
Abstract
Background
Marine benthic prokaryotic communities play crucial roles in material recycling within coastal environments, including coral reefs. Coastal sedimentary microbiomes are particularly important as potential reservoirs of symbiotic, beneficial, and pathogenic bacteria in coral reef environments, and therefore presumably play a core role in local ecosystem functioning. However, there is a lack of studies comparing different environments with multiple sites on the island scale, particularly studies focusing on prokaryotic communities, as previous investigations have focused mainly on a single site or on specific environmental conditions. In our study, we collected coastal sediments from seven sites around Okinawa Island, Japan, including three different benthic types; sandy bottoms, seagrass meadows, and hard substratum with living scleractinian corals. We then used metabarcoding to identify prokaryotic compositions and estimate enzymes encoded by genes to infer their functions.
Results
The results showed that the three substrata had significantly different prokaryotic compositions. Seagrass meadow sites exhibited significantly higher prokaryotic alpha-diversity compared to sandy bottom sites. ANCOM analysis revealed that multiple bacterial orders were differentially abundant within each substratum. At coral reef sites, putative disease- and thermal stress-related opportunistic bacteria such as Rhodobacterales, Verrucomicrobiales, and Cytophagales were comparatively abundant, while seagrass meadow sites abundantly harbored Desulfobacterales, Steroidobacterales and Chromatiales, which are common bacterial orders in seagrass meadows. According to our gene-coded enzyme analyses the numbers of differentially abundant enzymes were highest in coral reef sites. Notably, superoxide dismutase, an important enzyme for anti-oxidative stress in coral tissue, was abundant at coral sites. Our results provide a list of prokaryotes to look into in each substrate, and further emphasize the importance of considering the microbiome, especially when focusing on environmental conservation.
Conclusion
Our findings prove that prokaryotic metabarcoding is capable of capturing compositional differences and the diversity of microbial communities in three different environments. Furthermore, several taxa were suggested to be differentially more abundant in specific environments, and gene-coded enzymic compositions also showed possible differences in ecological functions. Further study, in combination with field observations and temporal sampling, is key to achieving a better understanding of the interactions between the local microbiome and the surrounding benthic community.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
Microbiomes play crucial roles in global biochemical cycles, influencing essential compounds like phosphate, sulfate, nitrate, carbon, and metals in marine habitats [1, 2]. Recent advancements in technologies, such as high throughput sequencers, allow researchers to better investigate marine microbial communities in various habitats and understand their responses to environmental factors with high resolution [2,3,4]. Exploring microbial compositions in the environment is not only essential for the study of biodiversity and for understanding species’ interactions and aggregations at local scales [5, 6], but is also useful for assessing the general health of environments, as microbes are highly sensitive to marine environmental pollution [7,8,9,10]. Environmental assessments using microbial communities as proxies have explored the biological impact of anthropogenic activities, for example of mariculture [11,12,13]. Sedimentary prokaryotes have been shown to potentially mitigate anthropogenic contamination in the sediment, such as from heavy metals, polyaromatic hydrocarbons (PAHs), petroleum, and excess nutrients [14,15,16,17]. However, in the long term, intense environmental disturbances can lead to the lack of recovery of prokaryotic communities or their functions [18, 19].
In coral reef ecosystems, the interactions between benthic organisms and their associated prokaryotes play critical and fundamental roles in maintaining the overall health of the entire ecosystem [20,21,22,23,24,25]. These interactions have become increasingly important as coastal coral reefs face numerous threats such as nutrient loading [26, 27], land reclamation [28], ocean acidification [29,30,31], coral disease outbreaks [32,33,34], outbreaks of various organisms [35,36,37,38], and rising sea surface temperatures [39,40,41]. These global scale disturbances have made coral reefs at risk to “phase-shifts” [42], or large scale substitution of benthic structures from a coral-dominated state to being dominated by other organisms (e.g. macroalgae [43,44,45,46], sponges [47, 48], soft corals [49, 50], zoantharians [51, 52], etc.) or even to primarily abiotic substrate (e.g. rocky substrate) as suggested by Bellwood et al. [42]. To prevent such drastic changes, and to facilitate coral recovery from damage, environmental microbiome-based methodologies are likely to be critical [53]. However, while there have been many studies on scleractinian coral-associated prokaryotes [54,55,56,57,58], sponge-associated prokaryotes [5, 59,60,61,62,63,64,65,66], seawater prokaryotes [4, 67, 68], and potentially free-living Symbiodiniaceae [69,70,71,72], research on sedimentary prokaryotes in different coral reef habitats remains generally quite limited (e.g., Dong et al. [73]).
Scleractinian corals are known to exude substantial amount of mucus, which can introduce their body surface microbes and/or trapped microbes into the water column and sediments, and this coral mucus may potentially influence the surrounding sedimentary prokaryotic community [74]. Sedimentary prokaryotic communities are known to be more diverse and/or more similar to coral-associated communities than to those found in seawater [5, 75,76,77,78]. Carlos et al. [75] suggested that sediments might serve as potential coral pathogen reservoirs. Indeed, resuspended sedimentary prokaryotes can migrate onto the surface of coral, and the coral’s mucus layer can provide a means for prokaryotes to hitch a ride via mucus shed from the coral’s body surface [76]. Monitoring prokaryotic compositions in sediments, therefore, can be a key task in tracking coral health at the local level. Likewise, in seagrass meadows, which are vital for carbon sequestration (also known as “blue carbon” [79, 80]), the rhizosphere microbiome plays a crucial role in maintaining meadow health, involving essential biochemical pathways such as nitrogen fixation, sulfate reduction, sulfide oxidation, and urea turnover [81,82,83]. Alterations in sedimentary chemical conditions, such as hypoxia and/or enriched sulfide, often combined with other stressors such as high/low salinities or temperatures, can lead to large-scale die-offs of seagrass [84,85,86], which also affect prokaryote composition [83, 87, 88]. Prokaryotic communities have been shown to have the ability to recover relatively quickly after such disturbances due to their composition plasticity [19, 89, 90]. However, the recovery of organisms that rely on important biochemical pathways provided by prokaryotes may take longer.
Taking all of these fact into consideration, monitoring sedimentary microbial communities in coral reef ecosystems is of great importance, especially when comparing multiple environments with different communities (e.g., scleractinian corals, seagrass, sandy bottoms) within a small geographic range that could be connected via local currents and tidal fluctuations. In this context, despite its small size, subtropical Okinawa Island, Japan is an excellent natural laboratory for such studies as it has different marine environments with varying levels of anthropogenic impacts. In this study, we characterized the bacterial taxa of different marine habitats, namely coral-abundant sites, seagrass-abundant sites, and sandy bottom sites using sediment samples, and then consider which protist taxa could be appropriate bioindicators for future monitoring of coral reefs environmental health. We further assess the potential functional diversity (also known as “enzymic capabilities”) of each community.
Materials and methods
Sample collection
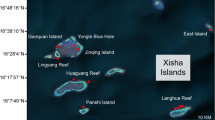
Sediment samples were collected from seven different sites around Okinawa Island; Sunabe, Kayou, Kin, Manza, Odo, Sesoko, and Uruma (see Fig. 1), from November 10 to December 25, 2020. Detailed descriptions of each sampling site are as follows.
Sunabe; near a well-populated and reclaimed land area, also with heavy coastal armoring and frequent visitors. Relatively close to the reef edge, presence of small river mouth and military base.
Kin; less populated natural beach with relatively more agriculture nearby. Large river mouth and mangrove forests within a kilometer.
Kayou; pristine site at a sparsely populated area with a small river mouth nearby. Zosteraceae, Hydrocharitaceae and Cymodoceaceae sp. growing abundantly.
Uruma; right next to an artificially constructed, reclaimed land causeway (“Kaichu-Doro Causeway”). No clear reef edge but some rocky substrates exist. Zosteraceae, Hydrocharitaceae and Cymodoceaceae sp. form assemblages.
Sesoko; near the southern edge of Sesoko Island, with relatively pristine environment, with small reef edge. Scleractinian corals abundant; Acropora spp. being most abundant. This is the only site which has soft corals nearby among our sampling sites.
Manza; the most natural site amongst our examined coral reef sites, with sharp and steep reef edge right in front of the sampling site. Scleractinian corals abundant, with Montipora spp. most abundant.
Odo; southernmost site with a large tide pool and beach nearby. Groundwater upwelling in front of the sampling site, as well as large civil engineering excavation ongoing roughly three hundred meters away on top of a hill nearby. Scleractinian corals abundant, Montipora spp. most abundant.
Sediment collection was done manually while snorkeling by scooping the sediment surface (< 0.5 cm) using 15 mL Falcon® tubes (ThermoFisher Scientific, Waltham, MA, USA). At each site, 20 sediment samples were collected, except at Sunabe where only 15 samples were collected due to rough sea surface conditions. Among these sites, Manza, Sesoko, and Odo were characterized by hard reefs with living scleractinian corals, and we hereafter refer to them as “coral reef” sites. Uruma and Kayou were characterized by abundant seagrass, and are referred to as “seagrass meadow” sites, whereas Sunabe and Kin were characterized by fine sand, and are referred to as “sandy bottom” sites in this study. The number of sampling sites was determined based on the available resources and the need to ensure comprehensive analyses. Under such conditions, considering the high biodiversity and our research priorities, we focused more on coral reef sites than on the other two ecosystems.
In coral reef sites, sediments were sampled at small distances (approximately < 1 m) from live scleractinian corals, and in seagrass meadows, sediments were sampled adjacent to seagrass while taking care to not include seagrass. Depths and GPS coordinates were recorded for all samples, except for one sample from Kayou (Ky_20) due to GPS failure (see Supplementary Table S1). After collection, sediment samples were immediately placed on dry ice, and kept frozen until further investigation.

Maps showing sites investigated in the present research. (A): Okinawa Islands in the Japanese Archipelago. (B): Okinawa Island in detail. Each dot represents a study site, and color indicates substratum. Blue = coral reef, red = sandy bottom, green = seagrass meadow sites, respectively
DNA extraction, amplification and sequencing
Each sample was split into two (equal) subsamples and was subjected to the following steps. Whole microbial genomic DNA was extracted using DNeasy® PowerSoil® Pro Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols except for pre-heating at 65℃ for 10 min right before vortexing, as well as reducing the final elution volume to 75 ml. Yields of the extracted DNA were measured using a Qubit 4 Fluorometer (ThermoFisher Scientific) following the manufacturer’s protocol and were subsequently diluted to 1 ng/µl. DNA was then used as template for targeting the V3-V4 regions of the prokaryotic 16 S rRNA gene using MightyAmp™ DNA Polymerase Ver.2 (Takara Bio Inc., Kusatsu, Japan) following the manufacturer’s protocol, with the universal primer set Pro341F/Pro805R [91]. After the first PCR and purification, indexing PCR was performed using TaKaRa Ex Taq® Hot Start Version (Takara Bio Inc.). All detailed amplification methods are available in supplementary materials. Sequencing libraries of 135 samples were adjusted to be 4 nM each and were pooled into two amplicon pools for next generation sequencing. The prepared amplicon pools were mixed with the Phix Control V3 (Illumina, San Diego, CA, USA) conditioned to be up to 30% of total volume, and then loaded into Miseq Reagent Kit V3 (600 cycles, Illumina). Finally, the samples were sequenced on Illumina Miseq (Illumina) using a 301 bp paired-end chemistry by two individual runs. All raw sequences are available online (see data availability sections for details).
