
Abstract
Houttuynia cordata Thunb (Family: Saururaceae) is well known as a traditional medicine plant and widely spread in China, Japan and India. It had been reported to have potential functions such as anti-bacterial, anti-cancer, and anti-inflammatory in human. Recently, it has been reported that the plant-derived microRNA(miRNA) possibly transported from one species to another and exerted a cross kingdom regulation, and miRNA has been considered as medicinal ingredents in herbs. However, knowledge is still rare about miRNAs in H. cordata. In this study we identified 163 conserved miRNAs and 30 novel miRNAs by high-throughput sequencing, and then randomly selected miRNA’s expression trend was identified by q-PCR, which was consistent with the sequencing result. Further bioinformatics analyses showed that the targets of H. cordata miRNAs were enriched in endocrine and other factor-regulated calcium reabsorption pathways, melanogenesis, insulin signaling pathway, and aldosterone -regulated sodium reabsorption pathways. These results will be helpful to understand new active components in H. cordata as food and traditional Chinese medicine. This study is the first report of miRNAs in H. cordata, and provides valuable data for further understanding the cross kingdom function of active components in H.cordata.
Similar content being viewed by others


Background
Houttuynia cordata Thunb (H.cordata), is a perennial native medicinal plant widely used in folk medicine in Japan, Korea, China and Southeast Asia [1]. Traditionally, H.cordata was used as a folk medicine for diuresis, anti-viral [2], anti-oxidant [3,4,5], anti-bacterial [6], anti-inflammatory [7,8,9], and anti-obesity [10, 11]. In 2014, Hyun Kang reported that H.cordata extract (HCE) significantly attenuated lipid accumulation in human HepG2 hepatocytes, and the hypolipidemic effects of HCE were induced by activating AMPK signaling which then inhibits lipid biosynthesis [12].
MicroRNAs (miRNAs) are a class small non-coding RNAs, which are 18–24 nucleotide in length and inhibit gene expression by mRNA cleavage or translation repression in the 3′ untranslated region (3’UTR) [13]. It is pervasive in human and plants, which have been acknowledge to play key roles in apoptosis, and developmental pattern [14, 15]. Recent studies suggested that the regulatory role of microRNAs is not only in the intracellular level, but also in the intercellular level, even in an inter-species manner [16, 17]. For instance, in 2012, plant microRNAs were discovered in serum and tissues of human and other animals [18]. Further studies showed these plant microRNAs were absorbed from food; one that was relatively high in serum, miR168a, directly targeted low-density lipoprotein receptor adaptor protein 1 (LDLRAP1) in liver cells and decreased the clearance of LDL from the blood. In 2015, Zhen et al. found plant miR2911 (Lonicera japonica, Honeysuckle) could directly target influenza A virus to inhibit H5N1 and H7N9 viral replication [19]. Moreover, diet-derived miR159 inhibited breast cancer cell proliferation via targeting transcription factor 7 (TCF7) [20]. Increasing evidence indicated that the mammalian digestive tract had the potential ability to absorb the plant miRNAs which were able to target mammalian genes and act as a biologically active molecules mediating cross-kingdom regulation [21,22,23].
Although it is well known that H. cordata has a wide range of biological activities, miRNAs of H.cordata still remain unknown. In the present study, we used high-throughput sequencing and qPCR, and identified conserved and novel miRNAs in H.cordata, and further analysed the miRNAs functions in H.cordata via bioinformatics analysis. This is the first reporte about miRNAs of H.cordata and will provide foundation for further understanding of active components in herbs.
Results
Construction of the H.cordata small RNA library by high-throughput sequencing
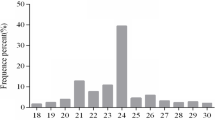
To identify the miRNAs in H. cordata, a small RNA library from commercial H. cordata was constructed and analyzed by high-throughput sequencing. After filtering low quality sequence and removal of adaptor sequences and contamination form raw data, a total of 7,713,807 clean reads in 18–30 nt length were collected for further study (Additional file 1: Table S1). Among them, the length distribution peaked at 28 nt in length (13.03%) (Fig. 1). All these 7,713,807 clean reads represented 2,105,956 unique reads. Next, 7,713,807 clean reads were mapped to H. cordata transcriptomic data (SRR7413372) using bowtie without mismatch. Among 7,713,807 clean reads, 3,728,613 (48.34%) reads were successfully aligned to the transcriptomic data. Subsequently, the matched unique sRNA were classified into different ncRNA categories by comparing to Rfam database (version 13.0). rRNA, snRNA, snoRNA and tRNA were abandoned and the remaining 3, 271,119 reads were subjected to further analysis (Additional file 1: Table S2).

