
Abstract
Cerebral malaria is still a deleterious health problem in tropical countries. The wide spread of malarial drug resistance and the lack of an effective vaccine are obstacles for disease management and prevention. Parasite and human genetic factors play important roles in malaria susceptibility and disease severity. The malaria parasite exerted a potent selective signature on the human genome, which is apparent in the genetic polymorphism landscape of genes related to pathogenesis. Currently, much genomic data and a novel body of knowledge, including the identification of microRNAs, are being increasingly accumulated for the development of laboratory testing cassettes for cerebral malaria prevention. Therefore, understanding of the underlying complex molecular basis of cerebral malaria is important for the design of strategy for cerebral malaria treatment and control.
Similar content being viewed by others


Background
Cerebral malaria (CM) is a life-threatening disease that represents a global health problem particularly in tropical countries. According to a report of the World Health Organization (WHO) in the year 2015, malaria transmission still occurs in approximately 97 countries and territories, mostly in Sub-Saharan Africa, Southeast Asia, and South America. In the year 2013, the estimated incidence of malaria infection was 198 million cases (range 124–283 million) worldwide. Over 575,000 cases of CM have been reported. Approximately 584,000 cases (range 367,000–755,000) died from malaria. African children are the most affected case of CM. Most of the malaria-related deaths, approximately 90 %, occurred in Africa [1]. In Sub-Saharan Africa, 575,000 children with CM have been annually reported and 110,000 cases died (approximately 19–25 % case fatality rate). Unfortunately, >2 % of survivors of CM experienced developmental and behavioral impairment lasting for 6 months. The disability, severity, and neurological duration are critical for CM management and essential for understanding of CM pathogenesis. Due to prevention and control programs, the morbidity and motility rates of malaria were reduced globally. In Thailand, the mortality rate decreased 50–74 % between 2000 and 2013 [1, 2].
Plasmodium falciparum is the causative organism leading to human CM development. The bite of a P. falciparum-infected female anopheline mosquito mediates the development of various disease severities ranging from uncomplicated malaria to severe malaria and CM. Uncomplicated malaria or mild malaria is defined as a febrile illness without any clinical or laboratory signs of severity or vital organ dysfunction. Complicated malaria or severe malaria involves the central nervous system (cerebral malaria), the pulmonary system (respiratory failure), the renal system (acute renal failure), and the hematopoietic system (severe anemia). According to the updated definition of severe falciparum malaria by the WHO (2015), severe falciparum malaria is defined as the presence of P. falciparum asexual parasitemia, with one or more clinical features or laboratory findings (Table 1) and without any identified alternative causes. The hallmarks of CM are coma (Glasgow Coma Scale <11, Blantyre coma score <3) or malaria with a persistent coma [3]. Clinical manifestations of severe malaria include but are not limited to CM (with incidence rate of 0.9–3.5 per 1000 child-year), severe malarial anemia (12–50 per 1000 child-year), and respiratory failure (1.4–5.4 per 1000 child-year) [1, 2].
Mechanism of CM
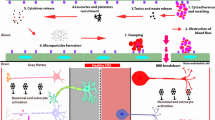
The mechanism of CM is not clearly understood. Researchers have exerted extensive efforts to elucidate the mechanism of CM using several approaches such as in vivo experimental mouse models [4, 5], in vitro co-cultures of parasitized red cells with human brain microvascular endothelial cells [6, 7], and postmortem tissue and clinical samples from patients in endemic areas [8, 9]. For the in vivo studies, Plasmodium berghei ANKA (PbA)-infected CBA and C57BL/6 mice have been used as a CM susceptible model. The mouse model manifests the neurological symptoms within 6–14 days after infection and then dies [10]. The differences in the characteristics between the murine model and humans are the host receptor and the absence of knob-like structures of the P. falciparum erythrocyte membrane protein 1 (PfEMP1) (Table 2) [11].