Bioinformatics
The demultiplexed paired-end sequence reads were analyzed in the QIIME2 v2021.11.0 [92] framework with several plug-in programs (All codes executed for this step is available in Additional file 1). Primers and the middle adaptor sequences were trimmed with Cutadapt v3.5 [93] (via q2-cutadapt) respectively from the 5’-end and 3’-end after applying a maximum of ten iterations. In case the primer sequences were not found or the trimmed length of sequences was less than 100 bp, reads were disposed. Quality control including quality filtering, denoising, correcting reading errors, merging paired-end reads and removal of non-biological sequences (including chimera) were performed by DADA2 v1.18.0 [94] (via q2-dada2) with the below custom values. For denoising, the maximum expected error values, ‘maxEE’ [95], were set to 2 for forward and 5 for reverse reads, respectively. The truncate length at 3’-end of both reads were independently decided by the base position, represented as a phred score of less than 20 at the first quartile of the total reads. After quality control, a count table of representative sequences was generated with dereplication of the same amplicon sequence variant (ASV). To standardize ASV richness of each sample biased by different numbers of reads, resampling, so called ‘coverage-based rarefaction’ [96], was performed in all samples equalizing the slope of the saturation curve to the least saturated sample. After the rarefaction, copy number correction was employed to justify the number of reads considering the copy numbers using the open-source software PICRUSt2 [97], as well as Integrated Microbial Genomes (IMG) as a reference database [98]. As well, Enzyme Commission numbers and abundance of each enzyme that is coded by bacteria detected in this research were obtained from IMG (all codes executed for copy number correction and obtaining Enzyme Commission numbers are available in Additional file 2). Taxonomic assignment to ASV was conducted with pre-trained Naive Bayes classifier [99] referring to the 16S ribosomal RNA sequence database SILVA v138.1 [100, 101], and then operational taxonomic units (OTUs) were determined against each ASV using scikit-learn [102], a machine-learning classifier plugin in Qiime2. ASVs assigned to mitochondria, chloroplasts, eukaryotes, or to none of the domains were removed from the dataset to focus on the target microbiome. The OTU count table obtained through these processes was used in subsequent community analyses. It should be noted that there are some bacterial taxa for which taxonomic classification is still under discussion; in this study we based our discussion on information from SILVA v138.1.
Distance among samples was calculated under Bray-Curtis configuration using “vegan” package [103], and then Unweighted Pair-group Method with Arithmetic Means (UPGMA) clustering was performed under “average” configuration using “pvclust” package [104] in R 4.1.2 [105]. We then conducted silhouette analysis to evaluate how many groups the samples could be divided into, and used these groups (i.e., “sandy bottom”, “seagrass meadow” and “coral reef”) to perform further analyses.
The Shannon diversity indices of the prokaryotic community in each sample were calculated with “vegan” package [103]. The Kruskal-Wallis test in “kruskal.test” function in “stats” package [105] was employed to confirm statistical significance of calculated Shannon indices among benthic components, in cooperation with “pairwise.wilcox.test” in the same package. Further, non-metric multidimensional scaling (nMDS) was performed using “metaMDS” function in “vegan” package [103]. To test how group similarity was affected by factors, we conducted PERMANOVA using “adonis2” function in “vegan” package [103] with setting benthic substrate types (“coral reef”, ”sandy bottom”, and “seagrass meadow”) as the factor. Then we assessed which pairs were significantly different using “pairwise.adonis” function in “pairwiseAdonis” package [106] with 999 times permutation and Bonferroni correction settings. Bacterial taxa that were differentially abundant in coral, sand or seagrass were examined using “ancombc2” command in “ANCOMBC” package [107]. For interpretation of results, bacterial orders for which names did not follow nomenclature or were not assigned in the SILVA database (e.g. “HOC36”,” NB1-j”, “KD4.96” etc.) were excluded. Linear and non-linear correlations between bacterial orders were tested using “secom_linear” and “secom_dist” commands in “ANCOMBC” package [107]. All R codes executed and metadata used are available in Additional files 3 and 4, respectively.
Enzymic composition analysis
Using estimated enzymic abundance based on the genes coded by the prokaryotic community obtained, the Shannon diversity index was calculated with “vegan” package [103] in R 4.1.2 [105]. The ANOVA test was employed to confirm statistical significance of calculated Shannon Indices among benthic components using “aov” and “TukeyHSD” functions in “stats” package [105]. Further, we analysed the enzymes differentially abundant in specific substrates using the “ancombc2” command in the “ANCOMBC” package [107].
Results
Number of OTUs and the most abundant bacterial phyla
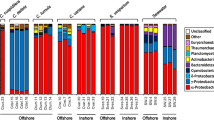
After sequence processing, 4,675 OTUs from 83,138 ASVs were obtained, and then applied in further analyses (in total 3,417,550 contigs, average of 25,315 contigs/sample; detailed information in Supplementary Table S2). The 4,675 OTUs were assigned into the following taxonomic ranks in SILVA: 79 phyla (Supplementary Table S3); 214 classes (Supplementary Table S4); 522 orders (Supplementary Table S5); 880 families (Supplementary Table S6); 1871 genera (Supplementary Table S7). Also original count table used for analyses are all available in Additional files 5 to 11. The prokaryotic community had only a few domain Archaea (0.77% of total counts), while domain Bacteria was predominant (99.23% of total counts). The most predominant phylum was Proteobacteria for all three benthic types (33.5% for corals, 33.2% for sandy bottoms and 37.0% for seagrass, respectively, Fig. 2, Table S8). The second- to fifth-most predominant phyla of coral reef sites were Bacteroidota, Cyanobacteria, Actinobacteriota and Verrucomicrobiota (15.7%; 14.0%; 11.0%, 5.5%, respectively). Sandy bottom and seagrass meadow sites were similar in terms of bacterial composition, sharing the second- to fourth-most predominant taxa in the same order; Actinobacteria (15.3% and 13.9%, respectively), Bacteroidota (11.5% and 12.5%) and Acidobacteriota (8.2% and 6.4%, respectively) with a difference in the fifth-most abundant phylum, namely Chloroflexi for sandy bottoms (= 5.3%) and Desulfobacterota for seagrass meadows (= 5.9%).

Bar plot showing the top 13 phyla in each benthic type. Different colors show different bacterial phyla. Columns show samples from coral reefs, seagrass meadows, and sandy bottom sites, from left to right. Color bar widths indicate relative abundance of each taxon
Structure of bacterial community
The hierarchical clustering (UPGMA) plot showed three major branches, composed of (i) Manza and Sesoko, (ii) Sunabe and Odo, and (iii) Uruma and Kayou clustered with more distant Kin (Supplementary Figure S1). The AU (Approximately Unbiased) p-values and BP (Bootstrap Probability) were low to moderate (ranging from 67 to 89%, and from 35 to 57% for AU and BP, respectively) for all branches except for the branch formed by Uruma and Kayou (AU = 99%, BP = 97%).
The result of the silhouette analysis based on OTU level data showed that the samples could be divided into three groups; sandy bottoms, seagrass meadows, and hard rocky substratum with living scleractinian corals (Supplementary table S9), and we subsequently used these categories in this research. In our results, most of the samples were assigned to their supposed groups (“sandy bottom” group (Kin and Sunabe), = 29/35 (82%); “seagrass meadow” group (Kayou and Uruma), 40/40 (= 100%); and “coral reef” group (Manza, Sesoko, and Odo), 45/60 (= 75%) (Supplementary tables S10).
The nMDS plot based on the Bray-Curtis dissimilarity indices result illustrates these three different groupings (Fig. 3).

nMDS plot of Bray-Curtis dissimilarity index of samples. Each circle, triangle or square represents a different sample. Sites have been abbreviated as follows: Kn: Kin, Sn: Sunabe, Ky: Kayou, Ur: Uruma, Sk: Sesoko, Mn: Manza, Od: Odo. Different colored mesh indicates groupings based on silhouette analysis results, mostly corresponding to red = sandy bottoms, blue = coral reefs, green = seagrass meadows
Prokaryotic richness and diversity
The highest value of prokaryotic Shannon diversity indices was observed at coral reef sites, whereas the lowest was found at sandy bottom sites (Supplementary Table S11). There was no significant difference in Shannon index between coral reef sites and sandy bottom sites or between coral reef sites and seagrass meadow sites (Fig. 4, Supplementary Table S12). However, seagrass meadow sites had significantly higher prokaryotic diversity when compared to sandy bottom sites (p < 0.01). When prokaryotic Shannon indices were compared among individual sites, Uruma showed significantly higher diversity compared to Kin (p < 0.01), Kayou (p < 0.01), and Sunabe (p < 0.05) (Supplementary Figure S2).

Box plots of prokaryotic (left) and enzymic (right) Shannon diversity indices among different types of substrata. Asterisks show statistical significance (*** p < 0.005)
Similarity and dissimilarity among coral reef, seagrass meadow and sandy bottom sites
The result of PERMANOVA showed that all three groups had significantly different bacterial compositions (p < 0.001, Table 1). Further, ANCOM analysis of the top 10% of the most abundant orders indicated that specific bacterial taxa were differentially abundant in each benthic substratum (Fig. 5). Orders Flavobacteriales, Rhodobacterales, Verrucomicrobiales, Cytophagales, Enterobacterales, and Eurycoccales were exclusively abundant in coral reef sites. Kiloniellales and Entomoplasmatales were prominent only in sandy bottom sites, and Steroidobacterales, Desulfobacterales, Chromatiales, and Thiotrichales were distinctly abundant only in seagrass meadow sites. Actinomarinales and Thermoanaerobaculales were distinctly less abundant in coral reef sites, Opitutales, Bacteroidales, and Sphingomonadales were less abundant in sandy bottom sites, and Dadabacteriales was distinctly less abundant in seagrass meadow sites.