Length distribution of small RNA. Sequence length distributions of small RNAs in H.cordata. Among these sequences 28 nt was the most abundant, accounting for 13.02% of the total reads
Statistics of conserved miRNAs in H.cordata
After comparing the remained 3,271,119 reads to miRBase database (version 21), and conjunction with the modified software miRDeep2 and sRNA-tools for the potential miRNAs and secondary structures analysis, about 163 conserved mature miRNAs were retained for their precursors’ hairpin structure (Additional file 1: Table S3). As shown in Fig. 2a, miR159a, miR166u, miR166a-3p, miR166h-3p, miR166e and other 10 miRNAs were highly expressed in H.cordata (reads number > 1000).

Characterization of conserved miRNAs in H.cordata. a Expression profile of conserved miRNAs. b Number of miRNAs length distribution and expression levels of conserved miRNAs with H.cordata. c First nucleotide bias for the first position of 18–30 nt miRNAs. Nucleotide U predominates. d Expression profile of novel miRNAs in H.cordata (sequence frequencies over 100). e The selected miRNAs’ expression by qPCR, novel-19 as control and taken as 1
The length of the identified conserved miRNAs ranges from 19 to 24 nt. Among them, the sequence of 21–22 nt dominated in the number of miRNAs with 91.8% and the sequencing frequency of miRNAs with 88.9% (Fig. 2b), which is consistent with the main length distribution [24]. It has been reported that uracil was the dominant nucleotide at the 5′ terminus, and mostly in 20–23 [25]. It is shown that the first position of mature miRNA sequence is uracil (Fig. 2c), which was consistent with previous reports [26, 27].
Statistics of novel miRNAs in H.cordata
The hairpin structure of miRNA precursor is a vital measure to predict novel miRNAs. The novel miRNAs were predicted by using the software programs miREvo and miRDeep2. In this way, we identified 30 novel miRNAs by deducing their characteristic hairpin secondary structures (Additional file 2: Figure S1). These 30 novel miRNAs come from 37 pre-miRNAs. As shown in Fig. 2d and Additional file 1: Table S4 in supporting information, only four novel miRNAs’ read frequencies are over 100 (novel-1, novel-11, novel-5, novel-6). We also investigated the length distribution of novel mature miRNAs, and showed the 20–24 nt was the major length and 21 nt possess a priority percentage (86.7%). Although the expression levels of novel miRNAs are lower than the conserved miRNAs, their specific functions may not be ignored.
Validation of conserved miRNAs and novel miRNAs in H.cordata
After a series of analyses of RNA sequencing, we randomly selected eleven miRNAs and then evaluated their expression in the H.cordata by qPCR. As shown in Fig. 2e, miR159 was the most highly expressed among selected miRNAs, followed by miR-166u, novel-5, miR166a-3p, miR166h-3p, miR166e, miR159b-3p, novel-6, novel-12, novel-59, and novel-19. The trend of selected miRNAs was inconsistent with the Illumina sequencing result. To determine whether those novel miRNAs are highly expressed in H.cordata, three plant materials sorghum (Sorghum bicolor), soybean (Glycine max) and maize (Zea mays) were tested for comparison. As shown in Fig. 3a and b, those miRNAs were highly expressed in H.cordata rather than other species.

characterization of novel miRNAs and RT-qPCR analysis of miRNAs in H.cordata. a The ct values of novel miRNAs in H.cordata, sorghum, zea maize and soybean. b Agarose gel electrophoresis of novel miRNAs (novel-5, novel-6, novel-9, novel-12); Lanes 1, 5, 9, 13 represented H.cordata; Lanes 2, 6, 10, 14 represented sorghum; Lanes 3, 7, 11, 15 represented maize; Lanes 4, 8, 12, 16 represented soybean
Bioinformatics analysis
The human gene database was applied for predicting targets of conserved and candidate novel miRNAs (reads> 1000). The criterion of H.cordata miRNAs’ binding sites in human mRNA 3′ UTR region was used for target prediction via miRanda. As shown in Additional file 3: Table S6, highly expressed conserved miRNAs were predicted to 2900 target genes. and novel miRNAs were predicted to target 1145 genes.