The development of CM is biologically complex and involves multiple mechanisms such as sequestration, immunopathology by the pro-inflammatory cytokine interferon-γ (IFN-γ), tumor necrosis factor alpha (TNF-α), and apoptosis. Red cell sequestration is an important step in the development of CM. The binding of PfEMP1 on infected red blood cells to host receptors such as intercellular adhesion molecule-1 (ICAM-1) and CD36 on brain endothelial cells mediates sequestration [12, 13]. Parasitized red blood cells (pRBCs) also form rosettes and clumps that impair microcirculation and cause hypoxia, which leads to neuronal tissue necrosis [14]. The production of IFN-γ from Th1 cells stimulates monocytes to express higher levels of the transmembrane form of TNF (memTNF) that interacts with tissue necrotic factor receptor 2 (TNFR2) expressed on brain endothelial cells causing the up-regulation of ICAM-1 on brain endothelial cells [15]. The up-regulation of ICAM-I on brain endothelial cells leads to an increase in platelets, pRBCs, and leukocyte adhesion. This contributes to the following clinical outcomes: (1) vessel obstruction and ischemia and (2) vessel disruption and brain hemorrhage. There are several reports of apoptotic mechanisms in endothelial cells leading to blood-brain barrier (BBB) dysfunctions in CM. The consequence of pRBC cytoadherence to the endothelial cell leads to (1) caspase 8 and 9 activation leading to apoptosis, (2) cytotoxic T cell activation leading to perforin-mediated cell death [16], (3) TNF-α overproduction as a result of the glycosylphosphatidylinositol (GPI activates apoptosis by inducing NO and oxidative stress) from the parasite, and (4) NF-kB activation in brain endothelial cells, neurons, and glial cells resulting in caspase 3 activation [8].
Microparticles are cellular membrane-derived vesicles generated by cytoskeletal alterations as a result of cellular membrane remodeling and loss of phospholipid asymmetry. Under physiological conditions, microparticles derived from platelets, white blood cells, red blood cells, and endothelial cells are expressed at normal levels. The overexpression of microparticles is associated with cell activation and apoptosis [17]. The ATP-binding cassette transporter A1 (ABCA1) gene is involved in the process of microparticle release. ABCA1 gene deletion or knockout is associated with protection from CM in mice [18, 19]. Moreover, endothelial microparticles (EMPs) and TNF are increased during coma episodes of CM in Malawian children compared to the uncomplicated malaria group [20]. This evidence highlights an important role of microparticles in the pathogenesis of CM [17].
The roles of CD8+ T cell in malaria infection are highlighted in many studies. CD8+ T cell acts as (1) effector cell in hepatic and erythrocytic phase for malaria vaccine development [21] and (2) pathogenic cell in experimental cerebral malaria (ECM) development [22]. In CM, it was found that dendritic cells (CD8α+ DCs) are responsible for malaria antigen presentation to CD8+ T cell. This leads to CD8+ T cell proliferation and cytotoxic T lymphocyte (CTL) production mediating ECM. The critical role of CD8α+ DCs as the major antigen-presenting subset in CM was further demonstrated in the protection against CM in CD8α+ DC-depleted mice [21]. The role of CD8+ T cell has been identified as the major mediator of lethality in ECM via the secretion of perforin and granzymes inducing brain endothelial apoptosis and disruption of BBB. In addition, the cytoadherence of pRBCs to brain endothelial cells could lead to malaria antigen uptake via a trogocytosis-like process [22]. Interestingly, brain endothelial cells of PbA-infected mice produced a large number of MP which finally resulted in CM pathogenesis [20].
Insight into gene regulation of malaria infection
Gene regulation is a process responsible for controlling the rate and manner of gene expression. In humans, dysfunctional gene regulation has been demonstrated in various pathological processes such as inflammatory responses and metabolic processes. Researchers have exerted extensive efforts to uncover the genetic regulation of infectious diseases to develop novel therapeutic agents that target the critical components, which are essential for the survival or amplification of the infective agents or which directly regulate the host immune system. Human gene expression is mainly regulated at the transcriptional and translational levels. Moreover, the mechanisms of epigenetic regulation of gene expression such as chromatin remodeling, histone modification, and non-coding RNAs, particularly microRNAs (miRNAs), have been shown to play important roles in the pathogenesis of complex diseases [20].
miRNAs are 21–25 nucleotide single-stranded non-coding RNA molecules. miRNAs are transcribed from miRNA genes by RNA polymerase II into long pri-miRNAs and then cleaved by the enzyme Drosha into smaller pre-miRNAs. This precursor is subsequently exported to the cytoplasm by the exportin-5/Ran-GTP complex and then further processed by Dicer into double-stranded RNAs. One of the strands is degraded, and the mature miRNA is incorporated with RISC (RNA-induced silencing complex). miRNAs regulate gene expression through complementary binding to the 3′ untranslated region (3′ UTR) of the targeted messenger RNA (mRNA). If the complementary paring pattern between the miRNA and the targeted mRNA is perfect, then the mRNA is cleaved by RISC, whereas if the paring is imperfect, then translational repression occurs [23].
