
Abstract
Objective
Melanoma accounts for 80% of skin cancer deaths. The pathogenesis of melanoma is regulated by gene networks. Thus, we aimed here to identify gene networks and hub genes associated with melanoma and to further identify their underlying mechanisms.
Methods
GTEx (normal skin) and TCGA (melanoma tumor) RNA-seq datasets were employed for this purpose. We conducted weighted gene co-expression network analysis (WGCNA) to identify key modules and hub genes associated with melanoma. Log-rank analysis and multivariate Cox model analysis were performed to identify prognosis genes, which were validated using two independent melanoma datasets. We also evaluated the correlation between prognostic gene and immune cell infiltration.
Results
The blue module was the most relevant for melanoma and was thus considered the key module. Intersecting genes were identified between this module and differentially expressed genes (DEGs). Finally, 72 genes were identified and verified as hub genes using the Oncomine database. Log-rank analysis and multivariate Cox model analysis identified 13 genes that were associated with the prognosis of the metastatic melanoma group, and RTP4 was validated as a prognostic gene using two independent melanoma datasets. RTP4 was not previously associated with melanoma. When we evaluated the correlation between prognostic gene and immune cell infiltration, we discovered that RTP4 was associated with immune cell infiltration. Further, RTP4 was significantly associated with genes encoding components of immune checkpoints (PDCD1, TIM-3, and LAG3).
Conclusions
RTP4 is a novel prognosis-related hub gene in cutaneous melanoma. The novel gene RTP4 identified here will facilitate the exploration of the molecular mechanism of the pathogenesis and progression of melanoma and the discovery of potential new target for drug therapy.
Similar content being viewed by others

Introduction
Melanoma and nonmelanoma represent the two main types of skin cancer. The most common nonmelanomas are basal cell and squamous cell carcinomas. Nonmelanoma skin cancers are the most frequently occurring skin cancers, with greater than one million diagnoses worldwide in 2018. Melanoma represents 21.6% of skin cancer cases. According to the GLOBOCAN database (gco.iarc.fr), in 2018, there were 287,723 new cases of melanoma worldwide. Melanoma is more aggressive and has a high mortality rate compared with other types of skin cancers, and its mortality rate is approximately 8-times higher compared with that of nonmelanoma skin cancer [1, 2]. The poor prognosis of patients with melanoma is associated with its high metastatic potential [3]. The main causes of death include extensive metastasis to the lung, liver, bone, and brain [4]. Early diagnosis and treatment of melanoma are therefore required to reduce its significant threat to human health.
Hyperplasia of abnormal melanocytes is a risk factor for melanoma, although most such hyperplasias are benign. However, some are associated with a significant risk of developing into a melanoma or already have developed its malignant phenotype. Among numerous studies aimed to identify the mechanism of melanoma, one study identified differentially expressed genes (DEGs) between metastatic and primary melanomas. These DEGs were enriched in mRNAs encoding cell adhesion and proliferation molecules. A protein–protein interaction network was also constructed, and some key genes with higher degrees in the network has been identified, such as PCNA, CDK1, and MAD2L1 [5]. As disruption of the epigenomic landscape is recognized as a widespread feature inherent in tumor development and progression, a study of DNA methylation identified markers associated with the melanoma. These identified methylation biomarkers involved in melanoma development (e.g., HOXA9 methylation) and tumor progression (e.g., TBC1D16 methylation). In addition, they determined PON3 DNA methylation as biomarkers with prognostic information independent of tumor thickness and ulceration [6]. Mutational analysis of melanoma tissue was another strategy to identify biomarkers between metastatic and primary melanomas. Numerous genes that have somatic mutations were identified, such as BRAF and TERT [7].
Weighted gene co-expression network analysis (WGCNA) is a systematic biological approach for constructing weighted correlation networks to identify key modules that are highly associated with clinical traits. Moreover, WGCNA is used to measure relationships between modules and genes as well as to rank genes in modules of interest with clinical data [8]. WGCNA can identify core-related genes, which may be involved in important pathological processes and have important clinical application. WGCNA is therefore widely used to conduct association analyses of gene sets with diseases and to identify candidate hub genes, particularly of patients with cancer [9, 10]. For example, a WGCNA study of pancreatic ductal adenocarcinoma identified five modules and found 10 hub genes that may indicate poor prognosis [11]. It has been established that the metastatic ability of melanoma is regulated by an intricate gene network. Thus, WGCNA method was also applied to investigate the relationship between the key module and hub genes associated with the metastasis ability of melanoma. PKP1 was identified as a new potential tumor suppressor in human melanoma, likely through regulating calcium signaling pathways [12]. Another study screened out SMARCA4 associated with metastasis melanoma, which in turn affects the signal transduction of the adherens junction [13]. Other genes CCNB2, ARHGAP30, and SEMA4D were also identified by WGCNA, which associated with survival as potential prognostic predictors and molecular targets of treatment [14].