Combined graph of the results obtained with ANCOM analysis (left to middle) and the total absolute abundance (right) of each bacterial order. For the ANCOM analysis, compared substratum types are shown at the top of each column. “W_statistics” stands for a log fold change value divided by standard error for effect size correction, and indicates to what type of substratum each bacterial order was differentially abundant. Asterisks show statistical significances (*p < 0.05, ** p < 0.01, ***p < 0.001)
Correlation between bacterial orders of each substratum
Most correlations of the top ten most abundant bacterial orders with a correlation coefficient exceeding 0.7 were detected by both Pearson and Distance methods (n = 16), whereas a few were detected by only Pearson’s method (n = 5, Fig. 6).

Combined graph showing the results of correlation among bacterial orders. From the top to bottom, rows show coral reef sites, sandy bottom sites, and seagrass meadow sites, respectively. Left column is based on the distance method, while right column is based on Pearson’s coefficient of correlation method, where warm colors mean positive correlation, and cold colors mean negative correlation
In coral reef sites, the abundance of Actinomarinales had a negative correlation with Cyanobacteriales, while showing positive correlations with the abundances of Desulfobacterales and Thermoanaerobaculales.
Secondly, in sandy bottom sites, Thermoanaerobaculales exhibited a positive correlation with Actinomarinales, as seen in coral reefs. However, Thermoanaerobaculales showed negative correlations with Verrucomicrobiales, Rhodobacterales and Flavobacteriales. Additionally, Rhodobacterales and Verrucomicrobiales had positive correlations, while Flavobacteriales and Rhizobiales showed negative correlations. Furthermore, Actinomarinales was positively correlated with Rhizobiales, and negatively correlated with Rhodobacterales and Verrucomicrobiales.
Thirdly, in seagrass meadow sites, Desulfobacterales and Microtrichales had negative correlations while Actinomarinales and Thermoanaerobaculales showed a positive correlation. As well, positive correlations between Caldilineales and Thermoanaerobaculales as well as between Actinomarinales and Caldilineales were detected by only Pearson’s method.
Enzymic capability estimation and analysis
In this study, a total of 2431 enzymes were estimated to be coded in the prokaryotic metagenome based on the IMG database (Supplementary Table S13, also original count table is available in Additional files 12 to 15). The Shannon diversity of enzymes was significantly higher in coral reef sites compared to the other two benthic types (Supplementary Table S14). When comparing individual sites, Kin showed significantly lower diversity compared to all other sites except for Kayou. Similarly, the diversity indices of Kayou and Uruma were significantly lower than those at Manza, Odo, Sesoko, and Sunabe (Supplementary Figure S2).
Among the top 5% of the most abundant enzymes in all samples, the ANCOM test revealed that 20 enzymes were significantly and exclusively abundant in coral reef sites, namely site-specific DNA-methyltransferase (adenine-specific) (EC 2.1.1.72), Cysteine desulfurase (EC 2.8.1.7), Ribonucleoside-diphosphate reductase (EC 1.17.4.1), Thioredoxin-disulfide (EC 1.8.1.9), C-terminal processing peptidase (EC 3.4.21.102), 23 S rRNA pseudouridine1911/1915/1917 synthase reductase (EC 5.4.99.23), Dihydroorotase (EC 3.5.2.3), N-acetylmuramoyl-L-alanine amidase (EC 3.5.1.28), Endopeptidase Clp (EC 3.4.21.92), 8-oxo-dGTP diphosphatase (EC 3.6.1.55), Protein-serine/threonine phosphatase (EC 3.1.3.16), Superoxide dismutase (EC 1.15.1.1), Mannose-1-phosphate guanylyltransferase (EC 2.7.7.13), Arsenate reductase (glutathione/glutaredoxin) (EC 1.20.4.1), 3-oxoacyl-[acyl-carrier-protein] reductase (EC 1.1.1.100), Threonine ammonia-lyase (EC 4.3.1.19), XTP/dITP diphosphatase (EC 3.6.1.66), Phosphoserine phosphatase (EC 3.1.3.3), Protein-glutamate methylesterase (EC 3.1.1.61), and GTP cyclohydrolase I (EC 3.5.4.16) in descending manner (Fig. 7). On the other hand, nine enzymes were more abundant in seagrass meadow sites, namely Histidine kinase (EC 2.7.13.3), Peptidylprolyl isomerase (EC 5.2.1.8), H+-transporting two-sector ATPase (EC 3.6.3.14), Dihydrolipoyl dehydrogenase (EC 1.8.1.4), Glutamate synthase (NADPH) (EC 1.4.1.13), Glutamate synthase (NADH)(EC 1.4.1.14), Type I site-specific deoxyribonuclease (EC 3.1.21.3), Citrate (Si)-synthase (EC 2.3.3.1), and Asparagine synthase (glutamine-hydrolysing) (EC 6.3.5.4) in the same manner as above. No enzymes were exclusively abundant in sandy bottom sites.

Combined graph of the results obtained with the ANCOM analysis (left to middle) and the total absolute abundance (right) of each coded enzyme by prokaryotes. For the ANCOM analysis, compared substratum types are shown at the top of each column. “W_statistics” stands for a log fold change value divided by standard error for effect size correction and indicates to what type of substratum each bacterial order was differentially abundant. Asterisks show statistical significance (*p < 0.05, ** p < 0.01, ***p < 0.001)
Discussion
While prokaryotic compositions in seawater and on the surfaces of benthic organisms have been extensively studied [e.g. 56, 58, 68], there is limited research available on sedimentary prokaryotes from different marine environments. In this study, we investigated the prokaryotic compositions in marine sediments from Okinawa Island and sought candidate prokaryotes as bioindicators to be used in future monitoring of coral reef ecosystem health. As a result, we revealed that different habitats have significantly different prokaryotic communities, and revealed bacterial taxa that characterize each environment that can potentially be candidates for environmental monitoring using metabarcoding methods. These approaches may help in preventing serious damage to corals caused by heat stress or diseases.
Dominant taxa and diversity indices
Among three substrata, the top four most abundant phyla were shared except for Cyanobacteria, which only appeared in coral reef sites, and Acidobacteriota which appeared in seagrass meadows and sandy bottom sites. The fifth-most abundant phylum was different among substrata, namely Verrucomicrobiota at coral reefs, Chloroflexi at sandy bottom sites, and Desulfobacterota in seagrass meadows. In all benthic types, Proteobacteria was the most dominant phylum, consistent with many previous studies focusing on various marine environmental samples such as shallow to deep sediments and water [108,109,110,111,112,113]. Cyanobacteria are suggested to be prominent in coral mucus when exposed to heat stress [21], thus its abundance only in coral reef sites may indicate such stress, although further investigations of the coral mucus microbiome and bleaching conditions are needed to verify this hypothesis. Chloroflexi has been reported to be abundant in marine sediments compared to seawater not only in coral reefs but worldwide, especially at anoxic sediments [114]. Therefore, our results may indicate that sandy bottom sites where Chloroflexi was abundant may have been experiencing deoxygenation, possibly due to the lower abundance of grazers when compared to coral reefs and seagrass meadows, as similarly observed in Okinawan sea cucumbers [115], as such organisms can increase O2 penetration depth in the sediment layer by plowing sediments via their feeding activities [116]. Desulfobacterota is a strict anaerobe, with chemoorganotrophic, chemolithoheterotrophic or chemolithoautotrophic bacteria, found across a wide range of aquatic environments [117]. This phylum is often predominant in coastal deep layer sediments where hypoxic conditions can occur [112, 118, 119]. It is also known that seagrass meadows occasionally become hypoxic possibly due to abnormally high temperatures [120], however water temperatures in November to December, when we conducted sampling for this study, were lower than summer maximum temperatures. Still, further investigations combined with continuous temperature measurements around Okinawa Island should assist to better understand the relationship of Desulfobacterota’s abundance and hypoxic environments caused by global warming.
The significantly lower alpha diversity at sandy bottom sites when compared to seagrass meadow sites may reflect the low surface complexity of sandy environments. Seagrass is known to have a unique bacterial community associated with its leaves, roots and rhizosphere [121,122,123], and these bacteria may contribute to the ecological function in seagrass meadow sites. While corals are also known to harbor symbiotic bacteria on their surfaces or inner skeletons [58, 76, 124, 125], there was no significant difference in diversity index between coral reef and sandy bottom. This could be attributed to the substantial variation in bacterial community diversity known to occur in corals [126, 127]. In fact, the second lowest and highest Shannon diversity indices in this study were obtained from Manza, one of the coral-rich sites (Shannon index = 4.62 and 5.66, respectively).
Microbial composition and differentially abundant prokaryotic orders in each substratum
The PERMANOVA analysis revealed the prokaryotic composition is significantly different among the three benthic types. This is the first report rigidly uncovering such distinct differences among different benthic components in coral reefs on a geographically small scale. Interestingly, both coral reef and sandy bottom sites formed individual groups despite individual sites in each group are largely distanced. This result suggests benthic type-specific prokaryotic compositions exist around Okinawa Island, which may have acclimated to their respective conditions over a long period of time. However, it is important to consider that human-caused impacts can also affect the local prokaryotic community in coral reefs [22], and thus what we observe here in each benthic type might be a result of anthropogenic disturbances. Prokaryotic communities are known to be highly plastic and capable of recovering relatively quickly after disturbances. Therefore, continuous monitoring of the prokaryotic components at each site is crucial to understand and track any changes that may occur over time, and such data should provide valuable insights into the resilience and stability of coral reef ecosystems in response to environmental changes and disturbances.
At sandy bottom sites, there were only two bacterial orders, Kiloniellales and Entomoplasmatales, that were differentially abundant exclusively. Although these bacterial orders have been reported from marine sediments in a few studies [128, 129], the ecological feature and environmental preference of these bacteria remain largely unknown. One fact that is shared between these orders is that they are both known to be symbiotic to other organisms. For example, Entomoplasmatales is a well-known symbiotic bacteria found the guts of crustaceans (e.g., [130, 131]), and has also been reported as a major bacterial symbiont of marine lake jellyfish Mastiguas sp. [132]. Kiloniellales is a marine-restricted bacterial order [133], that is commonly found as a symbiotic bacteria among cnidarians (jellyfish; Tripedalia cf. cystophora [132], Gonionemus vertens; [134], hydrozoan Millepora platyphylla but not found in scleractinians Porites lobata or Pocillopora meandrina; [127]); also known from juvenile sea star Mithrodia clavigera [135]). Therefore, the biased abundance of these bacterial orders in sandy bottom sites might reflect the high population density of such host organisms. However, comprehensive population density and species aggregation information of these taxa around Okinawa Island is currently lacking, as known for some other taxa (e.g., zoantharians [136], giant clams [137], and holothurians [115]).
