
Abstract
Molecular pathology is playing an increasingly important role in the treatment and overall management of patients with colorectal carcinoma. Three distinct genetic pathways have been identified that play a role in carcinogenesis: the chromosomal instability pathway, the microsatellite instability pathway, and the CpG island methylator phenotype pathway. Certain genetic mutations, some of which overlap with the aforementioned pathways, can also indicate that a carcinoma patient has a genetic predisposition syndrome, such as familial adenomatous polyposis, Lynch syndrome, and hamartomatous polyposis syndromes. A variety of advanced methods, including next-generation sequencing, are available to test for these and other mutations, such as targetable mutations that may allow tailoring of a treatment regimen to a patient’s specific cancer (e.g., KRAS and BRAF mutations). The possible future role of testing circulating tumor cells is also addressed. New mutations and syndromes continue to be discovered, ensuring that our knowledge of colorectal carcinoma and our ability to treat it will advance in the future.
Similar content being viewed by others


Background
Understanding of the molecular pathogenesis of colorectal carcinoma (CRC) began with the adenoma-carcinoma sequence, wherein tubular adenomas that arise within the colon accumulate additional molecular mutations over time, with oncogene activation and tumor suppressor inactivation, leading the adenoma to develop into adenocarcinoma (Fig. 1) [1]. A number of genes have been implicated in this sequence, including KRAS [2], TP53 [3], APC [4], and less commonly BRAF [5].

Three major pathways leading to colorectal cancers. (1) Conventional adenoma-carcinoma sequence with oncogene (e.g. KRAS) activation and tumor suppressor (e.g. APC, SMAD4 and TP53) inactivation, resulting in microsatellite stable (MSS) cancers; (2) Microsatellite instability (MSI) pathway with mismatch repair (MMR) protein deficiency in patients with Lynch syndrome, resulting in MSI-high (MSI-H) cancers; (3) Serrated pathway with CpG island methylation phenotype, resulting in either MSI-H cancers if methylation occurs in MLH1 promoter or MSS cancers if methylation occurs in tumor suppressor genes. HGD: high-grade dysplasia; LGD: low-grade dysplasia; SSA: sessile serrated adenoma
While this pathway of molecular oncogenesis, termed the chromosomal instability pathway (CIN), accounts for roughly 60% of CRCs, a variety of other insults can accumulate to cause the remaining proportion of the disease [6]. The most well-studied of these is the microsatellite instability (MSI) pathway (Fig. 1) [7]. Certain genes (most notably MLH1, MSH2, MSH6, and PMS2) encode proteins that repair mismatched DNA bases, preventing the formation of deleterious microsatellite sequences. If this functionality is lost due to gene mutation, an adenoma can form and may, within a few years, progress to malignancy as well; hypermethylation of MLH1 (see below) accounts for perhaps 12% of CRCs, with germline mutation of a mismatch repair gene accounting for about 3% [7].
A third pathway, the CpG island methylator phenotype (CIMP) or serrated pathway, causes 35% of CRCs and involves epigenetic hypermethylation of CpG islands in promoter regions of tumor suppressor genes, preventing them from undergoing transcription and therefore effectively inactivating them (Fig. 1). Such tumors typically arise from a sessile serrated adenoma, rather than a tubular adenoma (Fig. 1) [8]. This shares some overlap with the MSI pathway, as MLH1 undergoes hypermethylation in a large proportion of cases. Other potentially targeted genes include CDKN2A (which encodes p16) and THBS1 [9]. CIMP tumors are often proximally located and also harbor a BRAF mutation [10], which may have been present in the precursor sessile serrated adenoma.
These three pathways provide a basic but incomplete outline regarding the overall pathogenesis of CRC, as the molecular profile of each individual tumor is highly complex and variable. CRCs collectively have a median of 76 identifiable mutations, though only some truly contribute to oncogenesis [11]. Furthermore, the Cancer Genome Atlas Network recently reported that 16% of CRCs are hypermutated, with three-fourths of these demonstrating hypermethylation and the remaining one-fourth harboring mutations in POLE or in mismatch repair genes [12]. Their report also noted that ARID1A, SOX9, and FAM123B mutations are often identified in CRC. As additional molecular findings are cemented in the literature, classifications of colorectal carcinoma may become based primarily on molecular properties, rather than histologic subtype; indeed, such classifications are already being proposed [13, 14].
Identification of a CRC’s molecular underpinnings can impact a patient in several ways. Targeted therapy against the EGFR signaling pathway has become a mainstay in the treatment of metastatic disease [15]. Furthermore, several inborn syndromes exist that predispose patients to develop CRC more frequently and/or at a younger age than non-syndromic patients in the general population. The most famous of these are familial adenomatous polyposis (FAP) and Lynch syndrome (formerly termed hereditary non-polyposis colorectal cancer, or HNPCC), which respectively embody the CIN and MSI pathways of oncogenesis. There are other, less common syndromes that share some similarities with these two. Additionally, several hamartomatous polyposis syndromes exist as a side note. These syndromes will be discussed in the next section of this article, followed by an overview of the role of molecular testing in patient diagnosis and treatment. Finally, the future of molecular evaluation of CRC will be covered, with a look at promising upcoming targets.
Colorectal carcinoma syndromes
Although fewer than 10% of CRCs can currently be attributed to hereditary cancer syndromes, they are important for several reasons [16]. For one, they afford an opportunity to observe the key molecular factors leading to carcinogenesis. Second, their identification can have a lasting impact on the entire family of a cancer patient. Finally, as family history is a strong risk factor for CRC even in the absence of a known syndrome, there exists potential for discovering new syndromes and novel molecular associations [17]. For these and other reasons, the American Society of Clinical Oncology recently released clinical practice guidelines aimed at identifying syndrome-associated CRCs [18]. A brief review of important syndromes follows.
Familial adenomatous polyposis
Patients with FAP develop hundreds or thousands of adenomatous polyps throughout their colon. While each individual polyp has a very low risk of progressing to adenocarcinoma, the sheer number of polyps leads to a lifetime risk of CRC of effectively 100%, which occurs on average around 34 years of age [19]. Therefore, patients are generally offered a prophylactic colectomy, as early as in their teenage years. However, despite the fact that FAP is the most common gastrointestinal polyposis syndrome, it only accounts for 0.25% of CRC [20]. Extra-colonic manifestations are somewhat common but comparatively benign. They include gastric fundic gland polyps (which may harbor dysplasia), gastric adenomas, duodenal adenomas, and congenital hypertrophy of the retinal pigment epithelium [21]. Additionally, a rare form of papillary thyroid carcinoma known as the cribriform-morular subtype generally arises only in FAP patients [22].