To futher understand roles of miRNAs in H.cordata, a total of 3263 target genes were conducted GO and KEGG analyses. The top 30 enriched terms were shown in Fig. 4a and Additional file 4: Table S7. The predicted targets were classified into membrane-bounded organelle (or intracellular membrane-bounded organelle, 23.07%), cytoplasm (or cytoplasmic part, 19.24%), intracellular (or intracellular part, 27.26%), organelle (or intracellular organelle, 26.41%), cell (or cell part, 30.99%) and endomembrane system (4.22%), which would participated in regulation of cell communication, signaling, and localization, organic substance transport, cellular, single-organism cellular relative biological processes, and most of them exerted protein binding and bonding molecular functions. KEGG showed that all the targets were enriched in 277 categorize and 14 pathways were markedly enriched. The top 20 enriched pathways were in refer to multifarious substance reabsorption (endocrine and other factor-regulated calcium reabsorption, aldosterone-regulated sodium reabsorption), signaling pathway (cAMP signaling, prolaction signaling, thyroid hormone), glycolipid metabolism (insulin singaling, insulin secretion) and other disease (melanogenesis, ptoteoglycans cancer, colorectal cancer) pathways (Fig. 4b). Interestingly, we obtained melanogenesis and insulin relative pathways, which were consistent with the previous report that extract of H.cordata was involved in human melanoma cells with antiproliferative and pro-apoptosis activity [28], and anti-obesity via suppressing fatty acid uptake [11].

GO and KEGG pathway analysis of target genes. a the most enriched GO term in human. The x-axis shows the number of these target genes, the y-aixs shows the diverse biological functions. GO classification based on the target genes by miRNA over 1000 reads. b KEGG pathway analysis of target genes. The top 30 significant enriched pathways based on KEGG analysis
Discussion
Some studies have shown that diet-derived miRNAs can transport into the mammalian circulatory system via the gastrointestinal tract [20,21,22, 29, 30]. As reported by Chin et al., plant-derived miR159 entered into and inhibited breast cancer cells proliferation by targeting TCF7. Meaniwhile, we found the H.cordata has highly expressed miR159a (19,870 reads), and its predicted target is TCF7. Interestingly, Zhang’s group firstly found rice derived-miR168a with high level in serum and inhibited LDLRAP1 expression. Subsequently, they found that miR2911 (special nucleotide character with high GC content) inhibited influenza viruses and protect mice from influenza [18, 19]. Although the mechanism of the plant-derived miRNA absorption is still not clear, miRNAs may play crucial roles in affecting the absorbability of exogenous miRNAs. Whether miR159a of H.cordata shared the same mechanism to be absorbed and furtherly carry out functions need more expremental evidences.
As a traditional folk medicine, H.cordata is widely used in inflammation, pneumonia, cancer, anti-obesity, dysentery, enteritis and fever. Its bioactive components, including quercetin, afzelin, chlorogenic acid and rutin are present in H.cordata [31, 32]. Active ingredients, especially its miRNAs, responsible for its various effects as well as other beneficial applications continue to be identified. The present study identified miRNAs in H.cordata and predicted their target genes’ functions in the human. The result shows that many target genes are enriched in melanogenesis and insulin signaling by using KOBAS software. Melanoma is a dangerous skin cancer in the world [33], and with melanogenesis dysfuction, the excessive accumulation of melanin would cause diversified diseases such as malaise and cancer [34]. As shown in the Mongkol’s study, H.cordata extract could induce programmed cell death of the malignant melanoma cell line (A375) [28]. And microphthalamia-associated transcription factor(MITF) was involved in the expression and transport of melanosome component proteins [35, 36]. miRNAs revealed in H.cordata might be involved in melanogenesis via wnt/β-catenin signal pathways, and glycogen synthase kinase 3 beta (GSK3β) is an essential enzyme in the transcriptional and expression of melanogenic enzyme proteins, which inhibits the expression of MITF [37]. Interestingly, GSK3β in this study is the predicted target of miR159a, miR159b-3p, miR396a-5p, miR396g-3p and miR396h, so we deduce those miRNAs in H.cordata possibly regulate GSK3β and participate in melanogenesis.