There were four bacterial orders differentially abundant in seagrass meadow. In the order Steroidobacterales, the genus Woeseia predominantly occupied 99.8% of entire order in this research. Woeseia was originally described within Chromatiales by Du et al. [138] as a salt-dependent and facultative anaerobic bacteria, and Chromatiales is also one of the differentially abundant bacterial orders in seagrass meadows in this study. Woeseia is a globally distributed and ubiquitous marine bacteria with chemorganoheterotrophy and facultative chemolithoautotrophy based on inorganic sulfur and hydrogen compounds, often found in marine sediments [139, 140]. This genus is also a major symbiotic rhizosphere bacteria of seagrasses, such as Caulerpa and Cymodocea [110], as well as Zostera [141], seepweed Suaeda and common reed Phragmites [142], and the mangrove tree Kandelia candel [143]. Desulfobacterales, a sulfate-reducing and strictly anaerobic bacteria, is known to be abundant in coastal deep layer sediments, and was also differentially abundant in seagrass meadows. Seagrass meadows and mangrove habitats often have low oxygen concentrations [144,145,146,147]. Desulfobacterales is also thought to be a major player of nitrogen cycling, involved in an estimated average of 12% of the entire process in mangrove ecosystems [148]. Nie et al. [148] also suggested that the high abundance of nitrate reducing bacteria such as Desulfobacterales may contribute to the prevention of excessive nitrate-nitrogen accumulation, which could harm the local ecosystem in combination with some other bacterial orders such as Chromatiales, which was abundant in the current research. Chromatiales is known to be a symbiont of the sponge Stylissa carteri [149], and is also abundant in saltmarsh sediments covered by cordgrass Spartina alterniflora along with Thiotrichales [150].
Of potential importance, there are reports that some of these bacteria are related to prevalent environmental pollution. For instance, Woeseia is known to increase with iron and arsenic increases in seagrass meadows [151], and Desulfobacterales is influenced by heavy metal pollution in mangrove forests [152]. Desulfobacterales has also been reported to be prominent at both natural (e.g., estuarine [153]) and artificially eutrophicated sites (prawn mariculture pond [154] and salmon mariculture area [155]). Chromatiales has been suggested to be elevated by environmental stress (i.e., unseasonal heavy rainfall) in the seagrass Halophila ovalis when compared to healthy individuals [156]. Eutrophication in seagrass meadows is suggested to stimulate an unfavorable surge of heterotrophic bacteria, that occasionally can result in large-scale die-off events due to elevated sulfide concentrations in sediments [157]. Therefore, these bacteria can potentially be used as bioindicators of such disturbances, and further research combined with direct detection of biotic and abiotic, as well as natural and anthropogenic cues may assist to track local environmental stress better.
Our examined coral reef sites had six bacterial orders that were significantly and exclusively abundant. Order Rhodobacterales is a facultative anaerobic bacteria, known to be a primary surface colonizer in temperate coastal waters [158]. In tropical coastal areas, Rhodobacterales are known to be abundant on some coral species’ outer surfaces (e.g. Porites [127]), especially in aged surface mucus [76]. This order has been proposed to be prevalent in stressed or diseased coral individuals (pathogenic Vibrio infection [159], stony coral tissue loss disease [160], heat stress due to El Niño [161]). Notably, it has been implied that many of the bacteria within Rhodobacterales are capable of detecting and degrading dimethylsulphoniopropionate (DMSP) [162,163,164], which is mainly produced by Symbiodiniaeceae dinoflagellates symbiotic with coral [165]. Considering the fact that heat-stressed coral colonies express five-fold higher DMSP concentrations than healthy colonies [166], Rosales et al. [167] proposed that DMSP could chemoattract some kinds of bacteria and therefore is the cause of the prominence of Rhodobacterales in stressed coral colonies. Flavobacteriales was also abundant in coral reef sites, and this bacteria is known to encode diverse polymer-degrading enzymes, indicating their capacity to utilize a wide range of carbon sources [168]. This order has been suggested to be plentiful in coral mucus and tissue [167], being especially pronounced before and while host corals are exposed to environmental stress [169], such as disease infections [167]. Furthermore, it has been suggested that Flavobacteriales encodes similar functional genes as Rhodobacterales, although their expressions are substantially different [170]. Verrucomicrobiales, an order belongs to the phylum Verrucomicrobiota that has been reported as almost ubiquitous in marine habitats including coastal sediments (reported as Verrucomicrobia [171]), is also known to be abundant in coral mucus, and potentially pathogenic to corals [76]. Verrucomicrobiales increases in corals infected with black band disease [172] and also with growth anomalies [173]. Enterobacterales is a Gram-negative, rod shaped and facultative anaerobic bacteria [174], which occasionally has been reported to associate with aquatic organisms such as freshwater fish [175] and marine sponges [176]. This order has been suggested to be pronounced in bleached Acropora digitifera [177], although the genus Vibrio, a representative pathogenic taxon within this order, was not abundant in this research. Cytophagales is an order that prefers habitat where organic materials are abundant, and is capable of producing flexirubin-type pigments [178]. Cytophagales are often found in freshwater, and are also relatively common in coastal waters [178], and have been suggested to be a major decomposer of plant litter in marine wet land in China [179]. In tropical coral reefs, Cytophagales has been known to be an opportunistic bacteria that was observed to become more abundant on the body surface of the coral Montastraea cavernosa after infection of pathogenic Vibrio bacteria, presumably due to a decrease in predatory bacteria Halobacteriovorax [159]. Although Halobacteriovorax has been reported from marine sediments [180, 181], it was barely detected in this research. Cytophagales is also known to become more abundant once a coral host is subjected to continuous heat stress, e.g. by El Niño [161]. Eurycoccales is a photosynthetic cyanobacterial order, which has been previously reported from coastal sediments [155]. However, information about this order, such as its preferred habitat(s) or nutrient requirement(s), is still limited and more studies are needed to better interpret the meaning of their abundance in coral reef sites. Meanwhile, coral symbiotic bacteria, represented by family Endozoicomonaceae [24, 76, 77, 182,183,184] and a few other taxa (i.e. Oxalobacteraceae [76], Alteromonadales and Simkaniaceae [184, 185], Oceanospirillales [58]), were comparatively rare in the current research.
Overall, the current results may reflect high levels of environmental stressors such as heat stress, coral disease, and pathogenic bacteria in coral reef sites around Okinawa Island. Bacterial interactions between coral-ambient sediment and coral-ambient seawater have been reported in healthy [58, 76, 186] and diseased corals [187], and it has been suggested that coastal surface sediments serve as a possible pathogen reservoirs or “seed-banks” as proposed by Carlos et al. [75]. Although we did not conduct direct sampling of coral tissue or mucus, considering several putative stress-related bacterial orders (e.g., Flavobacteriales and Verrucomicrobiales) were relatively more abundant in this research compared to coral reef sediments in previous research (Supplementary Table S15), there is an urgent need to monitor coastal sedimentary microbial communities to track stress-related prokaryote demography around Okinawa. Simultaneously, there is yet only a few examples of research on coastal sedimentary microbiomes, especially from the western Pacific, making it harder to compare our results to previous research. Therefore, to take advantage of metabarcoding methods as environmental monitoring measures, it would be very helpful to create a local catalogue of conspicuous prokaryotic taxa depending on substratum type.
Correlation between bacterial orders at each environment
There were several correlations observed among bacterial orders in this research. However, due to the lack of essential information to interpret these relationships, such as habitat preferences, secondary metabolites and/or biochemical capabilities, many of these relationships remain unknown at this stage. Thanks to the recent technological advancements, further comparative details between some microbial groups could be provided; for example, the negative correlation observed between Actinomarinales and Cyanobacteriales at coral sites. Recently, López-Pérez et al. [188] explored the genome of Actinomarinales, a photoheterotrophic bacterial order, and revealed that it encodes cyanophycinase, enabling Actinomarinales to use cyanophycin as a source of arginine, which is one of the amino acids that they are auxotrophic for. Cyanophycin is an amino acid polymer produced by most Cyanobacteria that has arginine as a branched side chain and aspartic acid as its backbone [189]. Cyanophycin is predicted to work as a temporary storage of nitrogen, specifically accumulating fixed nitrogen at night and utilizing it during the day to uncouple nitrogen assimilation and photosynthesis, and hence cyanophycin is important in these species’ survival strategies [190].
Based on these facts, the negative correlations observed here might indicate the following scenario: increases of Actinomarinales raise its dependency on cyanophycin, and subsequently Cyanobacteria are consumed by Actinomarinales as a source of arginine. Actinomarinales is also known to work as a module-hub among small sized free-living planktonic bacteria in eutrophic estuaries [191]. Desulfobacterales, which showed a positive correlation with Actinomarinales, is one of the bacterial orders abundant in eutrophic environments such as estuaries [153]. Xie et al. [192] revealed that these two orders are abundant in plastic-polluted mangrove soil specifically in spring, while Actinomarinales become infrequent as water temperature rises. Therefore, abundant nutrient resources at coral sites or other condition (e.g. water temperatures, light intensity etc.) may lead to a positive correlation of these orders. Actinomarinales and Thermoanaerobaculales exhibited positive correlation in both coral sites and sandy bottom sites. According to our obtained data, family Thermoanaerobaculaceae occupied more than 90% of the entire Thermoanaerobaculales. Thermoanaerobaculaceae have been described as thermophilic, neutrophilic and chemoheterotrophic anaerobes that prefer some sugars, organic acids and proteinaceous compounds as growth substrates [193], and they have been suggested to be sulfate reducers [194, 195]. Thermoanaerobaculaceae is known to increase upon nitrate nitrogen contamination into the environment [196]. Actinomarinales is known to perform anammox (anaerobic ammonium oxidation) by reducing natural organic matter (NOM), and thus increase NOM availability [197]. Therefore, these orders might represent an increase in total nitrogen input into the environment. Flavobacteriales, which had a negative correlation with Rhizobiales and Thermoanaerobaculales at sandy bottom sites, was mostly occupied by a member of family Flavobacteriaceae in the current data (> 90%). Recent genome mining research of Flavobacteriaceae has revealed that they encode antimicrobial, antioxidant and cytotoxic compounds [168], and this might work to suppress Rhizobiales and Thermoanaerobaculales. To achieve a better understanding of such correlations among orders, however, more rigid information about their ecological and biochemical capabilities, as well as direct detection of abiotic variables such as salinity, dissolved oxygen, nitrate and phosphate concentrations, and pH are essentially needed.
Enzymic capability estimation
Although the actual amounts of enzymes were not investigated directly, the significantly higher enzymic diversity and the greater number (n = 20) of enzymes that were differentially abundant in coral reef sites may be explained by coral symbiotic prokaryotes’ complex enzymic reactions to various environmental stressors [198,199,200]. Likewise, although enzymic diversity was significantly lower than at coral rich sites, seagrass meadow sites had several enzymes exclusively abundant (n = 8), and this could also mirror diverse biochemical pathways of microbial symbionts in this habitat [87, 201, 202]. Comparing those two substrata, sandy bottom sites had poor three-dimensional rugosity, and this may potentially have resulted in the absence of exclusively abundant enzymes.