FAP has a few associated syndromes, which effectively consist of FAP plus an additional lesion or lesions [21]. In Gardner syndrome, patients develop skeletal osteomas, epidermoid cysts, and desmoid fibromatosis, usually of the abdomen. Turcot syndrome has been described as FAP combined with medulloblastoma. (Turcot syndrome is also considered a variant of Lynch syndrome; see next section.)
FAP, Gardner, and Turcot syndromes all result from a germline mutation in APC (adenomatous polyposis coli), a tumor suppressor gene located at 5q21 that encodes a protein responsible for degrading β-catenin within the Wnt signaling pathway; it also stabilizes microtubules [23]. The syndromes are inherited in an autosomal dominant fashion, though roughly one-fourth of cases arise from a de novo mutation [20]. Mutations are typically toward the 3′ end of exon 15. Mutations at the 5′ end of the gene have less severe manifestations than mutations at the 3′ end, leading to attenuated FAP (AFAP), wherein patients develop fewer than 100 adenomatous colon polyps and have a lifetime risk of CRC of approximately 70% [21].
CRC in FAP develops via the conventional CIN pathway. This sequence is initiated by activation of the Wnt signaling pathway, typically by mutation of one copy of the APC tumor suppressor gene in non-syndromic patients, or by the germline mutation in FAP patients. The second APC allele is then inactivated by deletion or additional mutation. These APC alterations lead to the development of dysplasia, first in aberrant crypt foci and then in true adenomatous polyps. As additional genes become mutated (including KRAS and TP53), malignancy develops. Overall, the CIN pathway causes gains or losses of large stretches of chromosome material, hence its name [24]. In general, CRC caused by CIN has an unfavorable prognosis [25].
In a side note, the overall molecular alterations in colitis-associated CRC (in patients with ulcerative colitis or Crohn’s disease) are similar but occur in a different order; for example, TP53 is affected early in the pathogenetic sequence, and APC is one of the final genes mutated [26, 27].
Lynch syndrome
Formerly known as hereditary non-polyposis colorectal cancer [28], Lynch syndrome accounts for roughly 3% of all CRC and therefore is the most common cause of syndrome-associated CRC [7]. Much as FAP embodies the CIN pathway of carcinogenesis, Lynch syndrome exemplifies the MSI pathway. Patients have a germline mutation in MLH1, MSH2, MSH6, or PMS2, and secondary loss of the functional allele is the first insult on the pathway to CRC [7]. Loss of one or more of these mismatch repair proteins allows errors in microsatellites to accumulate during DNA replication, ultimately leading to genome-wide microsatellite instability [29]. In rare cases, Lynch syndrome may occur in a patient with a germline mutation in EPCAM (which is adjacent to MSH2 on chromosome 2) [30].
Lynch syndrome is inherited in an autosomal dominant fashion. The lifetime risk of developing CRC is up to 53%, with a mean age at diagnosis of about 45–50 years [31]. CRC in Lynch syndrome patients develops from a tubular adenoma (unlike in non-syndromic patients with a sporadic MSI-high cancer, where it develops from a sessile serrated adenoma). These tumors have several identifiable characteristics; they are typically right-sided, they harbor a multitude of tumor-infiltrating lymphocytes, and they are more likely to be medullary, mucinous or signet ring cell in microscopic appearance (Fig. 2). They also have an improved prognosis compared to CRC arising via CIN, but they are less responsive to 5-fluorouracil [32, 33]. Lynch syndrome patients are also at increased risk for malignancy in a wide variety of other organs, including uterus, stomach, ovary, and brain [34]. While approximately 15% of all CRC are MSI-high [7], the majority of these are due to sporadic hypermethylation of MLH1 (i.e., the CIMP pathway), which has not been linked to a heritable cancer syndrome at this time [35], though certain methylation patterns have been linked to family history of CRC in some patients [36].

Representative microsatellite instability-high colorectal carcinomas. a Moderately differentiated adenocarcinoma with prominent tumor-infiltrating lymphocytes (original magnification 200X); b. Mucinous adenocarcinoma (original magnification 100X); c. Medullary carcinoma with a pushing border and prominent tumor-infiltrating lymphocytes f2:3 (original magnification 100X); d. Signet ring cell carcinoma (original magnification 200X)
As with FAP, there are several syndromes related to Lynch syndrome. Muir–Torre syndrome is also caused by germline mutation in a mismatch repair (MMR) gene (generally MLH1 or MSH2); patients are at increased risk of developing cutaneous sebaceous neoplasms, in addition to the sequelae of Lynch [37]. Roughly 28% of families with known Lynch syndrome have a member with Muir–Torre [38].
Patients with a biallelic germline loss in an MMR gene have constitutional mismatch repair-deficiency syndrome [39]. In addition to Lynch syndrome-related tumors, patients develop hematologic malignancies and brain tumors; often, a patient will develop a tumor from more than one of these categories. Café au lait spots are also common. Some patients with Lynch syndrome-related tumors and glioblastomas have been said to have a form of Turcot syndrome [40], in addition to those Turcot patients with FAP and medulloblastomas, as discussed above.
MUTYH-associated polyposis
Attenuated FAP is not the only syndrome characterized by the development of a modest number of colorectal adenomas. In MUTYH-associated polyposis (MAP), patients with biallelic MUTYH mutations develop colonic adenomas (usually more than 10, but varying from 1 to several hundred [41]) and are at increased risk for CRC [42, 43]. MAP appears to be responsible for fewer than 1% of CRC [44], but it may be underdiagnosed. By age 70, patients have an 80% risk of developing CRC [45], and testing for possible MUTYH mutation is therefore recommended in patients with more than 10 adenomatous polyps, a family history of CRC, and/or a known lack of APC mutations [46].
While two mutated alleles must be present in order for MAP to manifest, single-allele MUTYH mutations may be present in up to 1% of the people in certain populations [47]. Patients by definition lack a germline APC mutation. In additional to colorectal polyps and carcinoma, some patients may develop duodenal and gastric adenomas; extra-intestinal manifestations are rare [43].
MUTYH, located on 1p34, encodes for a base excision protein that repairs oxidative DNA damage [48]. MAP therefore shares some conceptual similarity with Lynch syndrome, as both involve malfunctioning repair of damaged DNA. While most patients harbor a c.536A > G or c.1187G > A mutation [47], more than 200 different gene mutations have been reported [49].
