
Abstract
Background
Plant elements and extracts have been used for centuries to treat a wide range of diseases, from cancer to modern lifestyle ailments like viral infections. These plant-based miRNAs have the capacity to control physiological and pathological conditions in both humans and animals, and they might be helpful in the detection and treatment of a variety of diseases. The present study investigates the miRNA of the well-known spice Curcuma Longa and its prospective targets using a variety of bioinformatics techniques.
Results
Using the integrative database of animal, plant, and viral microRNAs known as miRNEST 2.0, nine C. longa miRNAs were predicted. psRNA target service foretells the presence of 23 human target genes linked to a variety of disorders. By interacting with a variety of cellular and metabolic processes, miRNAs 167, 1525, and 756 have been found to be critical regulators of tumour microenvironment. SARS-cov2 and influenza A virus regulation have been connected to ZFP36L1 from miRNA 1525 and ETV5 from miRNA 756, respectively.
Conclusions
The current cross-kingdom study offers fresh knowledge about how to increase the effectiveness of plant-based therapies for disease prevention and serves as a platform for in vitro and in vivo research development.
Graphical abstract

Similar content being viewed by others
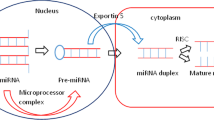
1 Background
In this revolutionary chorology, the appropriateness of plant natural products has a fructifying tendency in enhancing mankind's robustness by manifesting plants, plant parts, and plant products into daily life as active intake and dietary supplements. Bioprospecting identifies more significant and abundant plant-based compounds with pharmacological value. Modern herbal compounds such as saponins, alkaloids, and flavonoids may also be able to regulate the mechanisms of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) by inhibiting their main protease (Mpro) enzymes [1]. Because living plant species contribute more to the world's chemical diversity of bioactive compounds than any man-made synthetic library, finding novel plant molecules today would necessitate more advanced and powerful discovery perspectives. Curcuma Longa (C. longa), a plant belonging to the Zingiberaceae family, has been used for medicinal purposes since ancient times. Powder form of dry roots of C. longa is referred as turmeric and it is most common spice in various cuisines across the world. Medically turmeric has been used to treat gastrointestinal problems, biliary and hepatic disorders, diabetic sores, rheumatism, inflammation, sinusitis, anorexia, coryza, and cough. It has anti-cancer, anti-diabetic, antioxidant, hypolipidemic, anti-inflammatory, antibacterial, anti-fertility, anti-venom, hepatoprotective, nephroprotective, anticoagulant, and anti-HIV properties [2, 3]. The purpose of this study is to discover the potential impact of C. longa miRNAs on human target genes and SARS-CoV2 infection. This study differs from prior studies on C. longa miRNAs in terms of data identification, cross-kingdom interaction with human genes, and disease regulation pathways.
1.1 The achievements of plant miRNAs on human health
RNA interference (RNAi) is the most significant scientific achievement in the last two decades, and it is currently being used in clinical studies. With recently identified new class of RNAi molecules, microRNA (miRNA) is now playing an even well-known role in research and technology [4]. miRNAs are the most important genomic regulators, controlling 1–4% of all human genes. Small non-coding RNAs that have 21–25 nucleotides and are classified as miRNAs. miRNAs modulates mRNA content at the post-transcriptional stage to aid many important activities in biological systems, including embryonic development, cell maintenance, chemical signalling, and cell apoptosis. An anomaly in miRNA expression has been linked to the onset of a number of diseases ranging from viral to various cancers. miRNA can attach to viral RNA and inhibit viral genome processes by binding to the open reading frame (ORF) and slowing down the translation process. miRNAs have the ability to regulate cells in an autocrine, paracrine, and endocrine manner [5]. As a result, miRNAs has emerged as a possible marker for evaluating and diagnosing disease development [6]. miRNA continues to explore its path towards the prevention and treatment of human diseases. As an application, plant miRNAs could be used in the research and treatment of a variety of disorders. There are cutting-edge researches specifically demonstrating cross-kingdom gene transfer, mechanism of plant miRNAs absorbance, metabolism, and distribution on target site. Plant miRNAs absorption causes the intestine's epithelial cells to incorporate with miRNAs via a variety of processes, allowing plant miRNAs to reach gut cells, be delivered to specific physiological sub-compartments, and modulate gene expression in various body systems [7]. The first evidence of