Biomarkers for prognosis of cutaneous melanoma have drawn intensive attentions in recent years, and many studies have identified plenty of effective biomarkers and therapeutic targets in metastatic melanoma. To improve our understanding of the biological pathology of metastatic melanoma, we applied WGCNA to identify key modules and hub genes associated with melanoma. Prognostic genes were identified using multivariate Cox model analysis. We further examined the associations between prognostic genes, immune cell infiltration, and immune checkpoints.
Materials and methods
Data collection
GTEx (normal skin) and TCGA (melanoma tumor, SKCM) total gene RNA-seq datasets were obtained from UCSC Xena (https://xenabrowser.net/). TOIL was used to reprocess raw GTEx and TCGA counts data to correct for batch effects and to conduct merge-analysis of GTEx and TCGA datasets [15]. The combined data set included 469 melanoma samples and 812 normal samples. Basic clinical information of the 469 melanoma samples was downloaded from the UCSC Xena website, including sample type, Breslow depth, vital status, sex, ulceration, survival time, and TNM stage (Table S1). The samples comprised 367 metastatic and 102 primary melanomas, respectively.
Analyses of DEGs
mRNA expression data were extracted from the total gene expression data, yielding 19,521 mRNA genes for analysis. The DEGs were identified using the DESeq2 software package [16]. In this study, a |log2-fold change (log2FC)|> 2 and adjusted P < 0.01 were selected as the standard cutoffs to identify DEGs. Gene expression levels of normal skin and all tumor samples were determined, including comparisons of those of normal skin and all tumors, normal skin and primary tumors, normal skin and metastatic tumors, and primary and metastatic tumors.
WGCNA
We used the WGCNA package to construct gene co-expression networks. Raw mRNA gene counts were normalized to FPKM values, then converted to log2 (FPKM + 1) values. First, we excluded genes that were not detected using the expression profile and then calculated the variance of each included gene. Genes with standard deviations in the top 20% were subjected to further analysis. Certain samples were distant, and outliers were excluded according to their cluster distances.
To construct a weighted gene network, the soft threshold power β was defined as 6, which was the lowest power based on a scale-free topology [17]. After constructing a scale-free network, the expression matrix was transformed into an adjacency matrix and then transformed into a topological matrix. We used the average-linkage hierarchical clustering method, according to the topological overlap measure (TOM), to cluster genes, and we accordingly defined the minimum number of each gene network module as 30. The threshold for similar module combinations was defined as 0.25. Therefore, after identifying gene modules using dynamic shear, we calculated module eigengenes (MEs, the first principal component of one module), clustered the modules, and merged closer modules into new modules according to height = 0.25. To identify modules that were significantly associated with clinical traits, we generated a heat map of module-trait relationships according to the tutorial included in the WGCNA package of R. Genes in the most significant module were selected for subsequent analysis.
PPI network construction
Overlapping genes between significant modules and DEGs were selected for protein–protein interaction (PPI) analysis. The STRING tool provides information for this purpose [18]. PPI analysis may reveal protein function and identify cellular mechanisms through protein interactions. Here we established a PPI network of potential genes, and genes not significantly associated with other genes in the PPI network were excluded from subsequent analyses.
Functional enrichment analysis of network module genes
To analyze the functions of genes in the modules, we conducted Gene Ontology (GO) [19] and Kyoto Encyclopedia Gene and Genomes (KEGG pathway) [20] analyses using the cluster-Profiler package [21]. We identified overrepresented GO terms in the categories as follows: biological process (BP), molecular function (MF), and cellular component (CC). Those in KEGG pathway were similarly identified. The false discovery rate (FDR) P value < 0.05 was selected as the threshold.