POLE/POLD1-associated syndrome
The role of POLE and POLD1 in the development of colorectal polyposis and carcinogenesis has only recently been elucidated. These genes, which encode protein subunits of DNA polymerase complexes, may play a role in the development of hypermutated, microsatellite-stable CRC [50]. Patients with a germline POLE p.L424V mutation or one of a handful of POLD1 mutations appear to be at risk of developing CRC, as well as tumors of the brain, breast, and endometrium [51, 52]. Patients may develop an attenuated adenomatous polyposis, or may develop only a few polyps. To this end, the term “polymerase proofreading-associated polyposis” (PPAP) has been suggested [52]. POLE and POLD1 mutations may be inherited or arise de novo [53]. Germline mutations in these two genes appear to be fairly rare, and relatively few syndromic patients have been identified to date. Future reports on additional patients will likely help cement the clinicopathologic features associated with this syndrome.
Hamartomatous polyposis syndromes
There exist several syndromes that cause patients to develop multiple hamartomatous polyps throughout the colon (and often the rest of the gastrointestinal tract).
Peutz–Jeghers syndrome (PJS) is caused by germline mutation or deletion of STK11. This autosomal dominant syndrome leads to Peutz–Jeghers polyps in the stomach, small intestine, and colon, as well as characteristic mucocutaneous pigmentation [54]. These polyps have a characteristic “arborizing” architecture, with bundles of smooth muscle creating a framework that gives the polyps a tree-like appearance. These polyps are often harmless, though dysplasia and even carcinoma can occur within (Fig. 3a). Roughly half of patients develop a gastrointestinal cancer by age 70 [55]; they also are at increased risk for a variety of other neoplasms. Experts have argued that Peutz–Jeghers polyps only arise in syndromic patients, meaning that a patient diagnosed with one likely also has PJS [56].

Neoplastic changes in hamartomatous polyps. a Intramucosal carcinoma (yellow arrows) arising in a Peutz-Jegher’s polyp showing arborizing smooth muscle proliferation covered by hyperplastic colonic epithelium (original magnification 20X); b Low-grade dysplasia (blue arrows) arising in a juvenile polyp showing inflamed stroma and dilated glands (original magnification 20X)
Germline mutations in SMAD4 and BMPR1A can lead to juvenile polyposis syndrome (JPS), an autosomal dominant condition characterized by juvenile polyps growing through the gastrointestinal tract [54]. Microscopically, these polyps appear expanded and edematous, with dilated glands. As with Peutz–Jeghers polyps, they can develop dysplasia and give rise to malignancy (Fig. 3b); however, a lone juvenile polyp does not suggest that the patient is syndromic [57]. Up to 39% of patients with JPS will develop CRC in their lifetime, at a mean age of 44 years [58]. In at least some patients, a BMPR1A mutation instead leads to hereditary mixed polyposis syndrome, an autosomal dominant disorder causing a wide variety of colonic polyps to develop [59]. In other patients, this syndrome arises due to a SCG5 mutation [60].
Patients with a germline PTEN mutation may develop one of a number of syndromes. The most common of these is Cowden syndrome, which causes a wide variety of gastrointestinal hamartomas (including juvenile polyps and ganglioneuromas), along with characteristic skin lesions and a variety of visceral lesions [54]. In the majority of cases, more than 50 gastrointestinal polyps are found [61]. Patients have a 9% chance of developing CRC; the risk of breast cancer is much higher, at 85% [62]. The other syndromes caused by germline PTEN mutation are Bannayan–Riley–Ruvalcaba syndrome, Proteus syndrome, and Proteus-like syndrome, all of which manifest in childhood. Only the first is particularly associated with gastrointestinal polyposis, and none appear to confer an increased risk of malignancy [63].
Finally, Cronkhite–Canada syndrome is a rare, acquired hamartomatous polyposis syndrome characterized by unusual, juvenile-like polyps developing throughout the gastrointestinal tract [64]. Both these polyps and the background flat mucosa show stromal edema, gland dilation, and congestion. Patients also demonstrate ectodermal abnormalities. Up to 25% of patients develop CRC. While the pathophysiologic and genetic underpinnings of Cronkhite–Canada syndrome remain unclear, the disease may be autoimmune in origin [65].
Serrated polyposis
In serrated polyposis, patients develop a multitude of sessile serrated adenomas throughout their colon. This uncommon syndrome was originally termed “hyperplastic polyposis” prior to the discovery of sessile serrated adenomas, which bear histologic similarity to hyperplastic polyps [66]. Mean age at diagnosis is 55 years, and up to half of patients report a family history of CRC. Some patients develop numerous small left-sided polyps with KRAS mutations, while others develop fewer, larger right-sided polyps with BRAF mutations; a phenotype intermediate between these two has also been described [67]. One large cohort study detected CRC in 35% of patients with serrated polyposis, with an increased number of polyps conferring higher risk [68]. Despite these known phenotypes and molecular alterations, underlying germline mutations and a mode of inheritance have not yet been established for serrated polyposis, which may prove to be a rather heterogeneous disease or category of related diseases. In support of this is the finding that multiple serrated polyps have been described in some patients with MAP [69].
Base excision repair polyposis
This term has been suggested for patients with a germline mutation in the base excision repair gene NTHL1, which predisposes them to develop adenomatous polyposis and CRC. Only one report on this entity appears to exist so far [70].
Current molecular testing
Molecular testing can be used to identify patients with hereditary CRC syndromes or individuals genetically susceptible to developing CRCs. In addition, molecular testing of CRCs helps identify molecular biomarkers, which may improve patient care by individualizing cancer treatment.
Molecular testing in hereditary colorectal cancer syndromes
Lynch syndrome
Patients with Lynch syndrome may be identified clinically via use of the Amsterdam criteria and the revised Bethesda guidelines [71]. The revised Bethesda guidelines are more sensitive than the Amsterdam criteria in identification of these patients. However, it is now known that a significant portion of Lynch syndrome patients could still be missed by using the revised Bethesda guidelines [72, 73]. Therefore, the recently updated National Comprehensive Cancer Network (NCCN) guidelines recommend screening for Lynch syndrome in all individuals with CRC or individuals with CRC diagnosed at <70 years old and those ≥70 years old who meet the revised Bethesda guidelines.