cross-kingdom gene transfer was proclaimed by Zhang et al. [8] plant miRNA 168a have ability to regulate LDLRAP1 gene in mice model and found significant decrease in LDLRAP1 and increase level of miRNA 168a illustration that intake of plant miRNA can instantaneously alternate the LDL gene expression. Moreover, the miRNAs from strawberry perhaps customized the toll-like receptor adherence capacity that corresponds to autoimmune response and dendritic cell migration. G-protein subunit alpha 12 (GNA12) involved regulation m-TOR signalling cascade, a synthetic isoform of plant miRNA 171 suppress the activity of this compound in human embryo kidney cells (HEK293) and affecting m-TOR mechanism [9]. However, plant miRNAs mechanisms are differed from animal miRNAs because of its methyl and 2-hydroxyl group (2-OH) at 3’ terminus. These groups improve the integrity and stability of plant miRNA and hydroxyl group slower down the degradation rate in synthetic plant miRNAs [10]. The recent accumulating attestation on horizontal gene transfer indicates although, these evidences have criticism and scepticism concerning the dependability and sensitivity of the techniques applied for cross-kingdom miRNAs transmission. Cross-kingdom regulation of plant-derived miRNAs needs attention because of its potential to create novel therapeutic treatments for miRNA deregulation-related illnesses.
1.2 Computational approaches for studying miRNAs and its effects on disease prevention across the kingdoms
Plant miRNAs have a significant impact on horizontal gene transfer mechanisms and may become an important research topic to investigate. In terms of plant miRNAs' ability to regulate gene expression across kingdoms, very few studies have used in silico approaches to assess their role in human disease targets and disease regulation. When developing plant-based miRNAs for mammalian genes, there are four primary properties that are often exploited. These include base-pairing between the 'seed' region and the target gene, low free energy estimation (genuine paring with miRNA target), target prediction (possible binding sites necessary for cross-kingdom transfer) and site accessibility [7]. These principles are well-founded for predicting plant-based miRNAs transfer in mammalian genes. We have been working on bioinformatics for a long time and have a large collection of cross-kingdom systematic results. In our in silico research, we discovered that the miRNAs of Ocimum basilicum [11], Bacopa monnieri [12], Persea americana [13], and Prunus armeniaca [14] all might play a direct role in disease state and maintenance in our in silico research. The C. longa miRNAs are not been evaluated as well their human target genes are not known. Therefore, in the present study, we have identified miRNA of C. longa from various data sources and their human target genes are identified. Through this study, we would like to emphasize that some effects of turmeric may be mediated by its miRNAs.
2 Methods
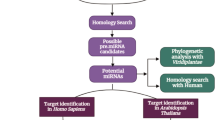
C. longa miRNA sequences were retrieved from the miRNEST 2.0 sequence prediction database (http://mirnest.amu.edu.pl). Arabidopsis thaliana (A. thaliana) mature miRNAs was used to set reference miRNAs for prediction. The first step was to create a database of matured A. thaliana miRNAs. C. longa ESTs were aligned with a reference sample of mature A. thaliana miRNAs using BLASTn software [15]. Following alignment, filtration criteria were used to extract unique miRNAs for predicting functional miRNAs (E-value, bit score, and mismatches, among others). Target human gene interactions were carried out using the psRNATarget software, and these projected miRNAs were then hybridized with the 3'-UTR of human transcripts [16]. Gene enrichment ontology analysis of functions of targets was investigated using the ShinyGO software. This is done in order to limit down possible targets for further investigation [17]. STRING and network analyst computational software were utilized to determine human gene–gene and gene-protein interaction for these discovered miRNA targets [18,19,20,21]. The bottleneck, stress, and betweenness algorithms were used to assess the target genes in Cytoscape v 3.9.1 cytohubba plugin [22]. Utilizing the Cofold web server, filters like the minimal free energy (MFE) and partition function are applied to prevent isolated base pairs and concentrated parameters. The most stable secondary structure of RNA can be predicted using this web server [23, 24]. The only stem loop structure of miRNAs was chosen for the study of miRNAs of C. longa. Because the stem loop structure is crucial for the secondary RNA structure because it gives miRNAs their structural integrity. Furthermore, stem loop topology may have an impact on enzymatic activities and aids in the provision of recognition sites for RNA binding proteins [25]. The purpose of this study is to evaluate the miRNAs that have been anticipated [26, 27]. A structural flow chart of methods is depicted in Fig. 1.

A structural flow chart of methods
3 Results
3.1 C. longa sequence prediction using miRNEST 2.0
miRNEST analysis provide the identification of miRNAs in plants animals and viruses. Using an EST database, this software discovered miRNAs in plants and animals. This application has identified a total of 10,004 miRNAs in 199 plant and 221 animal kingdoms. miRNEST 2.0 is an improved version that recognizes a total of 39,122 miRNAs [15]. The most recent edition of miRBase (version 22) has information on 38,589 pre-miRNAs from 271 organisms, including 1917 precursors and 2654 mature miRNAs in humans. It provides data for 326 hairpins and 428 mature sequences for A. thaliana and 258 hairpins and 469 mature sequences for Drosophila melanogaster as examples of other model species [28]. In the present study, we used this software to identify, predict and retrieve the sequences of C. longa miRNAs. The predicted miRNAs are listed in Table 1. Furthermore, these miRNAs were also tested using the miRBase in silico database. A total of 9 miRNAs were identified for further evaluation (Table 1).
3.2 Analysis and interaction of human targets of C. longa miRNA
The psRNATarget tool is effective for validating miRNA–miRNAs interactions [16]. The psRNATarget software was developed to identify the target genes of predicted plant miRNAs. This software uses a complementary mismatch-sensitive 'seed' region to identify potential targets for interested miRNAs. This software estimates mRNA target availability and energy to unwind the secondary structure throughout the target site to calculate target accessibility. This software's performance, most importantly, estimates mRNA target availability. This technique was used to investigate the C. longa plant miRNAs target gene for humans, and a total of 23 target genes were discovered, all of which play a key role in modulating human diseased conditions. In a recent study, the target gene ZFP36L1 from miRNA 1525 also had a role in the regulation of the influenza A virus through translational repression [29]. miRNA 167 and miRNA 1525 significantly inhibit mitogen-activated protein kinases (MAPK) and NF-kB signalling pathways in cross-kingdom analysis, indicating that these two miRNAs are likely beneficial for maintaining cell proliferation and angiogenesis and play a role in regulating the tumour microenvironment. The miRNA 756 target gene ETV5 plays an important role in maintaining lung homoeostasis, and its activity has been found to be reduced in COVID-19 patients [30, 31] (Table 2).
3.3 Gene enrichment analysis for targeted genes
Gene ontology research of C. longa miRNAs targets revealed diverse roles, including genes involved in transcriptional regulators, and pathway involvement in signalling and metabolic processes. Various enrichment pathways, molecular activities, biological processes, and cellular components associated to human target genes are detected by the ShinyGO database. ALPK1, ATRN, CYB5B, SETD7, ZFP36L1, ETV5, and SRXN1 were among the genes involved in stress reactions. ALPK1, GAB1, DLG2, GREM2, DCC, ZFP36L1, and ETV5 were discovered to be involved in the regulation of the multicellular developmental process. These target genes are also involved in cell adhesion and the immunological response. CYB5B, GREM2, TNFSF15, and PPP3R2 were shown to be involved in the control of molecular activities. Molecular transducer and oxidoreductase activity for CYB5B, PNPO SRXN1, CLEC2D, DCC, and ATRN were also reported. These genes were found significantly Distribution of the lengths of 3' UTRs in target genes versus other coding genes in the genome. Moreover, these genes also show significant GC content compared with the rest in the genome. The numerous relationships between target genes and associated GO functions are depicted in Fig. 2A–E.


Functional annotation of targeted genes of C. longa using Gene Ontology. List of targets that have been thoroughly functionally analysed in terms of A. biological processes, B. molecular functions, C. cellular components. (The chart displays the functional hits as well as the fold enrichment of genes), D. Enriched GO biological component terms visualized as a network. Darker nodes are more significantly enriched gene sets. Bigger nodes represent larger gene sets. Thicker edges represent more overlapped genes and E. Distribution of the lengths of 3’ UTRs in query genes versus other coding genes in the genome and % of GC content for target genes. Colour red shows high fold enrichment and blue low fold enrichment with an FDR limit of < 0.05
3.4 Gene interaction predictions for targeted genes
We intended to use STRING and Network analyst databases in our study to identify protein–protein, gene–gene interrelationships and also the KEGG pathway functions for these networks of interacting molecules attributed to our human target gene of C. longa miRNAs for linking with cross-kingdom analysis (Fig. 3A, B). Large databases and an online platform are available for the computational analysis of protein–protein linkage. Some well-known in silico tools in use include IMEX CONSORTIUM, UniProt Consortium, BioGRID, HINT, iRefWeb, APID, GeneMANIA, HumanNet, and FunCoup. The prediction of protein–protein interactions yields a wealth of information about functional connectivity, pathways, and high-throughput experimental interactions [18,19,20]. C. longa miRNAs predicted targets interacted with numerous pathways and other gene we can anticipate that single miRNA can regulate multiple gene, gene functions, and gene-associated pathways (Fig. 4). These gene alterations may lead to changes in normal physiology and cause disease which are major cause of cancerous malignancies. Changes in Cytochrome b target gene, which is involved in lipid metabolism and may cause chemical carcinogenesis. On chromosome 18, the DCC gene is located, and its inactivation is linked to the development and metastasis of colorectal cancer [32]. Furthermore, research indicates that upregulating Gab1 increases breast cancer (BCs) and metastasis by separating the PAR complex, which has been identified as a major regulator of EMT. This suggests that Gab1 may serve as a biomarker for BCa that has spread to other organs (Wang et al., 2019). In a study using zi rats to describe the function of the ATRN gene in the central nervous system (CNS), researchers discovered that when ATRN gene function is lost, reactive oxygen species (ROS) are induced, which causes neurodegeneration and demonstrates the importance of ATRN gene accumulation in the CNS [33]. TNFSF15, a member of the tumour necrosis factor ligand superfamily, modulates inflammatory disorders and MAPK/NF-κB/PI3K signalling pathway. Alteration in this pathway leads to many diseases condition including cancer, autoimmune disorders, etc. [34] (Table 3). This analysis concludes that C. longa miRNAs, on the other hand, have the ability to regulate transcription and can act as epigenetic modifiers. A single miRNA interacts with a number of genes, while a single gene interacts with numerous miRNAs. As a result, these miRNAs-gene screenplays adapt to control a variety of illnesses and disease regulatory pathways.

A The enrichment analysis of protein- protein annotation of target genes through STRING and B Gene–gene interaction of targeted genes network analyst algorithm. Red node shows the predicted human target gene and blue node shows the interaction with target genes

The Cytoscape plugin cytohubba was used to identify the top hub nodes using the bottleneck method. Hub nodes in an enrichment analysis network study of their target genes. A colour-coded systems used to denote them from highly necessary (red) to necessary hub nodes (yellow)
3.4.1 Hub node identification
The targeted genes connection was identified hubs with the highest numbers of interactions. Degree method was applied to identify top 30 hub nodes using centrality parameters such as bottleneck, Stress and betweenness concerning significance and biological processes. In the hub interaction score 23 and the highest number of interactions, the topmost hub node was LMO2 (LIM Domain Only 2) key regulator of hematopoietic stem cells and cancer malignancies. Other significant proteins such as TRIM27 (tripartite motif containing 27), AESV (amino-terminal enhancer of split), KRTAP10-7 (Keratin Associated Protein 10-7), WDYHV1(WDYHV motif containing 1), SDCBP (Syndecan Binding Protein), NOTCH2NL (Notch homolog 2 N-terminal-like PROTEIN), ZNF250 (Zinc Finger Protein 250), PPP1R18 (protein phosphatase 1 regulatory subunit 18) and ZBTB24 (Zinc Finger And BTB Domain Containing 24) were identified as the other top nodes with 1,3,4,4,6,7,7,7 and 7 rank, respectively (Fig. 5). This top node indicates the sophisticated network linkages and track down the important role of LMO2 and TRIM27 in the signalling cascade by interacting proteins in this network.

The structure prediction of C. longa miRNA by RNACofold
3.5 RNA secondary structure prediction of C. longa miRNAs for human target genes
In our study, the predicted secondary structure of nine C. longa miRNAs was identified. A dot in the dot-bracket plot depicts the unpaired position of two sequences, whereas the bracket represents the matching pair. Furthermore, RNA sequences are in a heterodimer structure with the least amount of free energy and separate the two sequences. According to the sequence structure shown in the table, C. longa miRNAs has an energy level of under − 20 kcal/mol, which is favourable for post-transcriptional gene silencing processes [8] (Fig. 4) (Table 4). The secondary structure of C. longa miRNAs was also calculated using thermodynamic approaches and Mfold, with the folding formula ΔG = ΔH − TΔS [27] (Table 5).
4 Discussion
Curcuma longa's cross-kingdom significance in humans can be compared by evaluating the effects of different miRNAs from the plant using computational approaches. The therapeutic potential of C. longa and its miRNAs in treating a variety of human ailments is determined by this study. C. longa is a notable medicinal plant that is frequently used to treat a variety of cellular and pathological diseases. Numerous phytochemical investigations have been run to identify the key pharmacological properties of this therapeutic plant [35]. The basic mechanism, through which medicinal plants alter the human DNA is yet unknown. Most eukaryotic genomes, including those of plants, now express miRNAs as a substantial governing element. We applied an in silico strategy in this study to predict miRNAs from C. longa data and to identify their targets in Homo sapiens, which has been made possible by the gene function of these miRNAs. We discovered nine miRNAs by using the data on recognized plant miRNAs that is available in the miRNEST database. The precursor miRNA constructs were examined for other energy factors like MFE and stability as well as their capacity to acquire hairpin loops. It is well known that the miRNA secondary structure is more stable the lower the MFE value [36]. The two main characteristics in this study that were applied in the psRNATarget tool to find possible human targets were cross-kingdom complementarity and target identification. Potential miRNAs targets for C. longa were predicted in this analysis. The majority of the anticipated miRNAs targets were transcription factor coding genes. The projected miRNAs' GC content and 3' UTR length were determined to be significant, respectively, at p- 0.00029 and p- 0.026. Predicted miRNAs had energy criteria that were negative and within the acceptable range for RNA-mediated gene encoding. Our findings show that nine miRNAs sequences, including miRNA1525, miRNA129, miRNA167 (1, 2, 3, and 4), miRNA756, andmiRNA1568, are associated with the transcriptome of the C. longa. Evidently, 23 crucial human genes involved in biological, physiological, and metabolic processes are influenced by all of these miRNAs. Four of the 23 target genes were discovered to play a regulatory role in the hallmarks of cancer, including angiogenesis, metastasis, apoptosis, and cell proliferation. Furthermore, it was discovered that several targets, such as DLG2, ETV5, PGM3, and ALPK1, were key regulators of neurological diseases (Table 2). The results of the cross-kingdom investigation show that all 23 C. longa predicted genes miRNAs were the primary regulators of human cellular, metabolic, and biological processes, as well as a number of signalling networks. Potential targets LMO2, TRIM27, AES, KRTAP10-7, WDYHV1, SDCBP, NOTCH2NL, ZNF250, PPP1R18 AND ZBTB24 are examples of genes regulated by cross-kingdom research that have been discovered to play a significant role in a number of disorders, including hematopoietic stem cell formation [37], oncogenic roles in various malignancies [38, 39], regulation of NF-κB signalling cascade [40], neurogenesis [41] and immunodeficiency [42]. These point to the potential anti-carcinomous properties of these miRNAs for disease prevention and therapeutic importance. The results accomplish that using bioinformatics resources, C. longa miRNAs and their predicted human target genes were identified. In upcoming cross-kingdom research, these anticipated miRNAs might be helpful in addressing the disease-centric investigations. These miRNAs from C. longa have potential as therapeutic markers in the interdisciplinary diagnosis of diseases that are associated to genes. These miRNAs will be crucial markers for the diagnosis, prognosis, and treatment of a wide spectrum of disorders soon. However, the potential utility of these identified miRNAs further validated through in vitro and in vivo studies are required for these in silico findings. This constructive approach may potentially prove useful in future inter-kingdom study on disease analysis.
5 Conclusions
Our findings reveal that, C. longa miRNAs have immune-modulatory, DNA-repairing, anti-tumour, antiviral, anti-inflammatory, and anti-oxidative properties. These characteristics are identical to those described for C. longa active phytochemicals. As a result, part of C. longa's health advantages may be mediated by its miRNAs. The outcomes of this study could help researchers better understand how dietary miRNAs affect consumer physiology across kingdoms. This research on C. longa miRNAs provides insight on the development of short RNA biomarker treatments for target prediction and identification for a variety of diseases that are riding the wave of new innovations in plant sciences.
Availability of data and material
On request, access to data will be available.
Abbreviations
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus-2
- Mpro:
-
Main protease
- C. longa :
-
Curcuma Longa
- AIDS:
-
Human immunodeficiency virus infection and acquired immune deficiency syndrome
- RNAi:
-
RNA interference
- miRNA:
-
Micro RNA
- siRNAs:
-
Small-interfering RNAs
- ORF:
-
Open reading frame
- GNA12:
-
G-protein subunit alpha 12
- HEK293:
-
Human embryo kidney cells
- 2-OH group:
-
2'-Hydroxyl group
- A. thaliana:
-
Arabidopsis thaliana
- MAPK:
-
Mitogen-activated protein kinases
- BCs:
-
Breast cancer
- NF-B:
-
Nuclear factor kappa B
- CNS:
-
Central nervous system
- ROS:
-
Reactive oxygen species
- MFE:
-
Minimum folding energy
- NMR:
-
Nuclear magnetic resonance
References
Bhuiyan FR, Howlader S, Raihan T, Hasan M (2020) Plants metabolites: possibility of natural therapeutics against the COVID-19 pandemic. Front Med (Lausanne). https://doi.org/10.3389/fmed.2020.00444
Akram M, Shahab-Uddin AA, Khan U, Abdul H, Mohiuddin E, Asif M (2010) Curcuma longa and curcumin: a review article. Rom J Biol Plant Biol 55(2):65–70
Rafieian-Kopaei M, Nasri H, Sahinfard N, Rafieian M, Rafieian S, Shirzad M (2014) Turmeric: a spice with multifunctional medicinal properties. J HerbMed Pharmacol 3:5–8
de Almeida SST, Horst CH, Soto-Sánchez C, Fernandez E, de Almeida RT (2018) Delivery of miRNA-Targeted oligonucleotides in the rat striatum by magnetofection with neuromag®. Molecules. https://doi.org/10.3390/molecules23071825
Fani M, Zandi M, Ebrahimi S, Soltani S, Abbasi S (2021) The role of miRNAs in COVID-19 disease. Future Virol 16:301–306. https://doi.org/10.2217/fvl-2020-0389
Kang H (2019) Molecular sciences MicroRNA-mediated health-promoting effects of phytochemicals. Int J Mol Sci. https://doi.org/10.3390/ijms20102535
Saiyed AN, Vasavada AR, Johar SRK (2022) Recent trends in miRNA therapeutics and the application of plant miRNA for prevention and treatment of human diseases. Futur J Pharm Sci 8:24. https://doi.org/10.1186/S43094-022-00413-9
Zhang L, Hou D, Chen X, Li D, Zhu L, Zhang Y et al (2011) Exogenous plant MIR168a specifically targets mammalian LDLRAP1: evidence of cross-kingdom regulation by microRNA. Cell Res 22:107–126. https://doi.org/10.1038/cr.2011.158
Chen X, Liu L, Chu Q, Sun S, Wu Y, Tong Z et al (2021) Large-scale identification of extracellular plant miRNAs in mammals implicates their dietary intake. PLoS ONE. https://doi.org/10.1371/JOURNAL.PONE.0257878
Dávalos A, Henriques R, Latasa MJ, Laparra M, Coca M (2018) Literature review of baseline information on non-coding RNA (ncRNA) to support the risk assessment of ncRNA-based genetically modified plants for food and feed. EFSA Support Publ. https://doi.org/10.2903/sp.efsa.2019.EN-1688
Patel M, Patel S, Mangukia N, Patel S, Mankad A, Pandya H et al (2019) Ocimum basilicum miRNOME revisited: a cross kingdom approach. Genomics 111:772–785. https://doi.org/10.1016/j.ygeno.2018.04.016
Gadhavi H, Patel M, Mangukia N, Shah K, Bhadresha K, Patel SK et al (2019) Transcriptome-wide miRNA identification of Bacopa monnieri: a cross-kingdom approach. Plant Signal Behav. https://doi.org/10.1080/15592324.2019.1699265
Bhatt DH, Jha N, Johar SRK, Pandya HA (2017) In silico exploration of miRNA from EST data of avocado and predicting its cross-kingdom effects on human. Pharma Innov 6(9, Part H):542
Bhatt D, Bhatt DH, Rawal RM, Kaid JSR, Pandya HA (2017) Identification of apricot microrna from EST data and their cross-kingdom targets in human evaluating plant mirna as a modulator of human gene expression view project identification of apricot microrna from est data and their cross-kingdom targets in human. Res J Pharm Biol Chem Sci. Doi: https://doi.org/10.26479/2017.0301.13
Szcześniak MW, Makałowska I (2014) MiRNEST 2.0: A database of plant and animal microRNAs. Nucleic Acids Res. https://doi.org/10.1093/nar/gkt1156
Dai X, Zhuang Z, Zhao PX (2018) psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res 46:49–54. https://doi.org/10.1093/nar/gky316
Ge SX, Jung D, Jung D, Yao R (2020) ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36:2628–2629. https://doi.org/10.1093/BIOINFORMATICS/BTZ931
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J et al (2018) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47:607–613. https://doi.org/10.1093/nar/gky1131
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S et al (2021) The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49:605–612. https://doi.org/10.1093/nar/gkaa1074
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J et al (2014) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43:447–452. https://doi.org/10.1093/nar/gku1003
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J (2019) NetworkAnalyst 30: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz240
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY (2014) cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. https://doi.org/10.1186/1752-0509-8-S4-S11
Lorenz R, Hofacker IL, Stadler PF (2016) RNA folding with hard and soft constraints. Algorithms Mol Biol. https://doi.org/10.1186/S13015-016-0070-Z
Lorenz R, Bernhart SH, Höner zu, Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA Package, 2
O’Brien J, Hayder H, Zayed Y, Peng C (2018) Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2018.00402
Waugh A, Gendron P, Altman R, Brown JW, Case D, Gautheret D et al (2002) RNAML: a standard syntax for exchanging RNA information. RNA 8(6):707-717
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. https://doi.org/10.1093/nar/gkg595
Kozomara A, Birgaoanu M, Griffiths-Jones S (2018) miRBase: from microRNA sequences to function. Nucleic Acids Res 47:155–162. https://doi.org/10.1093/nar/gky1141
Lin R-J, Huang C-H, Liu P-C, Lin I-C, Huang Y-L, Chen A-Y et al (2020) Zinc finger protein ZFP36L1 inhibits influenza A virus through translational repression by targeting HA, M and NS RNA transcripts. Nucleic Acids Res 48:7371–7384. https://doi.org/10.1093/nar/gkaa458
Zhang Z, Newton K, Kummerfeld SK, Webster J, Kirkpatrick DS, Phu L et al (2017) Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc Natl Acad Sci U S A 114:3903–3908. https://doi.org/10.1073/pnas.1621177114
Melms JC, Biermann J, Huang H, Wang Y, Nair A, Clausi MG et al (2021) Amit Dipak Amin 1,2,33, Denis Schapiro 10,11. André F Rendeiro 595:33. https://doi.org/10.1038/s41586-021-03569-1