Identification and validation of hub genes
Hub genes are highly connected within a module and are significantly associated with biological functions [22]. Here we defined hub genes with high module membership (MM) (|cor.weighted|> 0.85). We used the Oncomine database (https://www.oncomine.org) to verify the expression of hub genes, which included the melanoma datasets as follows: haqq, riker, and talantov. A meta-analysis was performed that employed analysis of P values to evaluate the significance of differences in gene expression between melanomas and normal skin. P < 0.05 represents significant difference.
Identification and validation of prognostic genes
Prognostic genes were screened according to prognostic information into metastatic melanoma and primary melanoma groups. Patients were stratified into a high-level group or a low-level group according to the median expression level, and the Kaplan–Meier method was used to analyze survival. The log-rank test was used to compare the survival curves of patients in different groups, and P < 0.05 indicates significant differences. Analysis of a Cox proportional hazards model was performed after adjusting for sex, age upon diagnosis, and tumor stage. P < 0.05 represents a significant difference. The GSE65904 [23] and GSE22153 [24] of the GEO database were used for validating potential prognostic genes. The GSE65904 and GSE22153 datasets included 188 and 57 metastatic melanoma samples, respectively. Analysis using a Cox proportional hazards model was performed after adjusting for sex and age upon diagnosis. P < 0.05 indicates a significant difference.
Immune-related analysis
The microenvironment of melanoma tissue comprises tumor and immune cells as well as their secreted molecules such as proinflammatory and anti-inflammatory factors, which ultimately determine the malignant phenotype of the tumor. We therefore used the Tumor Immune Estimation Resource (TIMER) tool (http://cistrome.org/TIMER/) to evaluate the correlation between prognostic genes and immune cell infiltration in SKCM-Metastasis, including B cells, CD8 + T cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells (DCs) [25]. Purity-corrected partial Spearman’s rho value and statistical significance were applied. For gene expression levels compared with tumor-cell purity, P < 0.05 indicates a significant difference. Further, gene expression data were evaluated for immune checkpoint genes, including PDCD1 (PD1), CD274 (PD-L1), PDCD1LG2 (PD-L2), CTLA4, TIM-3, and LAG3. |log2FC)|> 1 and P < 0.01 were considered significant. The relationship between prognostic genes and immune checkpoints such as PDCD1, CD274, PDCD1LG2, CTLA4, TIM-3, and LAG3 were evaluated using Pearson’s correlation analysis.
Druggable targets
The ChEMBL and Drugbank databases comprise pharmacodynamic and pharmacokinetic information for bioactive compounds and drugs [26]. We searched for immune-related genes among the two databases to identify genes as targets of approved drugs or bioactive compounds.
Results
Detection of DEGs
The procedures used in our study were illustrated in the flow chart in Fig. 1. The gene expression levels of normal skin and tumor samples were compared. We detected 3,834 DEGs. Compared with 812 normal samples, 2,212 downregulated genes and 1,622 upregulated genes were identified in the 469 melanoma samples (Fig. S1). We further compared normal skin and primary tumors, normal skin and metastatic tumors, and primary tumors and metastatic tumors (Fig. 2). We identified 18 significant DEGs among all comparisons, indicating that they may play continuous roles in tumor development (Table S2), including AGR3, CRP, HTN3, KRT26, KRT38, KRTAP10-3, KRTAP10-4, KRTAP10-5, KRTAP10-7, KRTAP10-8, KRTAP10-9, KRTAP1-1, KRTAP2-4, KRTAP4-5, PRH2, SAGE1, TBC1D3, and TKTL1. Among these genes, 11 encode keratins or related proteins. We further identified 11 DEGs in a comparison of metastatic tumors with primary tumors (Table S3), which were not detected in the comparisons between other groups, including HMX1, ALB, ORM2, PRB3, SFTA3, RTL1, CRABP1, OR1E1, APOH, FBN3, and IGFL1. These results indicate that these genes play an important role in tumor metastasis.

Analytical procedures

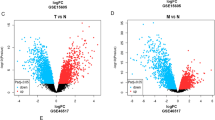
Common differentially expressed genes. A Venn diagram was utilized to screen the common genes. The comparison was made between normal skin and all tumor samples. Further comparisons included normal skin and primary tumors, normal skins and metastatic tumors, and primary and metastatic tumors
A weighted gene co-expression network
We defined β = 6 (scale-free R2 = 0.95) as the appropriate soft-thresholding value to satisfy the scale-free network criteria. We identified eight modules, which were assigned different colors (Fig. 3a). We calculated the correlation between modules and clinical traits (Fig. 3b). These analyses show that tumor (melanoma) significantly correlated with most of the modules in the eight clinical traits. According to the correlation between MEs and traits, the blue module (n = 438 genes) was the most relevant, and therefore, a key module associated with melanoma.