Immunohistochemistry for the four MMR proteins, PCR-based MSI testing, or both can be used as an initial screening test for Lynch syndrome. Immunohistochemical stains for the proteins MLH1, MSH2, MSH6, and PMS2 are a sensitive and specific way to detect MMR deficiency [72, 74]. Approximately 90% of MMR-deficient CRCs show loss of nuclear staining for one or more of the MMR proteins (Fig. 4a-b). The four proteins play a critical role in mismatch recognition and initiation of repair [29]. MLH1 and PMS2 form the hMutLα heterodimer, and loss of MLH1 invariably results in the degradation of PMS2. Similarly, MSH2 and MSH6 form the hMutSα heterodimer, and loss of MSH2 is consistently accompanied by loss of MSH6. However, the converse is not true, because loss of PMS2 or MSH6 does not always cause the degradation of MLH1 or MSH2, respectively. Therefore, when a tumor shows loss of MLH1 and PMS2 or loss of MSH2 and MSH6, it is likely due to defective MLH1 or MSH2, whereas isolated loss of PMS2 or MSH6 indicates a defect in PMS2 or MSH6, respectively.

Immunohistochemical stains for MLH1 and MSH2. a Loss of nuclear staining for MLH1 in the cells of a colorectal adenocarcinoma. The tumor also showed PMS2 loss. b Intact nuclear expression of MSH2 in the same tumor. It also showed intact MSH6 expression
PCR-based MSI testing can detect the genetic changes associated with an MMR defect, namely microsatellite instability. Approximately 90% of MSI-high tumors can be detected by MSI testing. Currently, most laboratories in the US use a fluorescence-based PCR assay from Promega (Madison, WI). The assay includes five mononucleotide repeat markers (BAT-25, BAT-26, NR-21, NR-24, and MONO-27) and two pentanucleotide repeat markers (Penta C and Penta D). Both normal tissue and tumor tissue are analyzed. The mononucleotide markers are used for detection of microsatellite instability, and the pentanucleotide markers are used to ensure that both normal tissue and tumor are from the same patient. The results are interpreted as MSI-high when 2 or more mononucleotide markers show instability, MSI-low when only 1 marker is unstable, and microsatellite stable (MSS) when all markers are stable (Fig. 5).

PCR-based MSI testing showing microsatellite instability in 5 of 5 mononucleotide markers from a colorectal carcinoma. Normal tissue from the same patient is shown for comparison
Both immunohistochemistry and MSI testing have their advantages and disadvantages. Technical failure is not uncommon in MMR immunohistochemistry. In addition, the stains can be falsely negative in treated rectal cancers [74]. The major advantage of the immunohistochemical assay is specific identification of possible defective MMR protein(s). On the other hand, the PCR-based MSI testing requires sufficient tumor tissue (at least 10–30% nucleated tumor cells in a sample). However, the results from the PCR-based assay are easy to interpret. In addition, it can be used in treated CRCs. Either MMR immunohistochemistry or PCR-based MSI testing can be used as an initial screening test. If one test indicates MSS in patients with a high suspicion for Lynch syndrome, the tumor should be tested using the second method to increase the sensitivity.
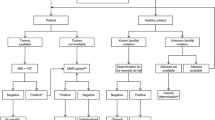
MSI-high CRCs can be sporadic or hereditary. In fact, most MSI-H CRCs are sporadic, caused by MLH1 promoter hypermethylation, and display loss of MLH1 and PMS2 by immunohistochemistry. In addition, sporadic MSI-high CRCs frequently harbor BRAF V600E mutation, which is extremely rare in Lynch syndrome-associated CRCs. If a tumor is MSI-high by PCR-based MSI testing or shows loss of MLH1 and PMS2 by immunohistochemistry, testing for the BRAF mutation and/or MLH1 promoter methylation assay should be followed. Such an algorithmic approach can be used to identify Lynch syndrome patients (Fig. 6).

An algorithmic approach used to identify Lynch syndrome patients at our institution. CRCs: colorectal cancers; MSI: microsatellite instability; MSS: microsatellite stable; ASaPCR: allele-specific PCR; IHC: immunohistochemistry; MMR: mismatch repair
As mentioned above, MMR immunohistochemistry is able to provide information regarding the relevant gene(s). When there is loss of MLH1/PMS2 with no BRAF mutation and no MLH1 promoter hypermethylation or loss of any other proteins (MSH2, MSH6, or PMS2), germline evaluation should be carried out for the genes corresponding to the absent proteins. For CRCs with loss of MSH2, a test for EPCAM deletions need to be included, since a deletion in the 3′ region causes somatic hypermethylation of MSH2 [75]. Mutations in the MMR genes include missense, nonsense, and splice site mutations as well as regulatory mutations. Large deletions are seen in 5–10% of MLH1 and 17–50% MSH2 gene mutations. DNA extracted from a blood sample is required for germline genetic testing. Sanger sequencing can be used to sequence all coding exons and intron/exon boundaries of the relevant MMR gene(s). Next-generation sequencing can also be used for that purpose. However, PMS2 cannot be sequenced by next-generation sequencing, due to high homology between the PMS2 functional gene and pseudogenes. Sanger sequencing on long-range PCR using functional gene-specific primers has been used to detect mutations in the PMS2 gene [76].
If MMR gene mutations are not detected by sequencing, analysis for large rearrangements or deletions of the MMR genes should be performed. Multiplex ligation-dependent probe amplification (MLPA) is commonly used to detect large deletions/rearrangements. Other methods used for large gene rearrangements include Southern blot hybridization, multiplex amplifiable probe hybridization, quantitative PCR analysis, and gene-targeted array-based comparative genomic hybridization [77].
FAP and attenuated FAP
The majority of germline mutations in APC are point mutations, short deletions, and short insertions, most of which introduce a stop codon, consequently resulting in a truncating APC protein. Two recurrent mutations at codons 1061 and 1309 are detected in approximately 30% of FAP cases. In addition, approximately 20% of the mutations are gross deletions, insertions, or complex rearrangements. Germline genetic testing of APC should be carried out for all patients suspicious for FAP or AFAP. Full sequencing of APC using Sanger sequencing or next-generation sequencing can be employed to detect the mutations. If no mutations are detected by sequencing, large rearrangement analysis of the gene should be performed [77].
MUTYH-associated polyposis
The presentation of MAP is similar to that of attenuated FAP. In individuals with ≥ 10 colonic adenomas but no germline alterations in APC, mutational analysis of MUTYH is recommended. Most mutations associated with MAP are missense mutations in the MUTYH gene. Two in particular, p.Y165C and p.G382D, are seen in 70–80% of the southern European Caucasian population [78]. Deletions, frameshift, and nonsense mutations have also been reported. Germline testing of MUTYH can be initiated by screening for the two most common mutations in the white population by PCR/restriction enzyme digestion-based techniques, or other technologies such as denaturing high-pressure liquid chromatography, pyrosequencing, Sanger sequencing, or allele-specific PCR [77]. If it is heterozygous for the mutations, full sequencing of the gene should be performed. Full sequencing should also be considered for nonwhite individuals suspected of having MAP [77].