Amongst the enzymes that were exclusively abundant in coral reef sites, cysteine desulfurase (EC 2.8.1.7) is known to be one of the most abundant and ubiquitous genes encoded in any organism’s genome [203], including bacteria [204]. This enzyme is involved in the conversion of L-cysteine into L-alanine and sulfane sulfur, and this process also involves iron-sulfur cluster synthesis [205]. So far, three types of cysteine desulfurase, namely NifS, IscS, and SufS, have been identified, with all of them considered important in oxidative stress meditation [206,207,208].
Another enzyme enriched in coral sites was superoxide dismutase (EC 1.15.1.1). Generally, superoxide dismutase is known to mediate superoxide compounds such as hydrogen peroxide [209], and have also been confirmed to be produced by various bacteria [210, 211]. One major and well-studied oxidative stressor within scleractinian corals are individual endosymbiotic dinoflgallete Symbiodiniaceae under heat-stressed conditions [212], and thus superoxide dismutase production capability is important for biochemical defense in coral tissue, especially under harsh condition such as high seawater temperatures during coral bleaching [213]. We must note, however, that we neither detected enzymes directly nor collected coral tissue sample directly. Still, considering the nature of microbial interactions between a coral’s surface and its surrounding environment (known as “the coral ecosphere” in [68]), our finding that coral sites have significantly higher production potential of superoxide dismutase than other substrate types needs to be followed up with more concrete future studies including metagenomic analyses or classical culturing to assess what bacteria are actually contributing to these differences.
The histidine kinase (EC 2.7.13.3) was abundant in seagrass meadow sites. In bacterial cells, histidine kinase is known as a part of membrane-anchored proteins forming the two-component regulatory system [214], and is essential for bacteria to detect and react to external stimuli by phosphorylating histidine as a signaling cue [215]. As well, this enzyme is suggested to be crucial for biofilm formation [216]. Therefore, the abundance of this enzyme may show dynamic environmental fluctuation in these sites. Coral reef sites are also presumed to have substantial environmental fluctuations, however the histidine kinase coding gene was not abundant in coral reef sites. This may indicate the ecological strategy of bacterial community in each substratum is different, and further investigation ideally employing other methods such as transcriptome analyses and enzyme assays are needed for a better understanding.
Conclusions
Our data showed that the different habitats in coral reefs have different prokaryotic communities, with characteristic taxa that are exclusively more abundant in specific environments. Although the main driver that determined community structure was not fully revealed in this study, some bacterial taxa that characterized our observed coral reef microbiome have previously been linked to environmental stressors such as heat stress, bleaching, and coral diseases. Considering the potential of benthic sediments to interact with coral surface prokaryotic communities as suggested previously, sedimentary prokaryote monitoring is extremely important to assess the potential risk of environmental degradation in coral reefs, and harmful pathogen prevalence in coral reefs. Therefore, further studies on the interactions between the microbial community and biotic (e.g., coral disease prevalences, corallivore abundances, organic content concentrations) and/or abiotic (e.g. water temperatures, hydrodynamics, solar radiation) parameters needs to be performed. In addition, our samples were from a single time period, and therefore continued sampling and longer-term monitoring are needed to track spatio-temporal variability. For now, our data from the current study provide a preliminary reference list of prokaryotic taxa we should pay attention to in order to better forecast coral reef health with higher resolution.
Data availability
All data used in this study, including codes, sampling metadata, and results of each analysis, are available in the supporting materials of this article. The raw sequences have been deposited in DDBJ with accession numbers DRR506213 - DRR506511, under bio-project ID PRJDB16769 (https://ddbj.nig.ac.jp/resource/bioproject/PRJDB16769). The data can also be accessed via NCBI at https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJDB16769.
References
Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320(5879):1034–9.
Fuhrman JA, Cram JA, Needham DM. Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol. 2015;13(3):133–46.
Ghiglione JF, Galand PE, Pommier T, Pedrós-Alió C, Maas EW, Bakker K, et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc Natl Acad Sci U S A. 2012;109(43):17633–8.
Sunagawa S, Acinas SG, Bork P, Bowler C, Eveillard D, Gorsky G, et al. Tara Oceans: towards global ocean ecosystems biology. Nat Rev Microbiol. 2020;18(8):428–45.
Cleary DF, Swierts T, Coelho FJ, Polónia AR, Huang YM, Ferreira MR, et al. The sponge microbiome within the greater coral reef microbial metacommunity. Nat Commun. 2019;10(1):1644.
Chiarello M, Auguet JC, Graham NA, Claverie T, Sucré E, Bouvier C, et al. Exceptional but vulnerable microbial diversity in coral reef animal surface microbiomes. Proc Royal Soc B. 2020;287(1927):20200642.
Soliman T, Reimer JD, Yang SY, Villar-Briones A, Roy MC, Jenke-Kodama H. Diversity of microbial communities and quantitative chemodiversity in layers of marine sediment cores from a causeway (Kaichu-Doro) in Okinawa Island, Japan. Front Microbiol. 2017;8:2451.
Astudillo-García C, Hermans SM, Stevenson B, Buckley HL, Lear G. Microbial assemblages and bioindicators as proxies for ecosystem health status: potential and limitations. Appl Microbiol Biotechnol. 2019;103:6407–21.
Chen J, McIlroy SE, Archana A, Baker DM, Panagiotou G. A pollution gradient contributes to the taxonomic, functional, and resistome diversity of microbial communities in marine sediments. Microbiome. 2019;7:104.
Ng EL, Lin SY, Dungan AM, Colwell JM, Ede S, Lwanga EH, et al. Microplastic pollution alters forest soil microbiome. J Hazard Mater. 2021;409:124606.
Keeley N, Wood SA, Pochon X. Development and preliminary validation of a multi-trophic metabarcoding biotic index for monitoring benthic organic enrichment. Ecol Indic. 2018;85:1044–57.
Laroche O, Wood SA, Tremblay LA, Ellis JI, Lear G, Pochon X. A cross-taxa study using environmental DNA/RNA metabarcoding to measure biological impacts of offshore oil and gas drilling and production operations. Mar Pollut Bull. 2018;127:97–107.
Aylagas E, Atalah J, Sánchez-Jerez P, Pearman JK, Casado N, Asensi J, et al. A step towards the validation of bacteria biotic indices using DNA metabarcoding for benthic monitoring. Mol Ecol Resour. 2021;21(6):1889–903.
Eggleton J, Thomas KV. A review of factors affecting the release and bioavailability of contaminants during sediment disturbance events. Environ Int. 2004;30(7):973–80.
Brooijmans RJ, Pastink MI, Siezen RJ. Hydrocarbon-degrading bacteria: the oil-spill clean-up crew. Microb Biotechnol. 2009;2(6):587.
Kostka JE, Prakash O, Overholt WA, Green SJ, Freyer G, Canion A, et al. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl Environ Microbiol. 2011;77(22):7962–74.
Dash HR, Mangwani N, Chakraborty J, Kumari S, Das S. Marine bacteria: potential candidates for enhanced bioremediation. Appl Microbiol Biotechnol. 2013;97(2):561–71.
Berga M, Székely AJ, Langenheder S. Effects of disturbance intensity and frequency on bacterial community composition and function. PLoS ONE. 2012;7(5):e36959.
Shade A, Peter H, Allison SD, Baho DL, Berga M, Bürgmann H, et al. Fundamentals of microbial community resistance and resilience. Front Microbiol. 2012;3:417.
Bourne DG, Morrow KM, Webster NS. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu Rev Microbiol. 2016;70:317–40.
Lee ST, Davy SK, Tang SL, Kench PS. Mucus sugar content shapes the bacterial community structure in thermally stressed Acropora muricata. Front Microbiol. 2016;7:371.
Glasl B, Webster NS, Bourne DG. Microbial indicators as a diagnostic tool for assessing water quality and climate stress in coral reef ecosystems. Mar Biol. 2017;164:91.
Peixoto RS, Rosado PM, Leite DCDA, Rosado AS, Bourne DG. Beneficial microorganisms for corals (BMC): proposed mechanisms for coral health and resilience. Front Microbiol. 2017;8:341.
Maher RL, Schmeltzer ER, Meiling S, McMinds R, Ezzat L, Shantz AA, et al. Coral microbiomes demonstrate flexibility and resilience through a reduction in community diversity following a thermal stress event. Front Ecol Evol. 2020;8:555698.
Gardner SG, Leggat W, Ainsworth TD. The microbiome of the endosymbiotic Symbiodiniaceae in corals exposed to thermal stress. Hydrobiologia. 2023;850:3685–704.
Bell PR, Elmetri I, Lapointe BE. Evidence of large-scale chronic eutrophication in the great barrier reef: quantification of chlorophyll a thresholds for sustaining coral reef communities. Ambio. 2014;43:361–76.
Iijima M, Yasumoto K, Yasumoto J, Yasumoto-Hirose M, Kuniya N, Takeuchi R, et al. Phosphate enrichment hampers development of juvenile Acropora digitifera coral by inhibiting skeleton formation. Mar Biotechnol. 2019;21:291–300.
Reimer JD, Yang SY, White KN, Asami R, Fujita K, Hongo C, et al. Effects of causeway construction on environment and biota of subtropical tidal flats in Okinawa, Japan. Mar Pollut Bull. 2015;94(1–2):153–67.
Kleypas JA, Buddemeier RW, Archer D, Gattuso JP, Langdon C, Opdyke BN. Geochemical consequences of increased atmospheric carbon dioxide on coral reefs. Science. 1999;284(5411):118–20.
Leclercq NC, Gattuso JP, Jaubert JEAN. CO2 partial pressure controls the calcification rate of a coral community. Glob Chang Biol. 2000;6(3):329–34.
Langdon C, Atkinson MJ. Effect of elevated pCO2 on photosynthesis and calcification of corals and interactions with seasonal change in temperature/irradiance and nutrient enrichment. J Geophys Res Oceans. 2005;110:C09S07.
Sutherland KP, Porter JW, Torres C. Disease and immunity in Caribbean and Indo-Pacific zooxanthellate corals. Mar Ecol Prog Ser. 2004;266:273–302.
Bourne DG, Garren M, Work TM, Rosenberg E, Smith GW, Harvell CD. Microbial disease and the coral holobiont. Trends Microbiol. 2009;17(12):554–62.
Das RR, Wada H, Masucci GD, Singh T, Tavakoli-Kolour P, Wada N, et al. Four-year field survey of black band disease and skeletal growth anomalies in encrusting Montipora spp. Corals around Sesoko Island Okinawa Divers. 2022;14(1):32.
Chesher RH. Destruction of Pacific corals by the sea star Acanthaster planci. Science. 1969;165(3890):280–3.
De’Ath G, Fabricius KE, Sweatman H, Puotinen M. The 27–year decline of coral cover on the great barrier reef and its causes. Proc Natl Acad Sci U S A. 2012;109(44):17995–9.