Identification of a module associated with clinical features. a Dendrogram of expressed genes clustered according to a dissimilarity measure (1-TOM). Dynamic Tree Cut corresponds to the original module, and Merged Dynamic corresponds to the final module. b Heat map of the correlation between modular significant and clinical features
PPI network construction
There were 350 intersecting genes between the blue module and DEGs, which were used to constructed a PPI network for validating gene co-expression network proteins. A node represents a gene in the network, and the edge represents the interaction between genes. Genes not linked to other genes in the PPI network were deleted, and a PPI network was reconstructed using the remaining genes. The network contained 264 nodes and 716 edges (Fig. S2). The PPI data indicated that these genes had a significant regulatory relationship and that 264 genes of the blue module may interact.
GO and pathway enrichment analysis
The genes in the blue module were selected for GO and KEGG pathway enrichment analysis. The most overrepresented GO terms in BP were associated with melanin, including pigmentation, melanin biosynthesis, and metabolism as well as developmental pigmentation. In the CC categories, the enriched GO terms in the blue module were primarily associated with melanosome and pigment granule (Fig. 4a). According to the KEGG database, the genes in the blue module were mainly enriched for transcriptional dysregulation in cancer, protein digestion and absorption, breast cancer, gastric cancer, and melanogenesis pathways (Fig. 4b).

Functional enrichment of genes in the blue module. a Gene Ontology (GO) functional enrichment of genes in the blue module. The x-axis represents the number of genes of each term and the y-axis shows the GO terms. BP: biological process, CC: cell component. b Kyoto Encyclopedia Gene and Genomes (KEGG) functional enrichment of genes in the blue module. The x-axis represents the number of genes of each term and the y-axis shows the KEGG terms
Hub gene identification and validation
The screening criteria of hub genes in the key module (|MM|> 0.85), identified 85 genes in the blue module as hub genes in the co-expression network. Meta-analysis of the Oncomine database employed the P value to validate expression differences between hub genes of melanoma and normal skin. Seventy-two genes were validated (P < 0.05) in this module. We generated a heat map to visualize the expressing of these 72 hub genes in DEG- discovery stage (Fig. S3).
Identification and validation of prognostic genes
We used log-rank analysis to evaluate the difference in overall survival between high and low expression levels of the 72 hub genes. The prognostic genes screened according to prognostic information were allocated into separate groups of metastatic melanoma or primary melanoma. This procedure found that 22 genes were significantly associated with the prognosis of patients in the metastatic group (Fig. 5), although a prognosis-associated gene was not identified in the primary group. Multivariate analysis revealed that 13 genes remained associated with prognosis (Table 1). Analysis of two independent datasets (GSE65904 and GSE22153) validated that RTP4 was a significant prognostic gene (Tables S4 and Table S5). Above results demonstrated that patients with metastatic melanoma with high expression of RTP4 experienced significantly longer overall survival than those with low expression. The RTP4 gene can act as an independent prognostic factor independent of other clinical traits.

Overall survival associated with the prognostic genes expressed by patients with metastatic melanoma. Patients were stratified into a high-level or low-level group according to median expression levels
Immune-related genes
Immune cell infiltration is highly associated with tumor development. Therefore, TIMER was utilized to examine the correlations between the expression levels of prognostic genes and immune cell infiltration. We discovered that RTP4 was associated with immune cell infiltration of samples of melanoma metastases, which negatively correlated with the cellular purity of melanomas and positively correlated with neutrophils (Fig. 6a). Gene expression levels were evaluated to identify immune checkpoint genes, including PDCD1, CD274, PDCD1LG2, CTLA4, TIM-3, and LAG3. PDCD1, CTLA4, TIM-3, and LAG3 were upregulated in melanomas compared with normal skin. Subsequently, correlations between immune checkpoints (PDCD1, CTLA4, TIM-3, LAG3) and RTP4 were evaluated. We found that RTP4 positively correlated with the genes mentioned above, including PDCD1 (r = 0.27, P = 4.2E-9), TIM-3 (r = 0.25, P = 9.4E-8), and LAG3 (r = 0.28, P = 8.5E-10) (Fig. 6b).

Effect of RTP4 on immune cell infiltration and immune checkpoint genes. a The correlation between RTP4 and immune cell infiltration. b Correlation between immune checkpoint genes and RTP4
Druggable targets
To determine whether the immune-related gene RTP4 expressed by patients with melanoma had predicted drug targets, we queried the ChEMBL and Drugbank databases. We were unable to identify such RTP4 targets. More efforts need to be made for drug development targeting this gene.
Discussion
The prognosis of patients with melanoma predominantly depends on the disease stage. The prognosis of patients with local disease is generally favorable, and the 5-year survival rate exceeds 90% [27]. Therefore, early diagnosis is critically important for administering effective treatment. The identification of diagnostic or prognostic biomarkers is therefore increasingly important. To this end, here we screened for potential biomarkers associated with melanoma using TCGA data. When we used WGCNA to identify gene modules significantly associated with melanoma, we identified a module (blue) as that most relevant to melanoma. This module comprised 85 genes with a correlation > 0.85. After validation using the Oncomine database, 72 hub genes were validated. The prognostic value of these biomarkers was evaluated using the information included in this dataset. Among them, 13 genes were significantly associated with the prognosis of patients assigned to the metastatic group. Two datasets were used to validate prognostic significance, and RTP4 was validated.
RTP4 was identified and validated as a prognostic gene. It is a novel gene associated with melanoma, which was not discovered in other melanoma studies. RTP4 has the potential to be an effective biomarkers and therapeutic targets in metastatic melanoma. RTP4 encodes a receptor transporter protein, which facilitates trafficking and functional cell surface expression of certain G protein-coupled receptors (GPCRs). RTP4 represents a novel targeting opportunity for many kinds of cancers. For example, two studies link RTP4 with prostate and breast cancer, respectively. One study found that RTP4 is a novel methylated biomarker for accurate diagnosis and treatment of prostate adenocarcinoma [28]. Another study discovered that the expression level of RTP4 independently predicts breast cancer outcomes of HER2( +) patients [29]. Further, RTP4 is required for antigen-dependent immune editing of cancer cells using CRISPR screens [30]. RTP4 plays a role in diverse viral infections such as those caused by HPV16 and influenza virus [31, 32]. Canonical interferons (IFNs) induce interferon-stimulated genes to exert their antiviral activities. RTP4 is induced by type I IFN (IFN-I) and binds the TANK-binding kinase (TBK1) complex to negatively regulate TBK1 signaling through interference with the expression and phosphorylation of TBK1 [33].
Immune cell infiltration is highly associated with the development of the tumor, and we therefore evaluated the correlation between prognostic gene expression and immune cell infiltration. As a result, we discovered that RTP4 was associated with immune cell infiltration of melanoma metastases. RTP4 expression negatively correlated with melanoma purity and highly correlated with neutrophils. Many types of immune cells migrate close to tumors and exert an anti-tumor effect [34]. Further, these infiltrating immune cells can induce tumor cells to produce immunosuppressive phenotypes, thereby promoting tumor growth. Thus, the role of neutrophils in tumors has attracted our attention. The close correlation between neutrophils and RTP4 indicates that RTP4 contributes to the regulation of neutrophil function. We further discovered that PDCD1, CTLA4, TIM-3, LAG3, and RTP4 were upregulated in melanomas. RTP4 was positively correlated with the genes mentioned above, including PDCD1, TIM-3, and LAG3, suggesting that immune checkpoint inhibitors may be considered for treating such patients.
Gene expression comparisons were made between normal skin and all tumor samples, normal skin and primary tumors, normal skin and metastatic tumors, and primary and metastatic tumors. Eighteen genes were significantly differentially expressed in all comparisons, indicating that these genes may play a continuous role in melanoma development. After reviewing the literature, no study on the relationship between these genes and melanoma was found; and the roles of these genes in melanoma therefore deserve further study. Among these genes, 11 encode keratins or related proteins. KRT26 and KRT38 are members of the keratin gene family and belong to type I keratins. Diseases associated with KRT38 include nodular basal cell carcinoma [35]. KRTAP10-3, KRTAP10-4, KRTAP10-5, KRTAP10-7, KRTAP10-8, KRTAP10-9, KRTAP1-1, KRTAP2-4, and KRTAP4-5 encode keratin-associated proteins, which are members of the keratin-associated protein (KAP) family. The KAP proteins form a matrix of keratin intermediate filaments, which contribute to the structure of hair fibers. Eleven genes were significantly differentially expressed between metastases and primary tumors, and ALB, ORM2, CRABP1, and APOH may be associated with the metastasis of melanoma. These genes are associated with several cancers, such as ALB that is associated with lymph node metastasis in lung squamous cell carcinoma [36]. The C-reactive protein (CRP)/albumin (ALB) ratio serves as a prognostic marker for several cancers [37]. There is no relevant functional study on the relationship between ALB, ORM2, CRABP1, and APOH and melanoma metastasis, which may represent future candidates.
Conclusions
Our aim here was to identify gene networks and hub genes associated with melanoma and to further identify the underlying mechanisms responsible for oncogenesis and tumor progression, including metastasis. WGCNA and survival analysis were used to identify key hub genes with prognostic value for melanoma. These identified prognostic genes were validated by two independent metastasis melanoma datasets. We also evaluated the correlation between prognostic gene and immune cell infiltration. RTP4 was significantly associated with the prognosis of patients with melanoma and was defined as a prognostic gene. We further discovered that RTP4 was associated with immune cell infiltration, which negatively correlated with the cellular purity of melanomas and positively correlated with neutrophils. Further, RTP4 was significantly associated with genes encoding components of immune checkpoints (PDCD1, TIM-3, and LAG3). Notably, RTP4 is a novel gene not previously associated with melanoma. Our research therefore provides more information for exploring the mechanisms responsible for the development of melanoma.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article. Materials are available from the corresponding author on reasonable request.
Abbreviations
- TCGA:
-
The Cancer Genome Atlas
- WGCNA:
-
Weighted gene co-expression network analysis
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia Gene and Genomes
- BP:
-
Biological process
- MF:
-
Molecular function
- CC:
-
Cellular component
- FDR:
-
False discovery rate
- PPI:
-
Protein–protein interaction
References
Finn L, Markovic SN, Joseph RW. Therapy for metastatic melanoma: the past, present, and future. BMC Med. 2012;10:23.
Liu-Smith F, Jia J, Zheng Y. UV-Induced Molecular Signaling Differences in Melanoma and Non-melanoma Skin Cancer. Adv Exp Med Biol. 2017;996:27–40.
Garbe C, Peris K, Hauschild A, et al. Diagnosis and treatment of melanoma. European consensus-based interdisciplinary guideline - Update 2016. Eur J Cancer. 2016;63:201–17.
Zhu Z, Liu W, Gotlieb V. The rapidly evolving therapies for advanced melanoma–Towards immunotherapy, molecular targeted therapy, and beyond. Crit Rev Oncol Hematol. 2016;99:91–9.
Qiu T, Wang H, Wang Y, Zhang Y, Hui Q, Tao K. Identification of genes associated with melanoma metastasis. Kaohsiung J Med Sci. 2015;31(11):553–61.
Wouters J, Vizoso M, Martinez-Cardus A, et al. Comprehensive DNA methylation study identifies novel progression-related and prognostic markers for cutaneous melanoma. BMC Med. 2017;15(1):101.
Yang S, Leone DA, Biswas A, et al. Concordance of somatic mutation profiles (BRAF, NRAS, and TERT) and tumoral PD-L1 in matched primary cutaneous and metastatic melanoma samples. Hum Pathol. 2018;82:206–14.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Tang J, Kong D, Cui Q, et al. Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis. Front Oncol. 2018;8:374.
Bao L, Guo T, Wang J, Zhang K, Bao M. Prognostic genes of triple-negative breast cancer identified by weighted gene co-expression network analysis. Oncol Lett. 2020;19(1):127–38.
Zhou Z, Cheng Y, Jiang Y, et al. Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis. Int J Biol Sci. 2018;14(2):124–36.
Wang HZ, Wang F, Chen PF, et al. Coexpression network analysis identified that plakophilin 1 is associated with the metastasis in human melanoma. Biomed Pharmacother. 2019;111:1234–42.
Liu SR, Yang X, Qi L, Zhu Z, Ji YZ. SMARCA4 promotes benign skin malignant transformation into melanoma through Adherens junction signal transduction. Clin Transl Oncol. 2021;23(3):591–600.
Chen S, Liu Z, Li M, et al. Potential Prognostic Predictors and Molecular Targets for Skin Melanoma Screened by Weighted Gene Co-expression Network Analysis. Curr Gene Ther. 2020;20(1):5–14.