Polymerase proofreading-associated polyposis
Missense mutations affecting the exonuclease domains of the polymerase genes POLE and POLD1 have recently been identified to be responsible for PPAP, which can present as a polyposis and/or a Lynch syndrome-like phenotype [51]. Individuals presenting with familial or early onset MMR-proficient CRC and/or APC-negative and MUTYH-negative polyposis should be screened for germline POLE or POLD1 exonuclease mutations. Sanger sequencing or next-generation sequencing can be used to detect the mutations.
Hamartomatous polyposis syndromes
Mutations in STK11 are responsible for approximately 90% of PJS cases. Most patients inherit the disease in an autosomal dominant manner; however, up to 45% of cases may be caused by a de novo STK11 mutation. Most of the genetic alterations are missense/nonsense and small deletion/insertion mutations. Others include large deletions/insertions and rearrangements [79]. Gene sequence analysis of the entire coding regions and the splicing sites by Sanger sequencing and gross deletion/duplication analysis of the gene by MLPA are used to characterize the mutations.
Up to 50% of JPS patients have a mutation in SMAD4 or BMPR1A. Single nucleotide variants explain about 40–45% of cases, and the remaining 10–15% of detectable cases are caused by gross deletions of either gene [80]. The initial test is concurrent sequence analysis of the BMPR1A and SMAD4 genes. If no causative mutations are identified by sequencing, gross deletion/duplication analysis of both genes should be carried out. PTEN mutations are identified in a small number of JPS patients. In addition, patients with a germline PTEN mutation may develop one of a number of other syndromes, such as Cowden syndrome. When no alterations are identified in the BMPR1A and SMAD4 genes, mutational analysis of PTEN may be considered.
Role of next-generation sequencing in hereditary colorectal cancer syndromes
Genetic testing for CRC susceptibility can either focus on well-characterized mutations based on a clinical suspicion or use panel testing to screen for multiple mutations in multiple genes simultaneously. Genetic panel testing can be achieved by next-general sequencing or Sanger sequencing. However, next-generation sequencing has demonstrated benefits on both costs and time required compared to Sanger sequencing [81]. Ambry Genetics (Aliso Viejo, CA) and GeneDx (Gaithersburg, MD) offer next-generation sequencing panels for hereditary CRCs. Similar testing is provided by the Mayo Clinic (Rochester, MN) and the University of Washington (Seattle, WA).
Molecular biomarker testing in colorectal cancers
MSI testing
MSI status may influence therapy decisions in stage II colorectal cancers. MSI is a good prognostic factor, though MSI-high cancers may not benefit from 5-FU-based adjunctive chemotherapy. Therefore, post-operative chemotherapy is not recommended for patients with stage II MSI-high CRC. The 2015 NCCN guidelines recommend that MSI testing or MMR immunohistochemistry should be performed in all patients with stage II CRC. In addition, MSI tumors contain significant more mutations then MSS tumors. They may be more likely to respond to immunotherapy, such as anti-PD1 agents [82]. Therefore, MSI or MMR testing should be performed in all patients with metastatic colorectal cancer if not previously done.
Extended RAS and BRAF mutational analysis for metastatic CRC patients
Mutations of KRAS and NRAS in exons 2, 3, and 4 result in constitutive activation of the EGFR signaling pathway. Clinical trials have provided evidence that these mutations are negative predictors of response to anti-EGFR therapy [83,84,85,86,87,88]. BRAF is an immediate downstream molecule of RAS. BRAF mutations also constitutively activate the enzyme activity, consequently persistently stimulating the EGFR signaling pathway. Although the results regarding the predictive role of BRAF mutations have been controversial, BRAF mutations in MSS cancers confer a poorer prognosis [83]. Therefore, the 2015 NCCN guidelines recommend that all patients with metastatic CRC should be tested for KRAS, NRAS and BRAF mutations. The NCCN guidelines also state that extended RAS mutations should be tested whenever possible.
Mutations in RAS and BRAF are missense single nucleotide substitutions (point mutations). Sanger sequencing, allelic-specific PCR, and pyrosequencing can be used to detect point mutations. However, these assays cannot be multiplexed, and Sanger sequencing requires high tumor cellularity. Many laboratories have developed multigene assays, which are more sensitive than Sanger sequencing and more efficient than the above assays. One example of a multigene assay is the SNapShot platform [89], which is a combination of multiplex PCR amplification of tumor DNA with single base extension of the PCR product following by capillary electrophoresis (Fig. 7). Recently, next-generation sequencing is receiving more widespread used for the same purpose.

A panel from the SNapShot assay showing a mutation in SMAD4 (p. R361C; 1081C > T)
Multiple gene mutation analysis by next-generation sequencing
In addition to KRAS and BRAF, some CRCs harbor mutations in other genes encoding for key intracellular molecular transducers of EGFR activation, such as PIK3CA and PTEN. Although their predictive and prognostic role is uncertain in metastatic CRC, they are potentially actionable genetic alterations. In fact, the PI3K/AKt/mTOR signaling pathway has been utilized as the therapeutic target for metastatic CRC in several clinical trials. Therefore, identification of mutational status of these genes, in addition to KRAS and BRAF, could help select better treatment for patients with metastatic CRC. Analysis of multiple mutations in multiple genes requires implementation of multi-target testing methodologies. Next-generation sequencing has been proven to be a robust and cost-effective tool to analyze a large number of gene alterations simultaneously [90, 91]. In addition, next-generation sequencing may identify uncommon genetic alterations, which could be potential actionable targets.
Although next-generation sequencing has been used for detection of gene mutations in malignancies for clinical implications in some centers, specific guidelines have not been developed. There is always a question regarding which sample should be used for analysis of gene mutations (metastasis versus primary tumor). Theoretically, it is more appropriate to test the metastatic lesions, because they are the cause of clinical morbidity and mortality. Multiple studies have compared KRAS alterations and have demonstrated rare discordance between primary cancer and metastasis [83]. However, significant discordance could occur when analyzing a larger panel. One study demonstrated a very different genotype in metastases from the primary tumor in about half of patients with synchronous metastatic CRCs [92]. Therefore, metastases may be a preferred specimen for next-generation sequencing of cancer genes, especially for patients with synchronous metastatic colorectal cancer. Nucleated tumor cellularity should be estimated by examination of an adjacent hematoxylin- and eosin-stained slide. Next-generation sequencing platforms have a minimum sensitivity of approximately 5%. Therefore, tumor cellularity of ≥ 10% is necessary for detection. During the data analysis, mutant-allele frequency (mutant/wild-type ratio) should be calculated. Both tumor cellularity and mutant-allele frequency help determine tumor heterogeneity and mutant allele-specific imbalance.