Kayal M, Vercelloni J, Lison de Loma T, Bosserelle P, Chancerelle Y, Geoffroy S, et al. Predator crown-of-thorns starfish (Acanthaster planci) outbreak, mass mortality of corals, and cascading effects on reef fish and benthic communities. PLoS ONE. 2012;7(10):e47363.
Pratchett MS, Caballes CF, Wilmes JC, Matthews S, Mellin C, Sweatman HP, et al. Thirty years of research on crown-of-thorns starfish (1986–2016): scientific advances and emerging opportunities. Diversity. 2017;9(4):41.
Hoegh-Guldberg O. Climate change, coral bleaching and the future of the world’s coral reefs. Mar Freshw Res. 1999;50(8):839–66.
Hughes TP, Kerry JT, Álvarez-Noriega M, Álvarez-Romero JG, Anderson KD, Baird AH, et al. Global warming and recurrent mass bleaching of corals. Nature. 2017;543(7645):373–7.
Le Nohaïc M, Ross CL, Cornwall CE, Comeau S, Lowe R, McCulloch MT, Schoepf V. Marine heatwave causes unprecedented regional mass bleaching of thermally resistant corals in northwestern Australia. Sci Rep. 2017;7(1):14999.
Bellwood DR, Hughes TP, Folke C, Nyström M. Confronting the coral reef crisis. Nature. 2004;429(6994):827–33.
Hughes TP. Catastrophes, phase shifts, and large-scale degradation of a Caribbean coral reef. Science. 1994;265(5178):1547–51.
Rogers CS, Miller J. Permanent ‘phase shifts’ or reversible declines in coral cover? Lack of recovery of two coral reefs in St. John, US Virgin Islands. Mar Ecol Prog Ser. 2006;306:103–14.
Adam TC, Burkepile DE, Holbrook SJ, Carpenter RC, Claudet J, Loiseau C, et al. Landscape-scale patterns of nutrient enrichment in a coral reef ecosystem: implications for coral to algae phase shifts. Ecol Appl. 2021;31(1):e2227.
Cannon SE, Aram E, Beiateuea T, Kiareti A, Peter M, Donner SD. Coral reefs in the Gilbert Islands of Kiribati: resistance, resilience, and recovery after more than a decade of multiple stressors. PLoS ONE. 2021;16(8):e0255304.
Aronson RBWF, Precht W, Toscano M, Koltes KH. The 1998 bleaching event and its aftermath on a coral reef in Belize. Mar Biol. 2002;141:435–47.
Maliao RJ, Turingan RG, Lin J. Phase-shift in coral reef communities in the Florida Keys National Marine Sanctuary (FKNMS), USA. Mar Biol. 2008;154:841–53.
Chou LM, Yamazato K. Community structure of coral reefs within the vicinity of Motobu and Sesoko, Okinawa, and the effects of human and natural influences. Galaxea. 1990;9:9–75.
Stobart B, Teleki K, Buckley R, Downing N, Callow M. Coral recovery at Aldabra Atoll, Seychelles: five years after the 1998 bleaching event. Phil Trans R Soc A. 2005;363(1826):251–5.
Cruz IC, de Kikuchi RK, Longo LL, Creed JC. Evidence of a phase shift to Epizoanthus Gabrieli Carlgreen, 1951 (Order Zoanthidea) and loss of coral cover on reefs in the Southwest Atlantic. Mar Ecol. 2015;36(3):318–25.
Reimer JD, Agostini S, Golbuu Y, Harvey BP, Izumiyama M, Jamodiong EA, et al. High abundances of zooxanthellate zoantharians (Palythoa and Zoanthus) at multiple natural analogues: potential model anthozoans? Coral Reefs. 2023;42:707–15.
Meirelles PM, Soares AC, Oliveira L, Leomil L, Appolinario LR, Francini-Filho RB, et al. Metagenomics of coral reefs under phase shift and high hydrodynamics. Front Microbiol. 2018;9:2203.
Sunagawa S, Woodley CM, Medina M. Threatened corals provide underexplored microbial habitats. PLoS ONE. 2010;5(3):e9554.
Blackall LL, Wilson B, Van Oppen MJ. Coral—the world’s most diverse symbiotic ecosystem. Mol Ecol. 2015;24(21):5330–47.
Glasl B, Bourne DG, Frade PR, Thomas T, Schaffelke B, Webster NS. Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome. 2019;7:94.
Caughman AM, Pratte ZA, Patin NV, Stewart FJ. Coral microbiome changes over the day–night cycle. Coral Reefs. 2021;40:921–35.
Ide K, Nakano Y, Ito M, Nishikawa Y, Fujimura H, Takeyama H. The effect of co-culture of two coral species on their bacterial composition under captive environments. Mar Biotechnol. 2022;24(5):871–81.
Simister RL, Deines P, Botté ES, Webster NS, Taylor MW. Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ Microbiol. 2012;14(2):517–24.
Fan L, Liu M, Simister R, Webster NS, Thomas T. Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 2013;7(5):991–1002.
Bayer K, Kamke J, Hentschel U. Quantification of bacterial and archaeal symbionts in high and low microbial abundance sponges using real-time PCR. FEMS Microbiol Ecol. 2014;89(3):679–90.
Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, et al. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol Ecol. 2014;23(6):1348–63.
Blanquer A, Uriz MJ, Cebrian E, Galand PE. Snapshot of a bacterial microbiome shift during the early symptoms of a massive sponge die-off in the western Mediterranean. Front Microbiol. 2016;7:752.
Engelberts JP, Robbins SJ, de Goeij JM, Aranda M, Bell SC, Webster NS. Characterization of a sponge microbiome using an integrative genome-centric approach. ISME J. 2020;14(5):1100–10.
Posadas N, Baquiran JIP, Nada MAL, Kelly M, Conaco C. Microbiome diversity and host immune functions influence survivorship of sponge holobionts under future ocean conditions. ISME J. 2022;16(1):58–67.
Webster NS, Thomas T. The sponge hologenome. MBio. 2016;7(2):e00135–16.
Jeunen GJ, Knapp M, Spencer HG, Lamare MD, Taylor HR, Stat M, et al. Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol Ecol Resour. 2019;19(2):426–38.
Weber L, Gonzalez-Díaz P, Armenteros M, Apprill A. The coral ecosphere: a unique coral reef habitat that fosters coral-microbial interactions. Limnol Oceanogr. 2019;64(6):2373–88.
Hirose M, Reimer JD, Hidaka M, Suda S. Phylogenetic analyses of potentially free-living Symbiodinium spp. isolated from coral reef sand in Okinawa, Japan. Mar Biol. 2008;155:105–12.
Sweet MJ. Symbiodinium diversity within Acropora muricata and the surrounding environment. Mar Ecol. 2014;35(3):343–53.
Nitschke MR, Davy SK, Ward S. Horizontal transmission of Symbiodinium cells between adult and juvenile corals is aided by benthic sediment. Coral Reefs. 2016;35:335–44.
Ali A, Kriefall NG, Emery LE, Kenkel CD, Matz MV, Davies SW. Recruit symbiosis establishment and Symbiodiniaceae composition influenced by adult corals and reef sediment. Coral Reefs. 2019;38:405–15.
Dong X, Lan H, Huang L, Zhang H, Lin X, Weng S, et al. Metagenomic views of microbial communities in sand sediments associated with coral reefs. Microb Ecol. 2023;85(2):465–77.
Wild C, Huettel M, Klueter A, Kremb SG, Rasheed MY, Jørgensen BB. Coral mucus functions as an energy carrier and particle trap in the reef ecosystem. Nature. 2004;428(6978):66–70.
Carlos C, Torres TT, Ottoboni LM. Bacterial communities and species-specific associations with the mucus of Brazilian coral species. Sci Rep. 2013;3(1):1624.
Glasl B, Herndl GJ, Frade PR. The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 2016;10(9):2280–92.
Pollock FJ, McMinds R, Smith S, Bourne DG, Willis BL, Medina M, et al. Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat Commun. 2018;9:4921.
Fifer JE, Bui V, Berg JT, Kriefall N, Klepac C, Bentlage B, Davies SW. Microbiome structuring within a coral colony and along a sedimentation gradient. Front Mar Sci. 2022;8:805202.
Duarte CM, Marbà N, Gacia E, Fourqurean JW, Beggins J, Barrón C, Apostolaki ET. Seagrass community metabolism: assessing the carbon sink capacity of seagrass meadows. Glob Biogeochem Cycles. 2010;24:GB4032.
Mcleod E, Chmura GL, Bouillon S, Salm R, Björk M, Duarte CM, et al. A blueprint for blue carbon: toward an improved understanding of the role of vegetated coastal habitats in sequestering CO2. Front Ecol Environ. 2011;9(10):552–60.
Hansen JW, Udy JW, Perry CJ, Dennison WC, Lomstein BA. Effect of the seagrass Zostera capricorni on sediment microbial processes. Mar Ecol Prog Ser. 2000;199:83–96.
Welsh DT. Nitrogen fixation in seagrass meadows: regulation, plant–bacteria interactions and significance to primary productivity. Ecol Lett. 2000;3(1):58–71.
Brodersen KE, Siboni N, Nielsen DA, Pernice M, Ralph PJ, Seymour J, Kühl M. Seagrass Rhizosphere microenvironment alters plant-associated microbial community composition. Environ Microbiol. 2018;20(8):2854–64.
Koch MS, Erskine JM. Sulfide as a phytotoxin to the tropical seagrass Thalassia testudinum: interactions with light, salinity and temperature. J Exp Mar Biol Ecol. 2001;266(1):81–95.
Borum J, Pedersen O, Greve TM, Frankovich TA, Zieman JC, Fourqurean JW, Madden CJ. The potential role of plant oxygen and sulphide dynamics in die-off events of the tropical seagrass, Thalassia testudinum. J Ecol. 2005;93(1):148–58.
Johnson CR, Koch MS, Pedersen O, Madden CJ. Hypersalinity as a trigger of seagrass (Thalassia testudinum) die-off events in Florida Bay: evidence based on shoot meristem O2 and H2S dynamics. J Exp Mar Biol Ecol. 2018;504:47–52.
Martin BC, Bougoure J, Ryan MH, Bennett WW, Colmer TD, Joyce NK, et al. Oxygen loss from seagrass roots coincides with colonisation of sulphide-oxidising cable bacteria and reduces sulphide stress. ISME J. 2019;13(3):707–19.
Szitenberg A, Beca-Carretero P, Azcárate-García T, Yergaliyev T, Alexander-Shani R, Winters G. Teasing apart the host-related, nutrient-related and temperature-related effects shaping the phenology and microbiome of the tropical seagrass Halophila Stipulacea. Environ Microbiome. 2022;17(1):1–17.