A role for miRNAs in malaria pathogenesis has been increasingly documented. Transcriptome-wide analysis of miRNAs in Anopheles gambiae showed the differential expression levels between blood meal and after infection by P. berghei [24]. In sickle cell anemia, miRNAs from sickle cells translocated into P. falciparum and inhibited parasite growth [25]. The study of miRNA expression in ECM and non-ECM revealed that miRNAs, e.g., let7i, miR-27a, and miR-150, are significantly differentially expressed in the brains of PbA-infected CBA mice [26]. miRNAs have been reported to control the expression of genes related to malaria pathogenesis in diverse diseases such as the inhibition of ICAM-1 by miR-17-3p, VCAM-1 by miR-126, and E-selection by miR-31 [27]. The apoptosis of endothelial cells, neuroglial cells, and erythroid cells is regulated by miR-29b, the miR-15-16 cluster, the let-7/miR-98 family, and the miR-17-92 cluster [28]. NF-kB signaling is inhibited by miR-181b and miR-146a [29–31]. Hypoxia is regulated by miR-210 [29]. In addition, miRNA array analysis of postmortem kidney samples from malaria patients with acute kidney injury has also been investigated [32]. Therefore, this evidence highlights the potential role of miRNAs in the pathogenesis of malaria. However, the study of miRNA in human CM is still limited. Ultimately, the application of miRNA as inhibitor of pathogenic gene expression should be further studied as a new approach for adjunct CM therapy.
Treatment and prevention of CM
The early diagnosis and urgent treatment of complicated malaria is a good practice for the prevention of CM. The complicated malaria case must be considered as a medical emergency and high priority of treatment. The recommended antimalarial treatment for complicated malaria including CM is water-soluble artemisinin derivative artesunate. The supportive management of malaria complication is essential such as fluid and electrolyte imbalance and convulsion. Many clinical trials studied on adjunctive therapies for complicated malaria using exchange blood transfusion, anti-TNF, etc. However, no effective adjunctive therapies demonstrated the improvement of severity outcome [33].
The control programs of malaria are environmental insecticide-spraying programs, insecticide-treated bed nets, drug treatment in the case of clinical infection, and prophylactic measure for travelers. However, the major problems of the control programs include multidrug resistance in parasites, insecticide (pyrethroids) resistance in mosquitos, and the lack of effective vaccines. P. falciparum resistance to multiple drugs has been widely detected in endemic areas. The failure of artemisinin treatment occurred in five countries of the Greater Mekong sub-region: Cambodia, Myanmar, Thailand, Vietnam, and the Lao People’s Democratic Republic [2]. Drug resistance in the malaria parasite is due to mutations of genes that encode the critical components of the drug target such as P. falciparum dihydrofolate reductase (Pfdhfr) [34–38].
The current approach for malaria vaccine development is based on the use of recombinant proteins or the attenuated whole organism. However, there is no practical or effective vaccine in clinical use. The key considerations are the delivery system, evolutionary changes in the parasites, and the development of resistance. After vaccination, humans have two main responses of antiparasite and antitoxic immunity. Vaccines are designed for various stages of the malaria life cycle: the pre-erythrocytic-stage vaccines for the prevention of infection, the blood-stage vaccines for the prevention of clinical illness and death, and the sexual stage vaccines for blocking transmission. The most advanced candidate vaccine for P. falciparum (RTS, S-AS01) is in phase III efficacy trials [39].