Bendardaf R, Lamlum H, Pyrhönen S (2004) Prognostic and predictive molecular markers in colorectal carcinoma. Anticancer Res 24:2519–2530
Ehara A, Sakakibara S, Ueda S (2016) The role of Attractin in neurodegeneration caused by oxidative stress. In: Free radicals and diseases. InTech, pp 139–147. https://doi.org/10.5772/63330
Hedl M, Abraham C (2014) A TNFSF15 disease-risk polymorphism increases pattern-recognition receptor-induced signaling through caspase-8–induced IL-1. Proc Natl Acad Sci 111:13451–13456. https://doi.org/10.1073/pnas.1404178111
Ahmad RS, Hussain MB, Tauseef M, Arshad MS, Waheed M, Shariati MA et al (2020) Review article biochemistry, safety, pharmacological activities, and clinical applications of turmeric: a mechanistic review. Evid-Based Complement Altern Med. https://doi.org/10.1155/2020/7656919
Kumar D, Kumar S, Ayachit G, Bhairappanavar SB, Ansari A, Sharma P et al (2017) Cross-kingdom regulation of putative miRNAs derived from happy tree in cancer pathway: a systems biology approach. Int J Mol Sci. https://doi.org/10.3390/ijms18061191
El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Vyas P, Patient R et al (2011) Structure of the leukemia oncogene LMO2: implications for the assembly of a hematopoietic transcription factor complex. J Amer Soc Hematol. 117(7):2146-2156. https://doi.org/10.1182/blood-2010-07
Du R, Huang C, Chen H, Liu K, Xiang P, Yao N et al (2020) SDCBP/MDA-9/syntenin phosphorylation by AURKA promotes esophageal squamous cell carcinoma progression through the EGFR-PI3K-Akt signaling pathway. Oncogene 39:5405–5419. https://doi.org/10.1038/s41388-020-1369-2
Czerwinska P, Mackiewicz AA (2021) Low levels of TRIM28-interacting KRAB-ZNF genes associate with cancer stemness and predict poor prognosis of kidney renal clear cell carcinoma patients. Cancers. https://doi.org/10.3390/cancers13194835
Xiao C, Zhang W, Hua M, Chen H, Yang B, Wang Y et al (2021) TRIM27 interacts with Iκbα to promote the growth of human renal cancer cells through regulating the NF-κB pathway. BMC Cancer. https://doi.org/10.1186/s12885-021-08562-5
Suzuki IK, Gacquer D, van Heurck R, Kumar D, Wojno M, Bilheu A et al (2018) Human-specific NOTCH2NL genes expand cortical neurogenesis through delta/notch regulation. Cell 173:1370-1384.e16. https://doi.org/10.1016/j.cell.2018.03.067
Helfricht A, Thijssen PE, Rother MB, Shah RG, Du L, Takada S et al (2020) Loss of ZBTB24 impairs nonhomologous end-joining and class-switch recombination in patients with ICF syndrome. J Exp Med. https://doi.org/10.1084/jem.20191688
Acknowledgements
The author Atiyabanu N. Saiyed graciously acknowledges The Indian Council of Medical Research Senior Research Fellowship Programme File No. 2019-3856/CMB/BMS.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
For the design of this study, AS went through the literature and performed computational analysis. The manuscript was reviewed by AV. SRKJ and AS made significant contributions to data management and manuscript assessment. The final manuscript was read and approved by all of the authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent of publication
Not applicable.
Competing interests
All the authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saiyed, A.N., Vasavada, A.R. & Johar, S.R.K. Employing in silico investigations to determine the cross-kingdom approach for Curcuma longa miRNAs and their human targets. Beni-Suef Univ J Basic Appl Sci 12, 3 (2023). https://doi.org/10.1186/s43088-022-00330-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43088-022-00330-z