Girvan MS, Campbell CD, Killham K, Prosser JI, Glover LA. Bacterial diversity promotes community stability and functional resilience after perturbation. Environ Microbiol. 2005;7(3):301–13.
Jones SE, Chiu CY, Krat TK, Wu JT, Shade A, McMahon KD. Typhoons initiate predictable change in aquatic bacterial communities. Limnol Oceanogr. 2008;53(4):1319–26.
Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE. 2014;9(8):e105592.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–2.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics. 2015;31(21):3476–82.
Chao A, Jost L. Coverage-based rarefaction and extrapolation: standardizing samples by completeness rather than size. Ecology. 2012;93(12):2533–47.
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–8.
Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(D1):D115–22.
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6(1):1–17.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590–6.
Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and all-species living Tree Project (LTP) taxonomic frameworks. Nucleic Acids Res. 2014;42(D1):D643–8.
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30.
Oksanen JF, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D et al. vegan: Community Ecology Package. R package version 2.5-7. 2020. https://CRAN.R-project.org/package=vegan.
Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22(12):1540–42.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. 2021. https://www.r-project.org/.
Arbizu PM, pairwiseAdonis. Pairwise Multilevel Comparison using Adonis. R package version 0.4. 2017. https://github.com/pmartinezarbizu/pairwiseAdonis.
Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11(1):3514.
Huang D, Zhang Z, Sun M, Feng Z, Ye M. Characterization and ecological function of bacterial communities in seabed sediments of the southwestern Yellow Sea and northwestern East China Sea, Western Pacific. Sci Total Environ. 2021;761:143233.
Zhang C, Liu Q, Li X, Wang M, Liu X, Yang J, et al. Spatial patterns and co-occurrence networks of microbial communities related to environmental heterogeneity in deep-sea surface sediments around Yap Trench, Western Pacific Ocean. Sci Total Environ. 2021;759:143799.
Aldeguer-Riquelme B, Rubio-Portillo E, Álvarez-Rogel J, Giménez-Casalduero F, Otero XL, Belando MD, et al. Factors structuring microbial communities in highly impacted coastal marine sediments (Mar Menor lagoon, SE Spain). Front Microbiol. 2022;13:937683.
Liu Y, Ji M, Yu T, Zaugg J, Anesio AM, Zhang Z, et al. A genome and gene catalog of glacier microbiomes. Nat Biotechnol. 2022;40(9):1341–8.
Salah-Tantawy A, Chang CSG, Liu MY, Young SS. Exploring the diversity and structural response of sediment-associated microbiota communities to environmental pollution at the siangshan wetland in Taiwan using environmental DNA metagenomic approach. Front Mar Sci. 2022;9:990428.
Hu X, Wang X, Zhao S, Cao L, Pan Y, Li F, et al. Uncovering the dynamic evolution of microbes and n-alkanes: insights from the Kuroshio Extension in the Northwest Pacific Ocean. Sci Total Environ. 2023;875:162418.
Hoshino T, Doi H, Uramoto GI, Wörmer L, Adhikari RR, Xiao N, et al. Global diversity of microbial communities in marine sediment. Proc Natl Acad Sci U S A. 2020;117(44):27587–97.
Hamamoto K, Poliseno A, Soliman T, Reimer JD. Shallow epifaunal sea cucumber densities and their relationship with the benthic community in the Okinawa Islands. PeerJ. 2022;10:e14181.
Lee S, Ferse S, Ford A, Wild C, Mangubhai S. Effect of sea cucumber density on the health of reef-flat sediments. In: Mangubhai S, Lalavanua W, Purcell SW, editors. Fiji’s Sea Cucumber Fishery: advances in Science for Improved Management. Fiji: Wildlife Conservation Society; 2017. pp. 54–61.
Waite DW, Chuvochina M, Pelikan C, Parks DH, Yilmaz P, Wagner M, et al. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int J Syst Evol Microbiol. 2020;70(11):5972–6016.
Begmatov S, Savvichev AS, Kadnikov VV, Beletsky AV, Rusanov II, Klyuvitkin AA, et al. Microbial communities involved in methane, sulfur, and nitrogen cycling in the sediments of the Barents Sea. Microorganisms. 2021;9(11):2362.
Markovski M, Najdek M, Herndl GJ, Korlević M. Compositional stability of sediment microbial communities during a seagrass meadow decline. Front Mar Sci. 2022;9:966070.
Koch MS, Johnson CR, Madden CJ, Pedersen O. Irradiance, water column O2, and tide drive internal O2 dynamics and meristem H2S detection in the dominant Caribbean-tropical atlantic seagrass, Thalassia testudinum. Estuaries Coast. 2022;45(8):2543–59.
Martin BC, Middleton JA, Fraser MW, Marshall IP, Scholz VV, Hausl B, Schmidt H. Cutting out the middle clam: lucinid endosymbiotic bacteria are also associated with seagrass roots worldwide. ISME J. 2020;14(11):2901–5.
Banister RB, Schwarz MT, Fine M, Ritchie KB, Muller EM. Instability and stasis among the microbiome of seagrass leaves, roots and rhizomes, and nearby sediments within a natural pH gradient. Microb Ecol. 2021;84:703–16.
Tarquinio F, Hyndes GA, Laverock B, Koenders A, Säwström C. The seagrass holobiont: understanding seagrass-bacteria interactions and their role in seagrass ecosystem functioning. FEMS Microbiol Lett. 2019;366(6):fnz057.
Le Campion-Alsumard T, Golubic S, Hutchings P. Microbial endoliths in skeletons of live and dead corals: Porites lobata (Moorea, French Polynesia). Mar Ecol Prog Ser. 1995;117:149–57.
Sweet MJ, Croquer A, Bythell JC. Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs. 2011;30:39–52.
Chen CP, Tseng CH, Chen CA, Tang SL. The dynamics of microbial partnerships in the coral Isopora palifera. ISME J. 2011;5(4):728–40.
Galand PE, Ruscheweyh HJ, Salazar G, Hochart C, Henry N, Hume BC, et al. Diversity of the Pacific Ocean coral reef microbiome. Nat Commun. 2023;14(1):3039.
Li C, Reimers CE, Chapman JW. Microbiome analyses and presence of cable bacteria in the burrow sediment of Upogebia pugettensis. Mar Ecol Prog Ser. 2020;648:79–94.
Chase AB, Bogdanov A, Demko AM, Jensen PR. Biogeographic patterns of biosynthetic potential and specialized metabolites in marine sediments. ISME J. 2023;17:976–83.
Zbinden M, Cambon-Bonavita MA. Occurrence of Deferribacterales and Entomoplasmatales in the deep-sea Alvinocarid shrimp Rimicaris exoculata gut. FEMS Microbiol Ecol. 2003;46(1):23–30.
Seabrook S, De Leo FC, Thurber AR. Flipping for food: the use of a methane seep by tanner crabs (Chionoecetes tanneri). Front Mar Sci. 2019;6:43.
Cleary DF, Becking LE, Polónia AR, Freitas RM, Gomes NC. Jellyfish-associated bacterial communities and bacterioplankton in Indonesian Marine lakes. FEMS Microbiol Ecol. 2016;92(5):fiw064.
Wiese J, Imhoff JF, Horn H, Borchert E, Kyrpides NC, Göker M, et al. Genome analysis of the marine bacterium Kiloniella laminariae and first insights into comparative genomics with related Kiloniella species. Arch Microbiol. 2020;202:815–24.
Hao W, Wang L, Li F, Sun T, Peng S, Li Y, et al. Bacterial communities associated with hydromedusa Gonionemus vertens in different regions in Chinese coastal waters. J Oceanol Limnol. 2022;40(4):1530–43.
Galac MR, Bosch I, Janies DA. Bacterial communities of oceanic sea star (Asteroidea: Echinodermata) larvae. Mar Biol. 2016;163:162.
Irei Y, Nozawa Y, Reimer JD. Distribution patterns of five zoanthid species in Okinawa Island, Japan. Zool Stud. 2011;50(4);426 – 33.
Neo ML, Lim KK, Yang SY, Soong GY, Masucci GD, Biondi P, et al. Status of giant clam resources around Okinawa-Jima Island, Ryukyu Archipelago, Japan. Aquat Conserv: Mar Freshw Ecosyst. 2019;29(6):1002–11.
Du ZJ, Wang ZJ, Zhao JX, Chen GJ. Woeseia Oceani gen. nov., sp. nov., a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. Nov. Int J Syst Evol Microbiol. 2016;66(1):107–12.
Mußmann M, Pjevac P, Krüger K, Dyksma S. Genomic repertoire of the Woeseiaceae/JTB255, cosmopolitan and abundant core members of microbial communities in marine sediments. ISME J. 2017;11(5):1276–81.
Tiralerdpanich P, Nasaree S, Pinyakong O, Sonthiphand P. Variation of the mangrove sediment microbiomes and their phenanthrene biodegradation rates during the dry and wet seasons. Environ Pollut. 2021;289:117849.
Zhang X, Shang S, Gao J, Liu J, Chen L, Sun C, et al. Microbial Community structure in the Rhizosphere of Zostera. Ecol Evol Biol. 2022;7(1):7–13.
Liu Y, Guo Z, Zhang P, Du J, Gao P, Zhang Z. Diversity and structure of vegetation rhizosphere bacterial community in various habitats of Liaohekou coastal wetlands. Sustainability. 2022;14(24):16396.
Meng S, Peng T, Liu X, Wang H, Huang T, Gu JD, Hu Z. Ecological role of bacteria involved in the biogeochemical cycles of mangroves based on functional genes detected through GeoChip 5.0. mSphere. 2022;7(1):e00936-21.
Taketani RG, Franco NO, Rosado AS, van Elsas JD. Microbial community response to a simulated hydrocarbon spill in mangrove sediments. J Microbiol. 2010;48:7–15.
Varon-Lopez M, Dias ACF, Fasanella CC, Durrer A, Melo IS, Kuramae EE, Andreote FD. Sulphur-oxidizing and sulphate-reducing communities in Brazilian mangrove sediments. Environ Microbiol. 2014;16(3):845–55.
Cleary DF, Coelho FJ, Oliveira V, Gomes NC, Polónia AR. Sediment depth and habitat as predictors of the diversity and composition of sediment bacterial communities in an inter-tidal estuarine environment. Mar Ecol. 2017;38(2):e12411.
Mohapatra M, Manu S, Dash SP, Rastogi G. Seagrasses and local environment control the bacterial community structure and carbon substrate utilization in brackish sediments. J Environ Manage. 2022;314:115013.
Nie S, Zhang Z, Mo S, Li J, He S, Kashif M, et al. Desulfobacterales stimulates nitrate reduction in the mangrove ecosystem of a subtropical gulf. Sci Total Environ. 2021;769:144562.