Future molecular testing in colorectal cancer
Characterization of biomarkers in circulating tumor DNA may be the future of personalized medicine in oncology. It is a non-invasive approach analyzing tumor genotypes in real time. A recent study demonstrated identification of tumor-associated mutations including KRAS, BRAF, and PIK3CA mutations in plasma DNA from the majority of 503 patients with metastatic CRC enrolled in the CORRECT trial [93]. Detection of genetic mutations using circulating tumor DNA may allow clinicians to follow the genetic evolution of the tumor noninvasively and may aid in predicting treatment response. However, to see widespread use in clinical practice, the technologies detecting circulating tumor cells need to be validated in large-scale studies.
Conclusions
Molecular testing has become of critical importance in the management of patients with CRC. It can identify the existence of a tumor predisposition syndrome, and it can help specify tumor-specific targets for patient therapy. Therefore, the genetic makeup of a particular CRC simply cannot be disregarded if the standard of care is to be met. As our knowledge of the genetics of CRC advances, new molecular targets and new genetic syndromes will almost certainly be discovered.
Abbreviations
- CIMP:
-
CpG island methylator phenotype
- CIN:
-
Chromosomal instability pathway
- CRC:
-
Colorectal cancer
- FAP:
-
Familial adenomatous polyposis
- HNPCC:
-
Hereditary non-polyposis colorectal cancer
- JPS:
-
Juvenile polyposis syndrome
- MAP:
-
MUTYH-associated polyposis
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MMR:
-
Mismatch repair
- MSI:
-
Microsatellite instability
- NCCN:
-
National Comprehensive Cancer Network
- PJS:
-
Peutz–Jeghers syndrome
References
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67.
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–32.
Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, Van Tuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–21.
Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–7.
Kalady MF, Dejulius KL, Sanchez JA, Jarrar A, Liu X, Manilich E, Skacel M, Church JM. BRAF mutations in colorectal cancer are associated with distinct clinical characteristics and worse prognosis. Dis Colon Rectum. 2012;55:128–33.
Ahnen DJ. The American College of Gastroenterology Emily Couric Lecture--the adenoma-carcinoma sequence revisited: has the era of genetic tailoring finally arrived? Am J Gastroenterol. 2011;106:190–8.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138:2073–87.
Langner C. Serrated and non-serrated precursor lesions of colorectal cancer. Dig Dis. 2015;33:28–37.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–6.
Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–45.
Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E. Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7.
Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30.
Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10(1):13–27.
Plesec TP, Hunt JL. KRAS mutation testing in colorectal cancer. Adv Anat Pathol. 2009;16:196–203.
Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–38.
Lindor NM. Familial colorectal cancer type X: the other half of hereditary nonpolyposis colon cancer syndrome. Surg Oncol Clin N Am. 2009;18:637–45.
Stoffel EM, Mangu PB, Gruber SB, Hamilton SR, Kalady MF, Lau MW, Lu KH, Roach N, Limburg PJ, American Society of Clinical Oncology; European Society of Clinical Oncology. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology Clinical Practice Guideline endorsement of the familial risk-colorectal cancer: European Society for Medical Oncology Clinical Practice Guidelines. J Clin Oncol. 2015;33:209–17.
Plawski A, Banasiewicz T, Borun P, Kubaszewski L, Krokowicz P, Skrzypczak-Zielinska M, Lubinski J. Familial adenomatous polyposis of the colon. Hered Cancer Clin Pract. 2013;11:15.
Legolvan MP, Taliano RJ, Resnick MB. Application of molecular techniques in the diagnosis, prognosis and management of patients with colorectal cancer: a practical approach. Hum Pathol. 2012;43:1157–68.
Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385–98.
Cameselle-Teijeiro J, Chan JK. Cribriform-morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma? Mod Pathol. 1999;12:400–11.
Leoz ML, Carballal S, Moreira L, Ocaña T, Balaguer F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl Clin Genet. 2015;8:95–107.
Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–72.
Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008;57:941–50.
Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–16.
Foersch S, Neurath MF. Colitis-associated neoplasia: molecular basis and clinical translation. Cell Mol Life Sci. 2014;71:3523–35.
Jass JR. Hereditary Non-Polyposis Colorectal Cancer: the rise and fall of a confusing term. World J Gastroenterol. 2006;12:4943–50.
Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98.
Ligtenberg MJ, Kuiper RP, Geurts van Kessel A, Hoogerbrugge N. EPCAM deletion carriers constitute a unique subgroup of Lynch syndrome patients. Fam Cancer. 2013;12:169–74.
Peltomaki P, Offerhaus GJA, Vasen HFA. Lynch syndrome. In: Bosman FT, Carniero F, Hruban RH, Thiese ND, editors. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon: IARC Press; 2010.
Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–26.
Saridaki Z, Souglakos J, Georgoulias V. Prognostic and predictive significance of MSI in stages II/III colon cancer. World J Gastroenterol. 2014;20:6809–14.
Watson P, Riley B. The tumor spectrum in the Lynch syndrome. Fam Cancer. 2005;4:245–8.
Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA, Zali MR. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol Bed Bench. 2013;6:120–8.
Frazier ML, Xi L, Zong J, Viscofsky N, Rashid A, Wu EF, Lynch PM, Amos CI, Issa JP. Association of the CpG island methylator phenotype with family history of cancer in patients with colorectal cancer. Cancer Res. 2003;63:4805–8.
Kruse R, Rütten A, Lamberti C, Hosseiny-Malayeri HR, Wang Y, Ruelfs C, Jungck M, Mathiak M, Ruzicka T, Hartschuh W, Bisceglia M, Friedl W, Propping P. Muir-Torre phenotype has a frequency of DNA mismatch-repair-gene mutations similar to that in hereditary nonpolyposis colorectal cancer families defined by the Amsterdam criteria. Am J Hum Genet. 1998;63:63–70.
South CD, Hampel H, Comeras I, Westman JA, Frankel WL, de la Chapelle A. The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst. 2008;100:277–81.
Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica. 2010;95:699–701.
Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, Booker SV, Petersen GM, Offerhaus GJA, Tersmette AC, Giardiello FM, Vogelstein B, Kinzler KW. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839–47.
Aretz S, Uhlhaas S, Goergens H, Siberg K, Vogel M, Pagenstecher C, Mangold E, Caspari R, Propping P, Friedl W. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer. 2006;119:807–14.
Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C → T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32.
Morreau H, Riddell RH, Aretz S. MUTYH-associated polyposis. In: Bosman FT, Carniero F, Hruban RH, Thiese ND, editors. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon: IARC Press; 2010.