Insulin signaling pathway involves many connected network cascades. Commonly, the actions of insulin are mediated via its receptors, followed by autophosphorylation of receptor by binding recruit insulin receptor substrates, and subsequently activate downstream pathways such as PI3K-Akt [38]. Insulin resistance is the disorder of insulin, and cause the type-2 diabetes T2D [39]. In our result, some miRNAs are predicted to target genes (IRS1, PIK3R3, PIK3CA, PIK3R2, PDPK1, AKT3, PPARGC1A, G6PC, G6PC3, FASN, and PRKAB2) (Fig. 5) involved ininsulin signaling pathway. IRS1 was found to attenuate insulin resistance [40]. The PIK3R2, PIK3R3 encode p85β and p85γ, isoforms of the p85 regulatory subunits [41], which regulate insulin signaling by generating PIP3 [42]. And FASN wasshown to connect with metabolic alterations in human such as insulin resistance and obesity [43]. In addition, H.cordata was reported to perform protective effects on mouse which consumed a high fat diet has been reported [11, 44]. As reported by Lin et al. and Miyata et al. studies, H.cordata aqueous extract significantly decreased epididymal fat, hepatic TC and TG via reducing hepatic activity of malic enzyme, fatty acid synthase (FAS). Furthermore, Kang’s group found that H.cordata ethyl acetate extract inhibited hepatic lipids accumulation via the activation of AMPK signaling, and inhibiting expression of FASN and SREBP-1c [12, 45]. The high-fat diet finally caused lipid accumulation and insulin resistance [46]. As shown in Additional file 1: Table S8, a total of 45 pedicted target genes were involved in insulin signaling pathways. FASN and PRKAB2 are putative targets of miR166e, miR159a, miR159a.1, miR159b, miR159b-3p and novel-6. Those results indicate that miRNAs in H.cordata may improve insulin resistance via promoting PRKAB2 and suppressing FAS expression, and they may be active components inH.cordata. For anti-obesity functions. However, the concrete mechanism of this process need more experimental evidences.

The miRNA-gene network. Relative gene of insulin signaling pathway, the green color is target gene, and bule is miRNA of H.cordata
Conclusions
In summary, we firstly identified 163 conserved miRNAs and 30 novel miRNAs from plant H.cordata, and predicted their potential target genes in human by bioinformatics analysis. It provides new information which may lead to better understanding of the H.cordata regulation pathways in human health and diseases as food and medical herb.
Methods
Plant materials
The mature H.cordata were collected from the supermarket, South China Agricultural University. The fresh root was immediately frozen in liquid nitrogen and stored at − 80 °C.
RNA extraction and library construction and RNA-Seq
Total RNA was isolated using cetyltrimethyl ammonium bromide (CTAB) as previously described [47]. In brief, a) 100-200 mg samples put into 1.5 mL tubes including 0.9 mL extarction buffer at 65 °C with 10 min. b) The mixture was centrifuged at 9000 rpm/min for 5 min at 4 °C. The supernatant was transferred to a new tube and 1/3 volume ph 3.5 KAC was added to it. Freeze it 30 min, and an equal volume of chloroform: isoamyl alcohol(24:1) was added to the homogenate and vortexing, which were centrifuged at 12,000 rpm/min 5 min. c) The supernatant was transferred to a new tube and an equal volume of phenol water(ph < 5.2) was then added to tube, which were centrifuged at 12,000 rpm/min 5 min. d) The supernatant was added an equal volume of isopropanol each tube. e) The deposit was RNA.
The Nano Photometer® spectrophotometer (IMPLEN, CA, USA) and 1% agarose gels were to check the quality of RNA. A total amount of 3 μg total RNA per sample was used as input material for the small RNA library. Sequencing libraries were generated using NEBNext® following manufacturer’s protocol. Briefly, after ligation 3′ and 5′ adapters to their bother ends. DNA fragments corresponding to 140~160 bp were recovered and dissolved in 8 μL elution buffer, and the cluster were applied by TruSeq SR Cluster Kit v3-cBot-HS (Illumia). At last, 50 bp single-end reads were generated by Illumina Hiseq™ 2000.
Identification of conserved and novel miRNAs
All small RNA tags were mapped to known database to identified known miRNAs. miRbase (version. 21) was used as reference, the known miRNAs and the secondary structures were obtained by software miRDeep2 [48] and srna-tools-cl. The base bias on the first position of identified konwn miRNA was also analysed. The characteristics of hairpin structures of miRNA precursors can be used to predict novel miRNAs [49]. The remaining small RNA in the former step were analyzed via intergrating miREvo [50] and miRDeep2 to predict novel miRNAs based on calculating the secondary structures, the Dicer cleavage sites and the minimum free energy (less than − 18 kcal/mol). A simple schematic description of bioinformatics analysis is shown in Additional file 5: Figure S2.
Human target gene prediction for H.cordata miRNAs
The miRNAs of H.cordata were used for human target prediction. MiRanda was employed to predict putative target genes, and then the target genes were mapped to GO and KEGG database to predicted their biological functions in the species.
Validation of miRNAs by stem-loop qRT-PCR
We randomly selected eleven (including five novel miRNAs and six conserved miRNAs)miRNAs for stem-loop qRT-PCR identification. The primers for PCR were listed in Additional file 1: Table S5. The stem-loop qRT-PCR steps as previously described [51]. The novel-19 miRNA was used as the internal control miRNA. The reverse primer for miRNAs was the Uni-miR qPCR Primer offered by the kit One Step PrimeScript miRNA cDNA Synthesis Kit (TaKaRa, Dalian). All reactions were performed in triplicate for each sample. Total RNA was exracted from sorghum, maize and soybean, respectively. PCR analysis was carried out accoding to described above.