Vivian J, Rao AA, Nothaft FA, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. 2017;35(4):314–6.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:17.
Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–13.
Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43(Database issue):D1049-56.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44(D1):D457–62.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Chen L, Yuan L, Wang Y, et al. Co-expression network analysis identified FCER1G in association with progression and prognosis in human clear cell renal cell carcinoma. Int J Biol Sci. 2017;13(11):1361–72.
Cirenajwis H, Ekedahl H, Lauss M, et al. Molecular stratification of metastatic melanoma using gene expression profiling: Prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget. 2015;6(14):12297–309.
Jonsson G, Busch C, Knappskog S, et al. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16(13):3356–67.
Li T, Fan J, Wang B, et al. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017;77(21):e108–10.
Gaulton A, Bellis LI, Bento AP, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(Database issue):D1100-7.
Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–206.
Xu N, Wu YP, Ke ZB, et al. Identification of key DNA methylation-driven genes in prostate adenocarcinoma: an integrative analysis of TCGA methylation data. J Transl Med. 2019;17(1):311.
Laurin M, Huber J, Pelletier A, et al. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci U S A. 2013;110(18):7434–9.
Wroblewska A, Dhainaut M, Ben-Zvi B, et al. Protein Barcodes Enable High-Dimensional Single-Cell CRISPR Screens. Cell. 2018;175(4):1141–55.
Klymenko T, Gu Q, Herbert I, et al. RNA-Seq Analysis of Differentiated Keratinocytes Reveals a Massive Response to Late Events during Human Papillomavirus 16 Infection, Including Loss of Epithelial Barrier Function. J Virol. 2017;91(24):e01001-e1017.
Zarei Ghobadi M, Mozhgani SH, Farzanehpour M, Behzadian F. Identifying novel biomarkers of the pediatric influenza infection by weighted co-expression network analysis. Virol J. 2019;16(1):124.
He X, Ashbrook AW, Du Y, et al. RTP4 inhibits IFN-I response and enhances experimental cerebral malaria and neuropathology. Proc Natl Acad Sci U S A. 2020;117(32):19465–74.
Wang D, He MQ, Fan DQ. RARRES1 is a novel immune-related biomarker in GBM. Am J Transl Res. 2019;11(9):5655–63.
Kurzen H, Esposito L, Langbein L, Hartschuh W. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23(6):501–9.
Dong M, Gong H, Li T, et al. Lymph node metastasis in lung squamous cell carcinoma and identification of metastasis-related genes based on the Cancer Genome Atlas. Cancer Med. 2019;8(14):6280–94.
Kudou K, Saeki H, Nakashima Y, et al. C-reactive protein/albumin ratio is a poor prognostic factor of esophagogastric junction and upper gastric cancer. J Gastroenterol Hepatol. 2019;34(2):355–63.
Acknowledgements
We thank the anonymous reviewers for their comments and suggestions which improved the manuscript.
Funding
The present study was supported by grants from the National Natural Science Foundation of China (No.81760565), Youth Top Talent project of High-level talent development support program of Yunnan Province, and Li Yunqing expert workstation of Yunnan Province (NO.202005AF150014).
Author information
Authors and Affiliations
Contributions
JY conceived this study; YL and JQ performed the analysis; YL, JQ and JY prepared the manuscript. All authors have read and approved the submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No competing financial interests exist.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Volcano plots reflecting significant differentially expressed genes by patients with melanoma compared with normal controls. Figure S2. Protein-protein interaction network of genes in the blue module. Figure S3. Heat map of 72 validated hub genes during the discovery stage of differentially expressed genes
Additional file 2: Table S1.
Basic clinical information of 469 melanoma samples. Table S2. Common differentially expressed genes in all comparisons. The gene expression comparison was made between normal skin and all tumor samples,normal skin and primary tumors, normal skin and metastatic tumors, and primary tumors and metastatic tumors. Table S3. Eleven genes were significantly differentially expressed in the comparison of metastatic tumors with primary tumors. Table S4. Prognostic genes in patients with metastatic melanoma identified using the GSE65904 dataset. Table S5. Prognostic genes in patients with metastatic melanoma identified using the GSE22153 dataset
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Qi, J. & Yang, J. RTP4 is a novel prognosis-related hub gene in cutaneous melanoma. Hereditas 158, 22 (2021). https://doi.org/10.1186/s41065-021-00183-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-021-00183-z