Cleary SP, Cotterchio M, Jenkins MA, Kim H, Bristow R, Green R, Haile R, Hopper JL, LeMarchand L, Lindor N, Parfrey P, Potter J, Younghusband B, Gallinger S. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009;136:1251–60.
Jenkins MA, Croitoru ME, Monga N, Cleary SP, Cotterchio M, Hopper JL, Gallinger S. Risk of colorectal cancer in monoallelic and biallelic carriers of MYH mutations: a population-based case-family study. Cancer Epidemiol Biomarkers Prev. 2006;15:312–4.
Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044–58.
Aretz S, Tricarico R, Papi L, Spier I, Pin E, Horpaopan S, Cordisco EL, Pedroni M, Stienen D, Gentile A, Panza A, Piepoli A, de Leon MP, Friedl W, Viel A, Genuardi M. MUTYH-associated polyposis (MAP): evidence for the origin of the common European mutations p.Tyr179Cys and p.Gly396Asp by founder events. Eur J Hum Genet. 2014;22:923–9.
Venesio T, Balsamo A, Errichiello E, Ranzani GN, Risio M. Oxidative DNA damage drives carcinogenesis in MUTYH-associated-polyposis by specific mutations of mitochondrial and MAPK genes. Mod Pathol. 2013;26:1371–81.
Out AA, Tops CM, Nielsen M, Weiss MM, van Minderhout IJ, Fokkema IF, Buisine MP, Claes K, Colas C, Fodde R, Fostira F, Franken PF, Gaustadnes M, Heinimann K, Hodgson SV, Hogervorst FB, Holinski-Feder E, Lagerstedt-Robinson K, Olschwang S, van den Ouweland AM, Redeker EJ, Scott RJ, Vankeirsbilck B, Grønlund RV, Wijnen JT, Wikman FP, Aretz S, Sampson JR, Devilee P, den Dunnen JT, Hes FJ. Leiden Open Variation Database of the MUTYH gene. Hum Mutat. 2010;31:1205–15.
Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, CORGI Consortium; WGS500 Consortium, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–44.
Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, Puente DA, Pons T, González S, Iglesias S, Darder E, Piñol V, Soto JL, Valencia A, Blanco I, Urioste M, Brunet J, Lázaro C, Capellá G, Puente XS, Valle L. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016;18:325–32.
Church JM. Polymerase proofreading-associated polyposis: a new, dominantly inherited syndrome of hereditary colorectal cancer predisposition. Dis Colon Rectum. 2014;57:396–7.
Valle L, Hernández-Illán E, Bellido F, Aiza G, Castillejo A, Castillejo MI, Navarro M, Seguí N, Vargas G, Guarinos C, Juarez M, Sanjuán X, Iglesias S, Alenda C, Egoavil C, Segura Á, Juan MJ, Rodriguez-Soler M, Brunet J, González S, Jover R, Lázaro C, Capellá G, Pineda M, Soto JL, Blanco I. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Hum Mol Genet. 2014;23:3506–12.
Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100:476–90.
van Lier MG, Westerman AM, Wagner A, Looman CW, Wilson JH, de Rooij FW, Lemmens VE, Kuipers EJ, Mathus-Vliegen EM, van Leerdam ME. High cancer risk and increased mortality in patients with Peutz-Jeghers syndrome. Gut. 2011;60:141–7.
Burkart AL, Sheridan T, Lewin M, Fenton H, Ali NJ, Montgomery E. Do sporadic Peutz-Jeghers polyps exist? Experience of a large teaching hospital. Am J Surg Pathol. 2007;31:1209–14.
Nugent KP, Talbot IC, Hodgson SV, Phillips RK. Solitary juvenile polyps: not a marker for subsequent malignancy. Gastroenterology. 1993;105:698–700.
Brosens LA, van Hattem A, Hylind LM, Iacobuzio-Donahue C, Romans KE, Axilbund J, Cruz-Correa M, Tersmette AC, Offerhaus GJ, Giardiello FM. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56:965–7.
Cao X, Eu KW, Kumarasinghe MP, Li HH, Loi C, Cheah PY. Mapping of hereditary mixed polyposis syndrome (HMPS) to chromosome 10q23 by genomewide high-density single nucleotide polymorphism (SNP) scan and identification of BMPR1A loss of function. J Med Genet. 2006;43:e13.
Jaeger E, Leedham S, Lewis A, Segditsas S, Becker M, Cuadrado PR, Davis H, Kaur K, Heinimann K, Howarth K, East J, Taylor J, Thomas H, Tomlinson I. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet. 2012;44:699–703.
Stanich PP, Owens VL, Sweetser S, Khambatta S, Smyrk TC, Richardson RL, Goetz MP, Patnaik MM. Colonic polyposis and neoplasia in Cowden syndrome. Mayo Clin Proc. 2011;86:489–92.
Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18:400–7.
Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11:687–94.
Sweetser S, Ahlquist DA, Osborn NK, Sanderson SO, Smyrk TC, Chari ST, Boardman LA. Clinicopathologic features and treatment outcomes in Cronkhite-Canada syndrome: support for autoimmunity. Dig Dis Sci. 2012;57:496–502.
Slavik T, Montgomery EA. Cronkhite–Canada syndrome six decades on: the many faces of an enigmatic disease. J Clin Pathol. 2014;67:891–7.
Torlakovic E, Snover DC. Serrated adenomatous polyposis in humans. Gastroenterology. 1996;110:748–55.
Guarinos C, Sánchez-Fortún C, Rodríguez-Soler M, Alenda C, Payá A, Jover R. Serrated polyposis syndrome: molecular, pathological and clinical aspects. World J Gastroenterol. 2012;18:2452–61.
Boparai KS, Mathus-Vliegen EM, Koornstra JJ, Nagengast FM, van Leerdam M, van Noesel CJ, Houben M, Cats A, van Hest LP, Fockens P, Dekker E. Increased colorectal cancer risk during follow-up in patients with hyperplastic polyposis syndrome: a multicentre cohort study. Gut. 2010;59:1094–100.
Boparai KS, Dekker E, Van Eeden S, Polak MM, Bartelsman JF, Mathus-Vliegen EM, Keller JJ, van Noesel CJ. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008;135:2014–8.
Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, van Zelst-Stams WA, Jongmans MC, Gilissen C, Hehir-Kwa JY, Hoischen A, Shendure J, Boyle EA, Kamping EJ, Nagtegaal ID, Tops BB, Nagengast FM, Geurts van Kessel A, van Krieken JH, Kuiper RP, Hoogerbrugge N. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668–71.