GO and KEGG enrichment analysis and Cytoscape network construction
All the candidate target genes were used for Gene Ontology (GO) enrichment analysis. GOseq was implemented for GO enrichment analysis [52]. We used KOBAS (http://kobas.cbi.pku.edu.cn) software to test the statistical enrichment of the target gene candidates in KEGG pathways [53]. The relative correlations among miRNAs and miRNA-target genes in H.cordata, Cytoscape networks were constructed by the Cytoscape 3.6.1.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CTAB:
-
Cetyltrimethyl ammonium bromide
- FAS:
-
Fatty acid synthase
- GO:
-
Gene ontology
- GSK3β:
-
Glycogen synthase kinase 3 beta
- H. cordata :
-
Houttuynia cordata Thunb
- HCE:
-
Houttuynia cordata Thunb extract
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LDLRAP1:
-
Low-density lipoprotein receptor adaptor protein 1
- miRNAs:
-
Micrornas
- qRT-PCR:
-
quantitative real time RT-PCR
- SREBP-1c:
-
Sterol regulatory element binding protein-1c
- TCF7:
-
Transcription factor 7
References
Lu HM, Liang YZ, Yi LZ, Wu XJ. Anti-inflammatory effect of Houttuynia cordata injection. J Ethnopharmacol. 2006;104(1–2):245–9. https://doi.org/10.1016/j.jep.2005.09.012 PubMed PMID: 16213118.
Lai KC, Chiu YJ, Tang YJ, Lin KL, Chiang JH, Jiang YL, et al. Houttuynia cordata Thunb extract inhibits cell growth and induces apoptosis in human primary colorectal cancer cells. Anticancer Res. 2010;30(9):3549–56 PubMed PMID: 20944136.
Kumar M, Prasad SK, Krishnamurthy S, Hemalatha S. Antihyperglycemic activity of Houttuynia cordata Thunb. In Streptozotocin-induced diabetic rats. Adv Pharmacol Sci. 2014;2014:809438. https://doi.org/10.1155/2014/809438 PubMed PMID: 24707284; PubMed Central PMCID: PMCPMC3953599.
Lee SB, Kang JW, Kim SJ, Ahn J, Kim J, Lee SM. Afzelin ameliorates D-galactosamine and lipopolysaccharide-induced fulminant hepatic failure by modulating mitochondrial quality control and dynamics. Br J Pharmacol. 2017;174(2):195–209. https://doi.org/10.1111/bph.13669 PubMed PMID: 27861739; PubMed Central PMCID: PMCPMC5192940.
Yang UJ, Maeng H, Park TS, Shim SM. Houttuynia cordata extract improves physical endurance performance by regulating endothelial production of nitric oxide. J Med Food. 2015;18(9):1022–31. https://doi.org/10.1089/jmf.2014.3371 PubMed PMID: 25923355.
Verma RS, Joshi N, Padalia RC, Singh VR, Goswami P, Kumar A, Iqbal H, Verma RK, Chanda D, Chauhan A, Saikia D. Chemical Composition and Allelopathic, Antibacterial, Antifungal, and Antiacetylcholinesterase Activity of Fish‐mint (Houttuynia cordataThunb.) from India. Chemistry & Biodiversity. 2017;14(10):e1700189. https://doi.org/10.1002/cbdv.201700189.
Xu YY, Zhang YY, Ou YY, Lu XX, Pan LY, Li H, et al. Houttuyniacordata Thunb. Polysaccharides ameliorates lipopolysaccharide-induced acute lung injury in mice. J Ethnopharmacol. 2015;173:81–90. https://doi.org/10.1016/j.jep.2015.07.015 PubMed PMID: 26190353.
Shin S, Joo SS, Jeon JH, Park D, Jang MJ, Kim TO, et al. Anti-inflammatory effects of a Houttuynia cordata supercritical extract. J Vet Sci. 2010;11(3):273–5 PubMed PMID: 20706037; PubMed Central PMCID: PMCPMC2924491.
Kim D, Park D, Kyung J, Yang YH, Choi EK, Lee YB, et al. Anti-inflammatory effects of Houttuynia cordata supercritical extract in carrageenan-air pouch inflammation model. Lab Anim Res. 2012;28(2):137–40. https://doi.org/10.5625/lar.2012.28.2.137 PubMed PMID: 22787488; PubMed Central PMCID: PMCPMC3389838.
Park HS, Shim SM. Preventive responses to a standardized extract of Houttuynia cordata supplemented diet in obesity induced by a high-fat diet in mice. J Korean Soc Appl Biol Chem. 2015;58(5):643–9. https://doi.org/10.1007/s13765-015-0087-x PubMed PMID: WOS:000360374100003.
Miyata M, Koyama T, Yazawa K. Water extract of Houttuynia cordata Thunb. Leaves exerts anti-obesity effects by inhibiting fatty acid and glycerol absorption. J Nutr Sci Vitaminol (Tokyo). 2010;56(2):150–6 PubMed PMID: 20495298.
Kang H, Koppula S. Houttuynia cordata attenuates lipid accumulation via activation of AMP-activated protein kinase signaling pathway in HepG2 cells. Am J Chin Med. 2014;42(3):651–64. https://doi.org/10.1142/S0192415X14500426 PubMed PMID: 24871657.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33. https://doi.org/10.1016/j.cell.2009.01.002 PubMed PMID: 19167326; PubMed Central PMCID: PMCPMC3794896.
Park H, Huang X, Lu C, Cairo MS, Zhou X. MicroRNA-146a and microRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J Biol Chem. 2015;290(5):2831–41. https://doi.org/10.1074/jbc.M114.591420 PubMed PMID: 25505246; PubMed Central PMCID: PMCPMC4317016.
Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17(4):196–203. https://doi.org/10.1016/j.tplants.2012.01.010 PubMed PMID: 22365280.
Liang H, Zen K, Zhang J, Zhang CY, Chen X. New roles for microRNAs in cross-species communication. RNA Biol. 2013;10(3):367–70. https://doi.org/10.4161/rna.23663 Epub 2013/02/01, PubMed PMID: 23364352; PubMed Central PMCID: PMCPMC3672279.
Chen X, Liang H, Zhang J, Zen K, Zhang CY. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22(3):125–32. https://doi.org/10.1016/j.tcb.2011.12.001 PubMed PMID: 22260888.
Zhang L, Hou D, Chen X, Li D, Zhu L, Zhang Y, et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res. 2012;22(1):107–26. https://doi.org/10.1038/cr.2011.158 Epub 2011/09/21, PubMed PMID: 21931358; PubMed Central PMCID: PMCPMC3351925.
Zhou Z, Li X, Liu J, Dong L, Chen Q, Liu J, et al. Honeysuckle-encoded atypical microRNA2911 directly targets influenza a viruses. Cell Res. 2015;25(1):39–49. https://doi.org/10.1038/cr.2014.130 PubMed PMID: 25287280; PubMed Central PMCID: PMCPMC4650580.
Chin AR, Fong MY, Somlo G, Wu J, Swiderski P, Wu X, et al. Cross-kingdom inhibition of breast cancer growth by plant miR159. Cell Res. 2016;26(2):217–28. https://doi.org/10.1038/cr.2016.13 PubMed PMID: 26794868; PubMed Central PMCID: PMCPMC4746606.
Yang J, Farmer LM, Agyekum AA, Elbaz-Younes I, Hirschi KD. Detection of an abundant plant-based small RNA in healthy consumers. PLoS One. 2015;10(9):e0137516. https://doi.org/10.1371/journal.pone.0137516 PubMed PMID: 26335106; PubMed Central PMCID: PMCPMC4559308.
Mlotshwa S, Pruss GJ, MacArthur JL, Endres MW, Davis C, Hofseth LJ, et al. A novel chemopreventive strategy based on therapeutic microRNAs produced in plants. Cell Res. 2015;25(4):521–4. https://doi.org/10.1038/cr.2015.25 PubMed PMID: PMC4387556.
Liang H, Zhang S, Fu Z, Wang Y, Wang N, Liu Y, et al. Effective detection and quantification of dietetically absorbed plant microRNAs in human plasma. J Nutr Biochem. 2015;26(5):505–12. https://doi.org/10.1016/j.jnutbio.2014.12.002 PubMed PMID: 25704478.
Gao F, Nan F, Feng J, Lv J, Liu Q, Xie S. Identification of conserved and novel microRNAs in Porphyridium purpureum via deep sequencing and bioinformatics. BMC Genomics. 2016;17(1):612. https://doi.org/10.1186/s12864-016-2985-7 PubMed PMID: 27516065; PubMed Central PMCID: PMCPMC4981961.
Bartel B, Bartel DP. MicroRNAs: at the root of plant development? Plant Physiol. 2003;132(2):709–17. https://doi.org/10.1104/pp.103.023630 PubMed PMID: 12805599; PubMed Central PMCID: PMCPMC523861.
Singh N, Srivastava S, Sharma A. Identification and analysis of miRNAs and their targets in ginger using bioinformatics approach. Gene. 2016;575(2):570–6. https://doi.org/10.1016/j.gene.2015.09.036 PubMed PMID: WOS:000366537600026.