Cleary DFR, Polónia ARM, Becking LE, de Voogd NJ, Purwanto, Gomes H, Gomes NCM. Compositional analysis of bacterial communities in seawater, sediment, and sponges in the Misool coral reef system, Indonesia. Mar Biodivers. 2018;48:1889–901.
Thomas F, Giblin AE, Cardon ZG, Sievert SM. Rhizosphere heterogeneity shapes abundance and activity of sulfur-oxidizing bacteria in vegetated salt marsh sediments. Fronti Microbiol. 2014;5:309.
Martin BC, Middleton JA, Skrzypek G, Kendrick GA, Cosgrove J, Fraser MW. Composition of seagrass root associated bacterial communities are linked to nutrients and heavy metal concentrations in an anthropogenically influenced estuary. Front Mar Sci. 2022;8:768864.
Wu S, Li R, Xie S, Shi C. Depth-related change of sulfate-reducing bacteria community in mangrove sediments: the influence of heavy metal contamination. Mar Pollut Bull. 2019;140:443–50.
Aylagas E, Borja Á, Tangherlini M, Dell’Anno A, Corinaldesi C, Michell CT, et al. A bacterial community-based index to assess the ecological status of estuarine and coastal environments. Mar Pollut Bull. 2017;114(2):679–88.
Lin G, Sun F, Wang C, Zhang L, Zhang X. Assessment of the effect of Enteromorpha prolifera on bacterial community structures in aquaculture environment. PLoS ONE. 2017;12(7):e0179792.
Rubin-Blum M, Sisma-Ventura G, Yudkovski Y, Belkin N, Kanari M, Herut B, Rahav E. Diversity, activity, and abundance of benthic microbes in the Southeastern Mediterranean Sea. FEMS Microbiol Ecol. 2022;98(2):fiac009.
Martin BC, Alarcon MS, Gleeson D, Middleton JA, Fraser MW, Ryan MH, et al. Root microbiomes as indicators of seagrass health. FEMS Microbiol Ecol. 2020;96(2):fiz201.
Ugarelli K, Chakrabarti S, Laas P, Stingl U. The seagrass holobiont and its microbiome. Microorganisms. 2017;5(4):81.
Dang H, Li T, Chen M, Huang G. Cross-ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Appl Environ Microbiol. 2008;74(1):52–60.
Welsh RM, Rosales SM, Zaneveld JR, Payet JP, McMinds R, Hubbs SL, Thurber RLV. Alien vs. predator: bacterial challenge alters coral microbiomes unless controlled by Halobacteriovorax predators. PeerJ. 2017;5:e3315.
Rosales SM, Clark AS, Huebner LK, Ruzicka RR, Muller EM. Rhodobacterales and Rhizobiales are associated with stony coral tissue loss disease and its suspected sources of transmission. Front Microbiol. 2020;11:681.
Sun F, Yang H, Zhang X, Tan F, Shi Q. Response characteristics of bacterial communities in multiple coral genera at the early stages of coral bleaching during El Niño. Ecol Indic. 2022;144:109569.
Miller TR, Hnilicka K, Dziedzic A, Desplats P, Belas R. Chemotaxis of Silicibacter sp. strain TM1040 toward dinoflagellate products. Appl Environ Microbiol. 2004;70(8):4692–701.
Curson ARJ, Rogers R, Todd JD, Brearley CA, Johnston AWB. Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine α-proteobacteria and Rhodobacter sphaeroides. Environ Microbiol. 2008;10(3):757–67.
Raina JB, Dinsdale EA, Willis BL, Bourne DG. Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol. 2010;18(3):101–8.
Broadbent AD, Jones GB, Jones RJ. DMSP in corals and benthic algae from the great barrier reef. Estuar Coast Shelf Sci. 2002;55(4):547–55.
Garren M, Son K, Raina JB, Rusconi R, Menolascina F, Shapiro OH, et al. A bacterial pathogen uses dimethylsulfoniopropionate as a cue to target heat-stressed corals. ISME J. 2014;8(5):999–1007.
Rosales SM, Huebner LK, Evans JS, Apprill A, Baker AC, Becker CC, et al. A meta-analysis of the stony coral tissue loss disease microbiome finds key bacteria in unaffected and lesion tissue in diseased colonies. ISME Commun. 2023;3(1):19.
Gavriilidou A, Gutleben J, Versluis D, Forgiarini F, van Passel MW, Ingham CJ, et al. Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genom. 2020;21:569.
McDevitt-Irwin JM, Baum JK, Garren M, Vega Thurber RL. Responses of coral-associated bacterial communities to local and global stressors. Front Mar Sci. 2017;4:262.
Kieft B, Li Z, Bryson S, Crump BC, Hettich R, Pan C, et al. Microbial community structure-function relationships in Yaquina bay estuary reveal spatially distinct carbon and nitrogen cycling capacities. Front Microbiol. 2018;9:1282.
Freitas S, Hatosy S, Fuhrman JA, Huse SM, Mark Welch DB, Sogin ML, Martiny AC. Global distribution and diversity of marine Verrucomicrobia. ISME J. 2012;6(8):1499–505.
Sekar R, Mills DK, Remily ER, Voss JD, Richardson LL. Microbial communities in the surface mucopolysaccharide layer and the black band microbial mat of black band-diseased Siderastrea siderea. Appl Environ Microbiol. 2006;72(9):5963–73.
Soffer N. What is killing the corals? Viral and bacterial interrogations using metagenomics and microscopy. Oregon State University. 2013. https://ir.library.oregonstate.edu/concern/graduate_thesis_or_dissertations/4m90f1364?locale=en Accessed 13 Sep 2023.
Adeolu M, Alnajar S, Naushad S, Gupta RS. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int J Syst Evol Microbiol. 2016;66(12):5575-99.
Sullam KE, Essinger SD, Lozupone CA, O’Connor MP, Rosen GL, Knight ROB, et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol. 2012;21(13):3363–78.
Oliveira BF, Lopes IR, Canellas AL, Muricy G, Dobson AD, Laport MS. Not that close to mommy: horizontal transmission seeds the microbiome associated with the marine sponge Plakina cyanorosea. Microorganisms. 2020;8(12):1978.
Xu M, Cheng K, Xiao B, Tong M, Cai Z, Jong MC, et al. Bacterial communities vary from different scleractinian coral species and between bleached and non-bleached corals. Microbiol Spectr. 2023;11(3):e04910–22.
Reichenbach H. The order Cytophagales. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt, editors. The Prokaryotes. Third Edition. Volume 7. New York: Springer; 2006. pp. 549–90.
Zhang Y, Li Y, Wang L, Tang Y, Chen J, Hu Y, et al. Soil microbiological variability under different successional stages of the Chongming Dongtan wetland and its effect on soil organic carbon storage. Ecol Eng. 2013;52:308–15.
Williams HN. A study of the occurrence and distribution of bdellovibrios in estuarine sediment over an annual cycle. Microb Ecol. 1988;15:9–20.
Williams HN, Chen H. Environmental regulation of the distribution and ecology of Bdellovibrio and like organisms. Front Microbiol. 2020;11:545070.
Bayer T, Neave MJ, Alsheikh-Hussain A, Aranda M, Yum LK, Mincer T, et al. The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl Environ Microbiol. 2013;79(15):4759–62.
Lee ST, Davy SK, Tang SL, Fan TY, Kench PS. Successive shifts in the microbial community of the surface mucus layer and tissues of the coral Acropora muricata under thermal stress. FEMS Microbiol Ecol. 2015;91(12):fiv142.
Ziegler M, Grupstra CG, Barreto MM, Eaton M, BaOmar J, Zubier K, et al. Coral bacterial community structure responds to environmental change in a host-specific manner. Nat Commun. 2019;10(1):3092.
Osman EO, Suggett DJ, Voolstra CR, Pettay DT, Clark DR, Pogoreutz C, et al. Coral microbiome composition along the northern Red Sea suggests high plasticity of bacterial and specificity of endosymbiotic dinoflagellate communities. Microbiome. 2020;8:8.
Sweet MJ, Croquer A, Bythell JC. Dynamics of bacterial community development in the reef coral Acropora muricata following experimental antibiotic treatment. Coral Reefs. 2011;30:1121–33.
Keller-Costa T, Lago-Lestón A, Saraiva JP, Toscan R, Silva SG, Gonçalves J, et al. Metagenomic insights into the taxonomy, function, and dysbiosis of prokaryotic communities in octocorals. Microbiome. 2021;9:72.
López-Pérez M, Haro-Moreno JM, Iranzo J, Rodriguez-Valera F. Genomes of the Candidatus Actinomarinales order: highly streamlined marine epipelagic actinobacteria. Volume 5. mSystems; 2020. pp. e01041–20.
Miyakawa T, Yang J, Kawasaki M, Adachi N, Fujii A, Miyauchi Y, et al. Structural bases for aspartate recognition and polymerization efficiency of cyanobacterial cyanophycin synthetase. Nat Commun. 2022;13(1):5097.
Watzer B, Forchhammer K. Cyanophycin synthesis optimizes nitrogen utilization in the unicellular cyanobacterium Synechocystis sp. strain PCC 6803. Appl Environ Microbiol. 2018;84(20):e01298–18.
Duan L, Li JL, Yin LZ, Luo XQ, Ahmad M, Fang BZ, et al. Habitat-dependent prokaryotic microbial community, potential keystone species, and network complexity in a subtropical estuary. Environ Res. 2022;212:113376.
Xie H, Chen J, Feng L, He L, Zhou C, Hong P, et al. Chemotaxis-selective colonization of mangrove rhizosphere microbes on nine different microplastics. Sci Total Environ. 2021;752:142223.
Dedysh SN, Yilmaz P. Refining the taxonomic structure of the phylum Acidobacteria. Int J Syst Evol Microbiol. 2018;68(12):3796–806.
Flieder M, Buongiorno J, Herbold CW, Hausmann B, Rattei T, Lloyd KG, et al. Novel taxa of Acidobacteriota implicated in seafloor sulfur cycling. ISME J. 2021;15(11):3159–80.
Banker RM, Lipovac J, Stachowicz JJ, Gold DA. Sodium molybdate does not inhibit sulfate-reducing bacteria but increases shell growth in the Pacific oyster Magallana Gigas. PLoS ONE. 2022;17(2):e0262939.
Lehosmaa K, Muotka T, Pirttilä AM, Jaakola I, Rossi PM, Jyväsjärvi J. Bacterial communities at a groundwater-surface water ecotone: gradual change or abrupt transition points along a contamination gradient? Environ Microbiol. 2021;23(11):6694–706.
Rios-Del Toro EE, Valenzuela EI, López-Lozano NE, Cortés-Martínez MG, Sánchez-Rodríguez MA, Calvario-Martínez O, et al. Anaerobic ammonium oxidation linked to sulfate and ferric iron reduction fuels nitrogen loss in marine sediments. