap junction (GJ) coupling is lost despite sustained expression of Cx isoforms [105]. Dysregulated Cx43 can result from genetic mutations, aberrant post-translational modifications, or environmental triggers, precipitating an array of pathological consequences [106, 107]. The dysregulation of Cx43 in astrocytes significantly impacts potassium buffering, a crucial function in the prevention of extracellular potassium accumulation [44]. Disrupted gap junctional communication due to Cx43 abnormalities compromises the spatial buffering capacity of astrocytes, leading to elevated extracellular potassium concentrations [44, 108]. This heightened potassium environment fosters neuronal hyperexcitability and synchronous firing, which are hallmark features of epileptic activity. Moreover, dysregulated Cx43 contributes to altered calcium wave propagation among astrocytes, influencing neurotransmitter release and perpetuating the epileptic milieu [109].
Astrocytes, with their extensive network, play a pivotal role in immune response modulation. Cx43 dysregulation further intertwines with neuroinflammation, a prominent aspect of epileptogenesis [110]. Dysfunctional Cx43 hampers astrocytic coordination in responding to inflammatory cues, potentially exacerbating neuroinflammatory processes [111]. In addition, impaired Cx43-mediated gap junctions hinder the formation of astrocytic scar tissue, crucial for containing aberrant neuronal activity [112, 113]. This deficiency may contribute to a persistent pro-epileptic microenvironment. Beyond localized effects, Cx43 dysregulation extends its impact to long-range network dynamics. Altered astrocytic connectivity disrupts the synchronization of neural networks, fostering hypersynchrony associated with epileptic seizures [36]. Compromised gap junctions may impede the propagation of antiepileptic signals, further tipping the balance towards hyperexcitability [114]. The intricate interplay of Cx43 dysregulation in both local and network-level astrocytic functions underscores its multifaceted role in the pathogenesis of epilepsy.
BBB and TGF-βR
BBB dysfunction and dysregulation of TGF-βR signaling contribute significantly to astrocyte dysfunction in epileptogenesis. The BBB, a dynamic interface between the blood and the CNS, maintains homeostasis by selectively restricting the passage of substances [115]. In epilepsy, compromised BBB integrity leads to the infiltration of blood-derived factors into the brain parenchyma [116]. This breach triggers a cascade of events, including activation of astrocytes, which respond to the altered microenvironment. Upon exposure to blood-derived factors, astrocytes undergo phenotypic changes characterized by increased reactivity and altered expression of various transporters and receptors [117, 118]. Notably, dysregulated TGF-βR signaling is implicated in these astrocytic alterations [33, 40]. TGF-βR modulates astrocyte function in response to BBB disruption. Dysfunctional TGF-βR signaling exacerbates astrocyte reactivity, contributing to a pro-inflammatory environment conducive to epileptogenesis [119]. This altered astrocytic state, in turn, can further compromise BBB integrity, creating a positive feedback loop [40, 120]. This altered state contributes to network hyperexcitability, excitatory–inhibitory (E/I) imbalance, and cognitive deficits, with implications for synaptic remodeling and increased expression of c1q, a complement protein associated with synapse elimination [121,122,123,124]. In addition, TGF-βR dysregulation may influence the expression of tight junction proteins at the BBB, exacerbating permeability changes and allowing increased entry of inflammatory mediators [125].
Several limitations must be acknowledged in the scope of this comprehensive review. First, the majority of evidence is derived from experimental models, primarily rodents, and translation to human epileptogenesis necessitates caution. Human astrocytic heterogeneity, particularly in pathological conditions, adds complexity, and the extent to which findings from animal models accurately reflect human astrocyte behavior remains a subject of ongoing investigation. Furthermore, the diversity of epilepsy etiologies and patient populations introduces variability that challenges the generalizability of specific astrocytic dysregulations across different forms of epilepsy. In addition, the interdependence of various astrocytic functions and the intricate crosstalk with other cell types in the brain necessitate further elucidation. The evolving landscape of astrocyte research might bring forth new insights beyond the current scope, prompting continual reassessment and refinement of our understanding.
Conclusions
Astrocytic dysfunctions in epilepsy encompass disruptions in potassium buffering, water homeostasis, glutamatergic transmission, calcium signaling, calcium-binding proteins, GABA transporters, gap junction proteins, and BBB integrity. These disruptions collectively contribute to epileptogenesis by fostering a hyperexcitable environment. Dysregulated potassium buffering and impaired water homeostasis exacerbate seizure susceptibility, while altered glutamatergic transmission and calcium signaling promote excitotoxicity and hyperexcitability. Dysfunctional GABA transporters disrupt inhibitory neurotransmission, and gap junction protein dysregulation impacts neuronal synchronization, contributing to epileptogenic circuitry. Compromised BBB integrity allows entry of epileptogenic factors into the brain parenchyma. A comprehensive understanding of these astrocytic dysfunctions is essential for unraveling epilepsy’s complex pathogenesis, emphasizing the need for holistic approaches, particularly in human studies, to show their interplay effectively.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- AQP4:
-
Aquaporin 4
- BBB:
-
Blood–brain barrier
- Cx43:
-
Connexin 43
- GABA:
-
Gamma-aminobutyric acid
- GLAST:
-
Glutamate aspartate transporter
- GLT-1:
-
Glutamate transporter 1
- Kir4.1:
-
Astrocytic Inward Rectifier Potassium Channels 4.1
- TGF-βR:
-
Transforming growth factor β receptor
References
Sarmast ST, Abdullahi AM, Jahan N. Current classification of seizures and epilepsies: scope, limitations and recommendations for future action. Cureus. 2020;12(9): e10549.
Milligan TA. Epilepsy: a clinical overview. Am J Med. 2021;134(7):840–7. https://doi.org/10.1016/j.amjmed.2021.01.038.
Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5(7):380–91.
Ding Y, Cheng X. Analysis of etiology and clinical characteristics of 1170 patients with symptomatic epilepsy in Jianghan plain. Yangtze Med. 2020;04:132–9.
Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, Dykeman J, et al. Prevalence and incidence of epilepsy: a systematic review and meta-analysis of international studies. Neurology. 2017;88(3):296–303.
Beghi E, Hesdorffer D. Prevalence of epilepsy–an unknown quantity. Epilepsia. 2014;55(7):963–7.
Beghi E, Giussani G, Nichols E, Abd-Allah F, Abdela J, Abdelalim A, et al. Global, regional, and national burden of epilepsy, 1990–2013;2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(4):357–75. https://doi.org/10.1016/S1474-4422(18)30454-X.
Hauser WA, Beghi E. First seizure definitions and worldwide incidence and mortality. Epilepsia. 2008;49(Suppl 1):8–12.
Bazhanova ED, Kozlov AA, Litovchenko AV. Mechanisms of drug resistance in the pathogenesis of epilepsy: role of neuroinflammation. A literature review. Brain Sci. 2021;11(5):663.
Purnell BS, Alves M, Boison D. Astrocyte-neuron circuits in epilepsy. Neurobiol Dis. 2023;179: 106058.
Henning L, Unichenko P, Bedner P, Steinhäuser C, Henneberger C. Overview article astrocytes as initiators of epilepsy. Neurochem Res. 2023;48(4):1091–9. https://doi.org/10.1007/s11064-022-03773-z.
Heuser K, Szokol K, Taubøll E. The role of glial cells in epilepsy. Tidsskr den Nor laegeforening Tidsskr Prakt Med ny raekke. 2014;134(1):37–41.
Hayatdavoudi P, Hosseini M, Hajali V, Hosseini A, Rajabian A. The role of astrocytes in epileptic disorders. Physiol Rep. 2022;10(6): e15239. https://doi.org/10.14814/phy2.15239.
Fattorusso A, Matricardi S, Mencaroni E, Dell’Isola GB, Di Cara G, Striano P, et al. The pharmacoresistant epilepsy: an overview on existant and new emerging therapies. Front Neurol. 2021;12: 674483.
Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, et al. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci Off J Soc Neurosci. 2015;35(8):3330–45.
Sumadewi KT, Harkitasari S, Tjandra DC. Biomolecular mechanisms of epileptic seizures and epilepsy: a review. Acta Epileptol. 2023;5(1):28. https://doi.org/10.1186/s42494-023-00137-0.
Heuser K, de Curtis M, Steinhäuser C. Editorial: glial dysfunction in epileptogenesis. Front Neurol. 2021. https://doi.org/10.3389/fneur.2021.716308.
David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, et al. Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci Off J Soc Neurosci. 2009;29(34):10588–99.
Kinboshi M, Shimizu S, Mashimo T, Serikawa T, Ito H, Ikeda A, et al. Down-regulation of astrocytic Kir4.1 channels during the audiogenic epileptogenesis in leucine-rich glioma-inactivated 1 (Lgi1) mutant rats. Int J Mol Sci. 2019;20(5):1013.
Méndez-González MP, Rivera-Aponte DE, Benedikt J, Maldonado-Martínez G, Tejeda-Bayron F, Skatchkov SN, et al. Downregulation of astrocytic Kir4.1 potassium channels is associated with hippocampal neuronal hyperexcitability in type 2 diabetic mice. Brain Sci. 2020;10:72.
Hubbard JA, Szu JI, Yonan JM, Binder DK. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp Neurol. 2016;283:85–96.
Heuser K, Nagelhus EA, Taubøll E, Indahl U, Berg PR, Lien S, et al. Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res. 2010;88(1):55–64.
Lu DC, Zador Z, Yao J, Fazlollahi F, Manley GT. Aquaporin-4 reduces post-traumatic seizure susceptibility by promoting astrocytic glial scar formation in mice. J Neurotrauma. 2011;38(8):1193–201. https://doi.org/10.1089/neu.2011.2114.
Szu JI, Chaturvedi S, Patel DD, Binder DK. Aquaporin-4 dysregulation in a controlled cortical impact injury model of posttraumatic epilepsy. Neuroscience. 2020;428:140–53. https://doi.org/10.1016/j.neuroscience.2019.12.006.
Sun D, Tan ZB, Sun XD, Liu ZP, Chen WB, Milibari L, et al. Hippocampal astrocytic neogenin regulating glutamate uptake, a critical pathway for preventing epileptic response. Proc Natl Acad Sci U S A. 2021;118(16): e2022921118.
Peterson AR, Garcia TA, Cullion K, Tiwari-Woodruff SK, Pedapati EV, Binder DK. Targeted overexpression of glutamate transporter-1 reduces seizures and attenuates pathological changes in a mouse model of epilepsy. Neurobiol Dis. 2021;157: 105443.
Ramandi D, Elahdadi Salmani M, Moghimi A, Lashkarbolouki T, Fereidoni M. Pharmacological upregulation of GLT-1 alleviates the cognitive impairments in the animal model of temporal lobe epilepsy. PLoS ONE. 2021;16(1): e0246068.
Zhang C, Tabatabaei M, Bélanger S, Girouard H, Moeini M, Lu X, et al. Astrocytic endfoot Ca(2+) correlates with parenchymal vessel responses during 4-AP induced epilepsy: an in vivo two-photon lifetime microscopy study. J Cereb blood flow Metab Off J Int Soc Cereb Blood Flow Metab. 2019;39(2):260–71.
Umpierre AD, West PJ, White JA, Wilcox KS. Conditional knock-out of mGluR5 from astrocytes during epilepsy development impairs high-frequency glutamate uptake. J Neurosci Off J Soc Neurosci. 2019;39(4):727–42.
Szokol K, Heuser K, Tang W, Jensen V, Enger R, Bedner P, et al. Augmentation of Ca(2+) signaling in astrocytic endfeet in the latent phase of temporal lobe epilepsy. Front Cell Neurosci. 2015;9:49.
Khamis M, El DNS, Nada MA, Afifi HEDM. Serum protein S-100B as a novel biomarker of diagnosis and prognosis of childhood epilepsy. Egypt J Neurol Psychiatry Neurosurg. 2023;59(1):19. https://doi.org/10.1186/s41983-023-00605-x.
Mermer F, Poliquin S, Zhou S, Wang X, Ding Y, Yin F, et al. Astrocytic GABA transporter 1 deficit in novel SLC6A1 variants mediated epilepsy: connected from protein destabilization to seizures in mice and humans. Neurobiol Dis. 2022;172: 105810.
Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, et al. TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain. 2007;130(2):535–47. https://doi.org/10.1093/brain/awl317.
Mazaud D, Kottler B, Gonçalves-Pimentel C, Proelss S, Tüchler N, Deneubourg C, et al. Transcriptional regulation of the glutamate/GABA/glutamine cycle in adult glia controls motor activity and seizures in drosophila. J Neurosci Off J Soc Neurosci. 2019;39(27):5269–83.
Pirttimaki T, Parri HR, Crunelli V. Astrocytic GABA transporter GAT-1 dysfunction in experimental absence seizures. J Physiol. 2013;591(4):823–33.
Kékesi O, Ioja E, Szabó Z, Kardos J, Héja L. Recurrent seizure-like events are associated with coupled astroglial synchronization. Front Cell Neurosci. 2015;9:215.
Yoon JJ, Green CR, O’Carroll SJ, Nicholson LFB. Dose-dependent protective effect of connexin43 mimetic peptide against neurodegeneration in an ex vivo model of epileptiform lesion. Epilepsy Res. 2010;92(2):153–62.
Deshpande T, Li T, Henning L, Wu Z, Müller J, Seifert G, et al. Constitutive deletion of astrocytic connexins aggravates kainate-induced epilepsy. Glia. 2020;68(10):2136–47.
Volnova A, Tsytsarev V, Ganina O, Vélez-Crespo GE, Alves JM, Ignashchenkova A, et al. The anti-epileptic effects of carbenoxolone in vitro and in vivo. Int J Mol Sci. 2022;23(2):663.
Bar-Klein G, Cacheaux LP, Kamintsky L, Prager O, Weissberg I, Schoknecht K, et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann Neurol. 2014;75(6):864–75.
Prager O, Kamintsky L, Hasam-Henderson LA, Schoknecht K, Wuntke V, Papageorgiou I, et al. Seizure-induced microvascular injury is associated with impaired neurovascular coupling and blood-brain barrier dysfunction. Epilepsia. 2019;60(2):322–36.
McNeill J, Rudyk C, Hildebrand ME, Salmaso N. Ion channels and electrophysiological properties of astrocytes: implications for emergent stimulation technologies. Front Cell Neurosci. 2021;15: 644126.
Seifert G, Henneberger C, Steinhäuser C. Diversity of astrocyte potassium channels: an update. Brain Res Bull. 2018;136:26–36.
Wallraff A, Köhling R, Heinemann U, Theis M, Willecke K, Steinhäuser C. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci Off J Soc Neurosci. 2006;26(20):5438–47.
Sibille J, Dao Duc K, Holcman D, Rouach N. The neuroglial potassium cycle during neurotransmission: role of Kir4.1 channels. PLoS Comput Biol. 2015;11(3): e1004137. https://doi.org/10.1371/journal.pcbi.1004137.
Li X, Lv J, Li J, Ren X. Kir4.1 may represent a novel therapeutic target for diabetic retinopathy (Review). Exp Ther Med. 2021;22(3):1021. https://doi.org/10.3892/etm.2021.10453.
Wang F, Qi X, Zhang J, Huang JH. Astrocytic modulation of potassium under seizures. Neural Regen Res. 2020;15(6):980–7.
Kinboshi M, Ikeda A, Ohno Y. Role of astrocytic inwardly rectifying potassium (Kir) 4.1 channels in epileptogenesis. Front Neurol. 2020. https://doi.org/10.3389/fneur.2020.626658.
Chever O, Djukic B, McCarthy KD, Amzica F. Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J Neurosci Off J Soc Neurosci. 2010;30(47):15769–77.
Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, et al. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia. 2011;59(11):1635–42.
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009;360(19):1960–70.
Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U S A. 2010;107(32):14490–5.
Bellot-Saez A, Kékesi O, Morley JW, Buskila Y. Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci Biobehav Rev. 2017;77:87–97.
Ohno Y, Kunisawa N, Shimizu S. Emerging roles of astrocyte Kir4.1 channels in the pathogenesis and treatment of brain diseases. Int J Mol Sci. 2021;22(19):10236.
Hubbard JA, Hsu MS, Seldin MM, Binder DK. Expression of the astrocyte water channel aquaporin-4 in the mouse brain. ASN Neuro. 2015. https://doi.org/10.1177/1759091415605486.
Salman MM, Kitchen P, Halsey A, Wang MX, Törnroth-Horsefield S, Conner AC, et al. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain. 2022;145(1):64–75.
Schwartzkroin PA, Baraban SC, Hochman DW. Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res. 1998;32(1–2):275–85.
Andrew RD, Fagan M, Ballyk BA, Rosen AS. Seizure susceptibility and the osmotic state. Brain Res. 1989;498(1):175–80.
Szu JI, Binder DK. Mechanisms underlying aquaporin-4 subcellular mislocalization in epilepsy. Front Cell Neurosci. 2022;16: 900588.
Alvestad S, Hammer J, Hoddevik EH, Skare Ø, Sonnewald U, Amiry-Moghaddam M, et al. Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res. 2013;105(1–2):30–41.
Skauli N, Savchenko E, Ottersen OP, Roybon L, Amiry-Moghaddam M. Canonical bone morphogenetic protein signaling regulates expression of aquaporin-4 and its anchoring complex in mouse astrocytes. Front Cell Neurosci. 2022. https://doi.org/10.3389/fncel.2022.878154.
Belmaati Cherkaoui M, Vacca O, Izabelle C, Boulay AC, Boulogne C, Gillet C, et al. Dp71 contribution to the molecular scaffold anchoring aquaporine-4 channels in brain macroglial cells. Glia. 2021;69(4):954–70. https://doi.org/10.1002/glia.23941.
Hoddevik EH, Khan FH, Rahmani S, Ottersen OP, Boldt HB, Amiry-Moghaddam M. Factors determining the density of AQP4 water channel molecules at the brain–blood interface. Brain Struct Funct. 2017;222(4):1753–66. https://doi.org/10.1007/s00429-016-1305-y.
Kitchen P, Salman MM, Halsey AM, Clarke-Bland C, MacDonald JA, Ishida H, et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell. 2020;181(4):784-799.e19.
Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells. 2019;8(2):184.
Peterson AR, Binder DK. Regulation of synaptosomal GLT-1 and GLAST during epileptogenesis. Neuroscience. 2019;411:185–201.
Karki P, Hong P, Johnson JJ, Pajarillo E, Son DS, Aschner M, et al. Arundic acid increases expression and function of astrocytic glutamate transporter EAAT1 Via the ERK, Akt, and NF-κB pathways. Mol Neurobiol. 2018;55(6):5031–46.
Rao P, Yallapu MM, Sari Y, Fisher PB, Kumar S. Designing novel nanoformulations targeting glutamate transporter excitatory amino acid transporter 2: implications in treating drug addiction. J Pers nanomedicine. 2015;1(1):3–9.
Pajarillo E, Rizor A, Lee J, Aschner M, Lee E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology. 2019;161: 107559.
Rao VL, Başkaya MK, Doğan A, Rothstein JD, Dempsey RJ. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem. 1998;70(5):2020–7.
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276(5319):1699–702.
Karki P, Lee E, Aschner M. Manganese neurotoxicity: a focus on glutamate transporters. Ann Occup Environ Med. 2013;25(1):4.
Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65(4):529–34.
Barnes JR, Mukherjee B, Rogers BC, Nafar F, Gosse M, Parsons MP. The relationship between glutamate dynamics and activity-dependent synaptic plasticity. J Neurosci Off J Soc Neurosci. 2020;40(14):2793–807.
Barker-Haliski M, White HS. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med. 2015;5(8): a022863.
Valtcheva S, Venance L. Control of long-term plasticity by glutamate transporters. Front Synaptic Neurosci. 2019. https://doi.org/10.3389/fnsyn.2019.00010.
Hussein AM, Ghalwash M, Magdy K, Abulseoud OA. Beta lactams antibiotic ceftriaxone modulates seizures, oxidative stress and connexin 43 expression in hippocampus of pentylenetetrazole kindled rats. J Epilepsy Res. 2016;6(1):8–15. https://doi.org/10.14581/jer.16002.
Matos-Ocasio F, Hernández-López A, Thompson KJ. Ceftriaxone, a GLT-1 transporter activator, disrupts hippocampal learning in rats. Pharmacol Biochem Behav. 2014;122:118–21.
Sha L, Wang X, Li J, Shi X, Wu L, Shen Y, et al. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J Exp Med. 2016;214(2):547–63. https://doi.org/10.1084/jem.20160667.
Torres A, Wang F, Xu Q, Fujita T, Dobrowolski R, Willecke K, et al. Extracellular Ca2+ acts as a mediator of communication from neurons to glia. Sci Signal. 2012;5(208):ra8.
de Liyis BG, Tandy SG, Endira JF, Putri KA, Utami DKI. Anti-high mobility group box protein 1 monoclonal antibody downregulating P-glycoprotein as novel epilepsy therapeutics. Egypt J Neurol psychiatry Neurosurg. 2022;58(1):121.
Heuser K, Nome CG, Pettersen KH, Åbjørsbråten KS, Jensen V, Tang W, et al. Ca2+ signals in astrocytes facilitate spread of epileptiform activity. Cereb Cortex. 2018;28(11):4036–48.
MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol. 2015;7(5): a020388.
Kumaria A, Tolias CM, Burnstock G. ATP signalling in epilepsy. Purinergic Signal. 2008;4(4):339–46.
Verkhratsky A, Trebak M, Perocchi F, Khananshvili D, Sekler I. Crosslink between calcium and sodium signalling. Exp Physiol. 2018;103(2):157–69.
Xu JH, Tang FR. Voltage-dependent calcium channels, calcium binding proteins, and their interaction in the pathological process of epilepsy. Int J Mol Sci. 2018;19(9):2735.
Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med. 2013;13(1):24–57.
Langeh U, Singh S. Targeting S100B protein as a surrogate biomarker and its role in various neurological disorders. Curr Neuropharmacol. 2021;19(2):265–77.
Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, et al. S100B’s double life: intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009;1793(6):1008–22.
Michetti F, Clementi ME, Di Liddo R, Valeriani F, Ria F, Rende M, et al. The S100B protein: a multifaceted pathogenic factor more than a biomarker. Int J Mol Sci. 2023;24(11):9605.
Bianchi R, Giambanco I, Donato R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging. 2010;31(4):665–77.
Bianchi R, Adami C, Giambanco I, Donato R. S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol. 2007;81:108–18.
Brozzi F, Arcuri C, Giambanco I, Donato R. S100B protein regulates astrocyte shape and migration via interaction with Src kinase: implications for astrocyte development, activation, and tumor growth. J Biol Chem. 2009;284(13):8797–811.
Villarreal A, Seoane R, González Torres A, Rosciszewski G, Angelo MF, Rossi A, et al. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: implications for its role in the propagation of reactive gliosis. J Neurochem. 2014;131(2):190–205.
Walls AB, Waagepetersen HS, Bak LK, Schousboe A, Sonnewald U. The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res. 2015;40(2):402–9.
Wu X, Fu Y, Knott G, Lu J, Di Cristo G, Huang ZJ. GABA signaling promotes synapse elimination and axon pruning in developing cortical inhibitory interneurons. J Neurosci Off J Soc Neurosci. 2012;32(1):331–43.
Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60(8):1215–26. https://doi.org/10.1002/glia.22341.
Khazipov R. GABAergic synchronization in epilepsy. Cold Spring Harb Perspect Med. 2016;6(2): a022764.
Ishibashi M, Egawa K, Fukuda A. Diverse actions of astrocytes in GABAergic signaling. Int J Mol Sci. 2019;20:2964.
Müller J, Timmermann A, Henning L, Müller H, Steinhäuser C, Bedner P. Astrocytic GABA accumulation in experimental temporal lobe epilepsy. Front Neurol. 2020. https://doi.org/10.3389/fneur.2020.614923.
Cherubini E, Di Cristo G, Avoli M. Dysregulation of GABAergic signaling in neurodevelomental disorders: targeting cation-chloride co-transporters to re-establish a proper E/I balance. Front Cell Neurosci. 2022. https://doi.org/10.3389/fncel.2021.813441.
Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15(12):1392–8.
Xing L, Yang T, Cui S, Chen G. Connexin hemichannels in astrocytes: role in CNS disorders. Front Mol Neurosci. 2019. https://doi.org/10.3389/fnmol.2019.00023.