Prakash P, Rajakani R, Gupta V. Transcriptome-wide identification of Rauvolfia serpentina microRNAs and prediction of their potential targets. Comput Biol Chem. 2016;61:62–74. https://doi.org/10.1016/j.compbiolchem.2015.12.002 PubMed PMID: WOS:000374368200007.
Yanarojana M, Nararatwanchai T, Thairat S, Tancharoen S. Antiproliferative activity and induction of apoptosis in human melanoma cells by Houttuynia cordata Thunb extract. Anticancer Res. 2017;37(12):6619–28. https://doi.org/10.21873/anticanres.12119 PubMed PMID: 29187437.
Liang G, Zhu Y, Sun B, Shao Y, Jing A, Wang J, et al. Assessing the survival of exogenous plant microRNA in mice. Food Sci Nutr. 2014;2(4):380–8. https://doi.org/10.1002/fsn3.113 Epub 2014/12/05, PubMed PMID: 25473495; PubMed Central PMCID: PMCPMC4221836.
Yang J, Farmer LM, Agyekum AA, Hirschi KD. Detection of dietary plant-based small RNAs in animals. Cell Res. 2015;25(4):517–20. https://doi.org/10.1038/cr.2015.26 PubMed PMID: 25721324; PubMed Central PMCID: PMCPMC4387557.
Chiow KH, Phoon MC, Putti T, Tan BK, Chow VT. Evaluation of antiviral activities of Houttuynia cordata Thunb. Extract, quercetin, quercetrin and cinanserin on murine coronavirus and dengue virus infection. Asian Pac J Trop Med. 2016;9(1):1–7. https://doi.org/10.1016/j.apjtm.2015.12.002 PubMed PMID: 26851778.
Chung MS, Bae WJ, Choi SW, Lee KW, Jeong HC, Bashraheel F, et al. An Asian traditional herbal complex containing Houttuynia cordata Thunb, Perilla frutescens Var. acuta and green tea stimulates hair growth in mice. BMC Complement Altern Med. 2017;17(1):515. https://doi.org/10.1186/s12906-017-2003-x PubMed PMID: 29197368; PubMed Central PMCID: PMCPMC5712091.
Rutkowski P. Introduction to the special issue of European journal of surgical oncology: new roads in melanoma management. Eur J Surg Oncol. 2017;43(3):513–6. https://doi.org/10.1016/j.ejso.2016.12.001 PubMed PMID: 28034500.
Unver N, Freyschmidt-Paul P, Horster S, Wenck H, Stab F, Blatt T, et al. Alterations in the epidermal-dermal melanin axis and factor XIIIa melanophages in senile lentigo and ageing skin. Br J Dermatol. 2006;155(1):119–28. https://doi.org/10.1111/j.1365-2133.2006.07210.x PubMed PMID: 16792763.
Ko GA, Shrestha S, Kim Cho S. Sageretia thea fruit extracts rich in methyl linoleate and methyl linolenate downregulate melanogenesis via the Akt/GSK3beta signaling pathway. Nutr Res Pract. 2018;12(1):3–12. https://doi.org/10.4162/nrp.2018.12.1.3 PubMed PMID: 29399291; PubMed Central PMCID: PMCPMC5792254.
Ho H, Ganesan AK. The pleiotropic roles of autophagy regulators in melanogenesis. Pigment Cell Melanoma Res. 2011;24(4):595–604. https://doi.org/10.1111/j.1755-148X.2011.00889.x PubMed PMID: 21777401.
Bellei B, Flori E, Izzo E, Maresca V, Picardo M. GSK3beta inhibition promotes melanogenesis in mouse B16 melanoma cells and normal human melanocytes. Cell Signal. 2008;20(10):1750–61. https://doi.org/10.1016/j.cellsig.2008.06.001 PubMed PMID: 18602000.
Valian N, Ahmadiani A, Dargahi L. Increasing methamphetamine doses inhibit glycogen synthase kinase 3beta activity by stimulating the insulin signaling pathway in substantia nigra. J Cell Biochem. 2018. https://doi.org/10.1002/jcb.27082 PubMed PMID: 30011098.
Seidell JC. Obesity, insulin resistance and diabetes — a worldwide epidemic. Br J Nutr. 2007;83(S1):S5–8. Epub 03/01. https://doi.org/10.1017/S000711450000088X.
Hu X, Wang M, Bei W, Han Z, Guo J. The Chinese herbal medicine FTZ attenuates insulin resistance via IRS1 and PI3K in vitro and in rats with metabolic syndrome. J Transl Med. 2014;12:47. https://doi.org/10.1186/1479-5876-12-47 PubMed PMID: 24555840; PubMed Central PMCID: PMCPMC3943467.
Engelman JA, Ji L, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19.
Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7(2):85.
Berndt J, Kovacs P, Ruschke K, Kloting N, Fasshauer M, Schon MR, et al. Fatty acid synthase gene expression in human adipose tissue: association with obesity and type 2 diabetes. Diabetologia. 2007;50(7):1472–80. https://doi.org/10.1007/s00125-007-0689-x PubMed PMID: 17492427.
Lin MC, Hsu PC, Yin MC. Protective effects of Houttuynia cordata aqueous extract in mice consuming a high saturated fat diet. Food Funct. 2013;4(2):322–7. https://doi.org/10.1039/c2fo30228d PubMed PMID: 23165792.
Kang H, Koppula S. Houttuynia cordata alleviates high-fat diet-induced non-alcoholic fatty liver in experimental rats. Pharm Biol. 2015;53(3):414–22. https://doi.org/10.3109/13880209.2014.923002 PubMed PMID: 25272018.
Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. 2014;67:79–87. https://doi.org/10.1016/j.nbd.2014.03.011 PubMed PMID: 24686304; PubMed Central PMCID: PMCPMC4083060.
Ouyang K, Li J, Huang H, Que Q, Li P, Chen X. A simple method for RNA isolation from various tissues of the tree Neolamarckia cadamba. Biotechnol Biotechnol Equip. 2014;28(6):1008–13. https://doi.org/10.1080/13102818.2014.981086 PubMed PMID: 26019587; PubMed Central PMCID: PMCPMC4434054.
Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52. https://doi.org/10.1093/nar/gkr688 PubMed PMID: 21911355; PubMed Central PMCID: PMCPMC3245920.
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, et al. A uniform system for microRNA annotation. RNA. 2003;9(3):277–9 PubMed PMID: 12592000; PubMed Central PMCID: PMCPMC1370393.
Wen M, Shen Y, Shi S, Tang T. miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics. 2012;13:140. https://doi.org/10.1186/1471-2105-13-140 PubMed PMID: 22720726; PubMed Central PMCID: PMCPMC3410788.
Chen T, Xi QY, Ye RS, Cheng X, Qi QE, Wang SB, et al. Exploration of microRNAs in porcine milk exosomes. BMC Genomics. 2014;15:100. https://doi.org/10.1186/1471-2164-15-100 PubMed PMID: 24499489; PubMed Central PMCID: PMCPMC4008308.
Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. https://doi.org/10.1186/gb-2010-11-2-r14 PubMed PMID: 20132535; PubMed Central PMCID: PMCPMC2872874.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–4. https://doi.org/10.1093/nar/gkm882 PubMed PMID: 18077471; PubMed Central PMCID: PMCPMC2238879.
Acknowledgements
None
Funding
This work was supported by grants from the National Key Research and Development Programme (No. 2016YFD0500503, 2016YFD0501205) and National Natural Science Foundation of China [No. 31802156, 31872435, 31802032].
Availability of data and materials
H. cordata root transcriptomic data was SRR7413372/SRX4284770. H. cordata raw sequence reads was deposited SRP157866/ SRS3667823/SRX4550658/SRR7691090 (https://www.ncbi.nlm.nih.gov/sra).
Author information
Authors and Affiliations
Contributions
JH and JS carried out the bioinformatics and analysis, JH and TC participated in drafted the manuscript. QX, JL and ML collected the sample and q-PCR validation. JW, BZ and JH were performed extract total RNA. YZ conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Statistics of reads by sequencing, Table S2. Categorization of small RNAs, Table S3. Expression profile of conserved miRNAs in H.cordata, Table S4. Expression profile of novel miRNAs in H.cordata, Table S5. Reverse transcript primers for miRNAs, Table S8. Highly enriched KEGG pathways for putative human targets. (DOCX 39 kb)
Additional file 2:
Figure S1. Secondary structures of novel miRNAs. (PDF 2683 kb)
Additional file 3:
Table S6. Potential target genes of miRNAs. (XLSX 304 kb)
Additional file 4:
Table S7. The top 30 GO enrichment results and top 30 KEGG statistics. (XLSX 705 kb)
Additional file 5:
Figure S2. Overview of experiments and bioinformatics analyses applied to H.cordata. (TIF 77 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
He, J., Chen, T., Xi, Q. et al. Identification of microRNA in Houttuynia cordata Thunb and prediction of cross kingdom functions. ExRNA 1, 34 (2019). https://doi.org/10.1186/s41544-019-0028-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41544-019-0028-7