Lipton LR, Johnson V, Cummings C, Fisher S, Risby P, Eftekhar Sadat AT, Cranston T, Izatt L, Sasieni P, Hodgson SV, Thomas HJ, Tomlinson IP. Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. J Clin Oncol. 2004;22:4934–43.
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman JA, Panescu J, Fix D, Lockman J, LaJeunesse J, Comeras I, de la Chapelle A. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–8.
Pérez-Carbonell L, Ruiz-Ponte C, Guarinos C, Alenda C, Payá A, Brea A, Egoavil CM, Castillejo A, Barberá VM, Bessa X, Xicola RM, Rodríguez-Soler M, Sánchez-Fortún C, Acame N, Castellví-Bel S, Piñol V, Balaguer F, Bujanda L, De-Castro ML, Llor X, Andreu M, Carracedo A, Soto JL, Castells A, Jover R. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut. 2012;61:865–72.
Samowitz WS. Evaluation of colorectal cancers for Lynch syndrome: practical molecular diagnostics for surgical pathologists. Mod Pathol. 2015;28 Suppl 1:S109–13.
Ligtenberg MJ, Kuiper RP, Chan TL, Goossens M, Hebeda KM, Voorendt M, Lee TY, Bodmer D, Hoenselaar E, Hendriks-Cornelissen SJ, Tsui WY, Kong CK, Brunner HG, van Kessel AG, Yuen ST, van Krieken JH, Leung SY, Hoogerbrugge N. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat Genet. 2009;41:112–7.
Clendenning M, Hampel H, LaJeunesse J, Lindblom A, Lockman J, Nilbert M, Senter L, Sotamaa K, de la Chapelle A. Long-range PCR facilitates the identification of PMS2-specific mutations. Hum Mutat. 2006;27:490–5.
Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A, Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med. 2014;16:101–16.
Gismondi V, Meta M, Bonelli L, Radice P, Sala P, Bertario L, Viel A, Fornasarig M, Arrigoni A, Gentile M, Ponz de Leon M, Anselmi L, Mareni C, Bruzzi P, Varesco L. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer. 2004;109:680–4.
McGarrity TJ, Amos CI, Frazier ML, Wei C. Peutz-Jeghers Syndrome. 2001 Feb 23 [Updated 2013 Jul 25]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1266. Accessed 17 Mar 2016.
Larsen Haidle J, Howe JR. Juvenile Polyposis Syndrome. 2003 May 13 [Updated 2015 Dec 3]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1469. Accessed 17 Mar 2016.
Simbolo M, Mafficini A, Agostini M, Pedrazzani C, Bedin C, Urso ED, Nitti D, Turri G, Scardoni M, Fassan M, Scarpa A. Next-generation sequencing for genetic testing of familial colorectal cancer syndromes. Hered Cancer Clin Pract. 2015;13:18.
Gatalica Z, Snyder C, Maney T, Ghazalpour A, Holterman DA, Xiao N, Overberg P, Rose I, Basu GD, Vranic S, Lynch HT, Von Hoff DD, Hamid O. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014;23:2965–70.
Pillai RK, Lopategui JR, Dhall D, Guindi M, Slavin T, Lofton-Day CE, Patterson SD. The State of the Art in Colorectal Cancer Molecular Biomarker Testing. Adv Anat Pathol. 2016;23:92–103.
Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, Giuliani F, Barone C, Cartenì G, Rachiglio AM, Montesarchio V, Tonini G, Rizzi D, Cinieri S, Bordonaro R, Febbraro A, De Vita F, Orditura M, Fenizia F, Lambiase M, Rinaldi A, Tatangelo F, Botti G, Colucci G. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol. 2014;25:1756–61.
Hecht JR, Douillard JY, Schwartzberg L, Grothey A, Kopetz S, Rong A, Oliner KS, Sidhu R. Extended RAS analysis for anti-epidermal growth factor therapy in patients with metastatic colorectal cancer. Cancer Treat Rev. 2015;41:653–9.
Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, He P, Yu H, Koukakis R, Terwey JH, Jung AS, Sidhu R, Patterson SD. Analysis of KRAS/NRAS Mutations in a Phase III Study of Panitumumab with FOLFIRI Compared with FOLFIRI Alone as Second-line Treatment for Metastatic Colorectal Cancer. Clin Cancer Res. 2015;21:5469–79.
Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol. 2015;26:13–21.
Allegra CJ, Rumble RB, Schilsky RL. Extended RAS Gene Mutation Testing in Metastatic Colorectal Carcinoma to Predict Response to Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Therapy: American Society of Clinical Oncology Provisional Clinical Opinion Update 2015 Summary. J Oncol Pract. 2016;12:180–1.
Su Z, Dias-Santagata D, Duke M, Hutchinson K, Lin YL, Borger DR, Chung CH, Massion PP, Vnencak-Jones CL, Iafrate AJ, Pao W. A platform for rapid detection of multiple oncogenic mutations with relevance to targeted therapy in non-small-cell lung cancer. J Mol Diagn. 2011;13:74–84.
Haley L, Tseng LH, Zheng G, Dudley J, Anderson DA, Azad NS, Gocke CD, Eshleman JR, Lin MT. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol. 2015;28:1390–9.
Malapelle U, Pisapia P, Sgariglia R, Vigliar E, Biglietto M, Carlomagno C, Giuffrè G, Bellevicine C, Troncone G. Less frequently mutated genes in colorectal cancer: evidences from next-generation sequencing of 653 routine cases. J Clin Pathol. 2016 Jan;21 [Epub ahead of print].
Balschun K, Haag J, Wenke AK, von Schönfels W, Schwarz NT, Röcken CKRAS, NRAS. PIK3CA exon 20, and BRAF genotypes in synchronous and metachronous primary colorectal cancers diagnostic and therapeutic implications. J Mol Diagn. 2011;13:436–45.
Tabernero J, Lenz HJ, Siena S, Sobrero A, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Yoshino T, Goldberg RM, Sargent DJ, Wagner A, Laurent D, Teufel M, Jeffers M, Grothey A, Van Cutsem E. Analysis of circulating DNA and protein biomarkers to predict the clinical activity of regorafenib and assess prognosis in patients with metastatic colorectal cancer: a retrospective, exploratory analysis of the CORRECT trial. Lancet Oncol. 2015;16:937–48.
Acknowledgements
None.
Funding
None.
Availability of data and material
Not applicable.
Authors’ contributions
All authors have contributed to the writing and editing. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gonzalez, R.S., Washington, K. & Shi, C. Current applications of molecular pathology in colorectal carcinoma. Appl Cancer Res 37, 13 (2017). https://doi.org/10.1186/s41241-017-0020-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41241-017-0020-1