Walrave L, Vinken M, Leybaert L, Smolders I. Astrocytic Connexin43 channels as candidate targets in epilepsy treatment. Biomolecules. 2020;10(11):1578.
Bedner P, Steinhäuser C. Role of impaired astrocyte gap junction coupling in epileptogenesis. Cells. 2023;12(12):1669.
Aasen T, Johnstone S, Vidal-Brime L, Lynn KS, Koval M. Connexins: synthesis, post-translational modifications, and trafficking in health and disease. Int J Mol Sci. 2018;19(5):1296.
Leithe E, Mesnil M, Aasen T. The connexin 43 C-terminus: a tail of many tales. Biochim Biophys Acta - Biomembr. 2018;1860(1):48–64.
Xu L, Zeng LH, Wong M. Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2009;34(2):291–9.
Çarçak N, Onat F, Sitnikova E. Astrocytes as a target for therapeutic strategies in epilepsy: current insights. Front Mol Neurosci. 2023. https://doi.org/10.3389/fnmol.2023.1183775.
Yin X, Feng L, Ma D, Yin P, Wang X, Hou S, et al. Roles of astrocytic connexin-43, hemichannels, and gap junctions in oxygen-glucose deprivation/reperfusion injury induced neuroinflammation and the possible regulatory mechanisms of salvianolic acid B and carbenoxolone. J Neuroinflamm. 2018;15(1):97.
Boal AM, Risner ML, Cooper ML, Wareham LK, Calkins DJ. Astrocyte networks as therapeutic targets in glaucomatous neurodegeneration. Cells. 2021;10(6):1368.
De Bock M, Decrock E, Wang N, Bol M, Vinken M, Bultynck G, et al. The dual face of connexin-based astroglial Ca2+ communication: a key player in brain physiology and a prime target in pathology. Biochim Biophys Acta - Mol Cell Res. 2014;1843(10):2211–32.
Mylvaganam S, Ramani M, Krawczyk M, Carlen PL. Roles of gap junctions, connexins, and pannexins in epilepsy. Front Physiol. 2014;5:172.
Volman V, Perc M, Bazhenov M. Gap junctions and epileptic seizures–two sides of the same coin? PLoS ONE. 2011;6(5): e20572.
Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1): a020412.
Marchi N, Granata T, Ghosh C, Janigro D. Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia. 2012;53(11):1877–86.
Khakh BS, Sofroniew MV. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 2015;18(7):942–52.
Price BR, Johnson LA, Norris CM. Reactive astrocytes: the nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res Rev. 2021;68: 101335.
Luo J. TGF-β as a key modulator of astrocyte reactivity: disease relevance and therapeutic implications. Biomedicines. 2022;10(5):1206.
Senatorov VVJ, Friedman AR, Milikovsky DZ, Ofer J, Saar-Ashkenazy R, Charbash A, et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci Transl Med. 2019;11(521):8283.
Preininger MK, Kaufer D. Blood-brain barrier dysfunction and astrocyte senescence as reciprocal drivers of neuropathology in aging. Int J Mol Sci. 2022;23(11):6217.
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78.
Weissberg I, Wood L, Kamintsky L, Vazquez O, Milikovsky DZ, Alexander A, et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-β/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol Dis. 2015;78:115–25.
Holmes GL. Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Disord. 2015;17(2):101–16.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–96.
Acknowledgements
We thank every party involved in the making of this manuscript. We would like to thank the Doctoral Study Program, Faculty of Medicine, Universitas Udayana for their support. The final text has been read by all of the writers and have all given their consent publication. All figures are original.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
The initial concept for this literature review was hatched by KTS. The text was written by BGL, and KTS with guidance from INMA, IPEW, and NML. KTS, BDL, INMA, IPEW and NML completed, copyedited and revised the manuscript. All authors assisted in reviewing, composing the manuscript, creating the figures and reviewing the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sumadewi, K.T., de Liyis, B.G., Linawati, N.M. et al. Astrocyte dysregulation as an epileptogenic factor: a systematic review. Egypt J Neurol Psychiatry Neurosurg 60, 69 (2024). https://doi.org/10.1186/s41983-024-00843-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41983-024-00843-7