Human genetics of CM
The risk factors of CM are age, immunological status, overall health status of the host, variation in host genotype, and parasite diversity [40]. The specific population risk groups are young children in stable transmission areas, non-immune pregnant women, semi-immune pregnant women in areas of high transmission, people with HIV/AIDS, and international travelers from non-endemic areas [3]. Human genetic factors influence the outcome of malaria severity. CM predisposition could be explained by genetic polymorphisms of the genes involved in malaria pathobiology. Most of the malaria-related genes involve immunological responses and cell receptors. Single nucleotide polymorphisms (SNPs) in the promoter region of the TNF-α gene are significantly associated with susceptibility to CM in the Gambia, Africa, Myanmar, and Thailand [41, 42]. However, variation in the genetic background of each ethnic population contributes to the differences in the level of disease predisposition and severity. Candidate gene-based genetic association studies have been extensively analyzed in various populations (Table 3).
The human genetic variations/phenotypes associated with malaria infection are mainly demonstrated in the host red blood cells, and the immunological components include (1) red blood cell/hemoglobin defects such as thalassemia, sickle cell trait, HbC, and HbE; (2) enzyme defects such as G6PD deficiency and PK deficiency; (3) membrane defects such as ovalocytosis; (4) the Duffy blood group; (5) immunogenetic variants such as HLA alleles; and (6) immunological components such as complement receptor 1, NOS2, TNF-α, and the chromosome 5q31–q33 region (the cytokine region). These genetic variants are highly detected in malaria-endemic areas as a result of natural selection. The mechanism of protection is not yet clear. The reduction or failure of parasite invasion/multiplication in the red blood cells, induction of clearance by the immune system, and increased oxidative stress are responsible for malaria protection [43, 44].
Parasite genetics of CM
Parasite genetics may also be involved in CM because P. falciparum transforms host red cell membranes by incorporation of parasite-derived proteins with the erythrocyte cytoskeleton. The key virulence proteins are P. falciparum erythrocyte membrane protein 1 (PfEMP1) and knob-associated histidine-rich protein (KAHRP), which involve the formation of a knob structure linked to the red blood cell cytoskeleton. The development of PfEMP1 initiates from the trophozoite stage, and the knob structure that is formed adheres to the endothelial cell of the host [45, 46]. The parasite gene called “var” is extremely diverse and encodes PfEMP1 that binds to various receptors on the host. The var gene contains two exons encoding Duffy binding-like (DBL), cysteine-rich interdomain region (CIDR), N-terminal sequence (NTS), and acidic terminal sequence (ATS) domains [47, 48]. The different domains of PfEMP1 contribute to the variety of antigenic adhesion molecules and are responsible for different clinical consequences (Table 4). The parasite evades the host immune system by intergenic recombination to generate genetic variants. A substantial level of polymorphisms was observed in Csp (circumsporozoite protein) and Msp-1 and Msp-2, which encode the merozoite surface proteins 1 and 2, respectively [49, 50]. Moreover, mutations in genes related to treatment failure enhance the risk of CM.
Conclusions
CM is a fatal multifactorial disease, and approximately 20 % of patients will die from coma and seizure if no effective treatment is available. Multidrug resistance is still a problem in endemic areas, and effective vaccines are not fully developed. Now, geneticists are exerting extensive efforts to investigate the causative genes/markers of CM. Because more than 60 % of human protein-coding genes are predicted to be regulated by miRNAs, miRNAs may be involved in the mechanism of CM. The utilization of quantitative and qualitative analyses of miRNAs in CM will facilitate the discovery of new interventions for diagnostic and therapeutic purposes.
Abbreviations
- ATS:
-
Acidic terminal sequence
- CIDR:
-
Cysteine-rich interdomain region
- CM:
-
Cerebral malaria
- Csp :
-
Circumsporozoite protein
- DBL:
-
Duffy binding-like
- GPI:
-
Glycosylphosphatidylinositol
- ICAM-1:
-
Intercellular adhesion molecule-1
- IFN-γ:
-
Interferon-γ
- KAHRP:
-
Knob-associated histidine-rich protein
- memTNF:
-
Transmembrane form of TNF
- miRNAs:
-
MicroRNAs
- Msp-1:
-
Merozoite surface protein 1
- Msp-2:
-
Merozoite surface protein 2
- NTS:
-
N-terminal sequence
- PbA:
-
P. berghei ANKA
- PfEMP1:
-
P. falciparum erythrocyte membrane protein 1
- pRBCs:
-
Parasitized red blood cells
- SNPs:
-
Single nucleotide polymorphisms
- TNFR2:
-
Tissue necrotic factor receptor 2
- TNF-α:
-
Tumor necrosis factor alpha
References
World Health Organization. WHO malaria report Geneva. 2015.
Murphy SC, Breman JG. Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am J Trop Med Hyg. 2001;64(1–2 Suppl):57–67.
World Health Organization. Management of severe malaria: a practical handbook. 3rd ed. 2012.
Ball EA, Sambo MR, Martins M, Trovoada MJ, Benchimol C, Costa J, et al. FNAR1 controls progression to cerebral malaria in children and CD8+ T cell brain pathology in plasmodium berghei-infected mice. J Immunol. 2013;190(10):5118–27.
Lovegrove FE, Gharib SA, Patel SN, Hawkes CA, Kain KC WCL. Expression microarray analysis implicates apoptosis and interferon-responsive mechanisms in susceptibility to experimental cerebral malaria. Am J Pathol. 2007;171(6):1894–903.
Barbier M, Faille D, Loriod B, Textoris J, Camus C, Puthier D, et al. Platelets alter gene expression profile in human brain endothelial cells in an in vitro model of cerebral malaria. PLoS One. 2011;6(5):e19651.
Tripathi AK, Sullivan DJ, MF S. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74(6):3262–70.
Punsawad C, Maneerat Y, Chaisri U, Nantavisai K, Viriyavejakul P. Nuclear factor kappa B modulates apoptosis in the brain endothelial cells and intravascular leukocytes of fatal cerebral malaria. Malar J. 2013;12:260.
Combes V, Coltel N, Faille D, Wassmer SC, GE G. Cerebral malaria: role of microparticles and platelets in alterations of the blood–brain barrier. Int J Parasitol. 2006;36(5):541–6.
Lou J, Lucas R, GE G. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin Microbiol Rev. 2001;14(4):810–20.
Franke-Fayard B, Fonager J, Braks A, Khan SM CJJ. Sequestration and tissue accumulation of human malaria parasites: can we learn anything from rodent models of malaria? PLoS Pathog. 2010;6(9):e1001032.
Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol. 1999;29(6):927–37.
MacPherson GGWM, White NJ, Looareesuwan S, Warrell DA. Human cerebral malaria. A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985;119(3):385–401.
Dondorp AM, Pongponratn E, NJ W. Reduced microcirculatory flow in severe falciparum malaria: pathophysiology and electron-microscopic pathology. Acta Trop. 2004;89(3):309–17.
Pino P, Vouldoukis I, Kolb JP, Mahmoudi N, Livage ID, Bricaire F, et al. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis. 2003;187(8):1283–90.
Pottera S, Lingb TC, Balla HJ, Mansourb H, Mitchella A, Maluisha L, et al. Perforin mediated apoptosis of cerebral microvascular endothelial cells during experimental cerebral malaria. Int J Parasitol. 2006;36(4):485–96.
Combes V, El-Assaad F, Faille D, Jambou R, Hunt NH GEG. Microvesiculation and cell interactions at the brain-endothelial interface in cerebral malaria pathogenesis. Prog Neurobiol. 2010;91(2):140–51.
Grau GE ICG. Immunopathological consequences of the loss of engulfment genes: the case of ABCA1. J Soc Biol. 2005;199(2):199–206.
Combes V, Coltel N, Alibert M, Eck MV, Raymond C, Vague IJ, et al. ABCA1 gene deletion protects against cerebral malaria: potential pathogenic role of microparticles in neuropathology. Am J Pathol. 2005;166(1):295–302.
Combes V, Taylor TE, Vague IJ, Mège JL, Mwenechanya J, Tembo M, et al. Circulating endothelial microparticles in Malawian children with severe falciparum malaria complicated with coma. Jama. 2004;291(21):2542–4.
Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, et al. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A. 2008;105:14509–14.
Shanshan WH, Carla C, Chek MP, Sin YG, Laurent R. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol. 2015;37:221–31.
Albert B, Johnson A, Lewis J, Raff M, Roberts K, P W. Molecular biology of the cell. 5th ed. New York: Garland Science; 2008.
Dp B. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Biryukova I, Ye T, Levashina E. Transcriptome-wide analysis of microRNA expression in the malaria mosquito Anopheles gambiae. BMC Genomics. 2014;15:557.
LaMonte G, Philip N, Reardon J, Lacsina JR, Majoros W, Chapman L, et al. Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe. 2012;12(2):187–99.
El-Assaad F, Hempel C, Combes V, Mitchell AJ, Ball HJ, Kurtzhals JA, et al. Differential microRNA expression in experimental cerebral and noncerebral malaria. Infect Immun. 2011;79(6):2379–84.
Suarez Y, Wang C, Manes TD JSP. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J Immunol. 2010;184(1):21–5.
Wang Y, CG L. MicroRNA and cancer—focus on apoptosis. J Cell Mol Med. 2009;13(1):12–23.
Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481–6.
Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, et al. MicroRNA-181b regulates NF-kappaB-mediated vascular inflammation. J Clin Invest. 2012;122(6):1973–90.
Wang F, Xiong L, Huang X, Zhao T, Wu LY, Liu ZH, et al. miR-210 suppresses BNIP3 to protect against the apoptosis of neural progenitor cells. Stem Cell Res. 2013;11(1):657–67.
Day N, AM D. The management of patients with severe malaria. Am J Trop Med Hyg. 2007;77(6 Suppl):29–35.
Prapansilp P. Molecular pathological investigation of the pathophysiology of fatal malaria. UK: University of Oxford; 2012.
Atroosh WM, Al-Mekhlafi HM, Mahdy MA, Surin J. The detection of pfcrt and pfmdr1 point mutations as molecular markers of chloroquine drug resistance, Pahang, Malaysia. Malar J. 2012;11:251.
Triglia T, Wang P, Sims PF, Hyde JE, AF C. Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J. 1998;17(14):3807–15.
Peterson DS, Walliker D, TE W. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci U S A. 1988;85(23):9114–8.
Shandilya A, Chacko S, Jayaram B, Ghosh I. A plausible mechanism for the antimalarial activity of artemisinin: a computational approach. Sci Rep. 2013;3:2513.
Av H. Vaccines against malaria. Philosophical transactions of the Royal Society of London Series B. Biological Sci. 2011;366(1579):2806–14.
Mackinnon MJ, Mwangi TW, Snow RW, Marsh K, TN W. Heritability of malaria in Africa. PLoS Med. 2005;2(12):e340.
Gimenez F, Barraud de Lagerie S, Fernandez C, Pino P, Mazier D. Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003;60(8):1623–35.
Hananantachai H, Patarapotikul J, Ohashi J, Naka I, Krudsood S, Looareesuwan S, et al. Significant association between TNF-α (TNF) promoter allele (−1031C, −863C, and −857C) and cerebral malaria in Thailand. Tissue Antigens. 2007;69(3):277–80.
Lopez C, Saravia C, Gomez A, Hoebeke J, MA P. Mechanisms of genetically-based resistance to malaria. Gene. 2010;467(1–2):1–12.
Wilkinson RJ, Pasvol G. Host resistance to malaria runs into swampy water. Trends Microbiol. 1997;5(6):213–5.
Kyes S, Horrocks P, Newbold C. Antigenic variation at the infected red cell surface in malaria. Annu Rev Microbiol. 2001;55:673–707.
Pologe LG JVR. A chromosomal rearrangement in a P. falciparum histidine-rich protein gene is associated with the knobless phenotype. Nature. 1986;322(6078):474–7.
Goldberg DE AFC. Moving in and renovating: exporting proteins from Plasmodium into host erythrocytes. Nat Rev Microbiol. 2010;8(9):617–21.
Su XZ, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, et al. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82(1):89–100.
Rich SM, FJ A. Population structure and recent evolution of Plasmodium falciparum. Proc Natl Acad Sci U S A. 2000;97(13):6994–7001.
Beeson JG, GV B. Pathogenesis of Plasmodium falciparum malaria: the roles of parasite adhesion and antigenic variation. Cell Mol Life Sci. 2002;59(2):258–71.
Sahu U, Mohapatra BN, Kar SK, Ranjit M. Promoter polymorphisms in the ATP binding cassette transporter gene influence production of cell-derived microparticles and are highly associated with susceptibility to severe malaria in humans. Infect Immun. 2013;81(4):1287–94.
Panda AK, Panda M, Tripathy R, Pattanaik SS, Ravindran B, BK D. Complement receptor 1 variants confer protection from severe malaria in Odisha, India. PLoS One. 2012;7(11):e49420.
Teeranaipong P, Ohashi J, Patarapotikul J, Kimura R, Nuchnoi P, Hananantachai H, et al. A functional single-nucleotide polymorphism in the CR1 promoter region contributes to protection against cerebral malaria. J Infect Dis. 2008;198(12):1880–91.
McGuire W, Knight JC, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D. Severe malarial anemia and cerebral malaria are associated with different tumor necrosis factor promoter alleles. J Infect Dis. 1999;179(1):287–90.
Ubalee R, Suzuki F, Kikuchi M, Tasanor O, Wattanagoon Y, Ruangweerayut R, et al. Strong association of a tumor necrosis factor-alpha promoter allele with cerebral malaria in Myanmar. Tissue Antigens. 2001;58(6):407–10.
Nuchnoi P, Ohashi J, Kimura R, Hananantachai H, Naka I, Krudsood S, et al. Significant association between TIM1 promoter polymorphisms and protection against cerebral malaria in Thailand. Ann Hum Genet. 2008;72(Pt3):327–36.
Fry AE, Auburn S, Diakite M, Green A, Richardson A, Wilson J, et al. Variation in the ICAM1 gene is not associated with severe malaria phenotypes. Genes Immun. 2008;9(5):462–9.
Fernandez-Reyes D, Craig AG, Kyes SA, Peshu N, Snow RW, Berendt AR, et al. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Hum Mol Genet. 1997;6(8):1357–60.
Kuesap J, Hirayama K, Kikuchi M, Ruangweerayut R, K N-B. Study on association between genetic polymorphisms of haem oxygenase-1, tumour necrosis factor, cadmium exposure and malaria pathogenicity and severity. Malar J. 2010;9:260.
Takeda M, Kikuchi M, Ubalee R, Na-Bangchang K, Ruangweerayut R, Shibahara S, et al. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to cerebral malaria in Myanmar. Jpn J Infect Dis. 2005;58(5):268–71.
Kikuchia M, Looareesuwan S, Ubaleea R, Tasanorc O, Suzukia F, Wattanagoon F, et al. Association of adhesion molecule PECAM-1/CD31 polymorphism with susceptibility to cerebral malaria in Thais. Parasitol Int. 2001;50(4):235–9.
Omi K, Ohashi J, Naka I, Patarapotikul J, Hananantachai H, Looareesuwan S, et al. Polymorphisms of CD36 in Thai malaria patients. Southeast Asian J Trop Med Public Health. 2002;33 Suppl 3:1–4.
Omi K, Ohashi J, Patarapotikul J, Hananantachai H, Naka I, Looareesuwan S, et al. CD36 polymorphism is associated with protection from cerebral malaria. Am J Hum Genet. 2003;72(2):364–74.
Acknowledgements
We sincerely thank two anonymous reviewers and the Associated Editor for the valuable comment and suggestion. We appreciate Dr. Oranun Kerdpin from the Faculty of Pharmacy, Naresuan University, for editing the language of this manuscript.
Funding
This work was supported by the Thailand Research Fund, Office of the Higher Education Commission, and the Faculty of Medical Technology, Mahidol University, to PN (MRG5480062) and The Norwegian Ministry of Foreign Affairs, in collaboration with Mahidol University providing the Norwegian scholarship for Capacity Building for Institutes in Myanmar to STW. This work was also partially supported by the Office of the Higher Education Commission and Mahidol University under the National Research University Initiatives.
Availability of data and materials
None.
Authors’ contributions
STW and PN drafted the manuscript. PN, HH, UK, CP, and VP edited and critically revised the manuscript. All authors read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
None.
Ethics approval and consent to participate
None.
Disclosure statement
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wah, S.T., Hananantachai, H., Kerdpin, U. et al. Molecular basis of human cerebral malaria development. Trop Med Health 44, 33 (2016). https://doi.org/10.1186/s41182-016-0033-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41182-016-0033-6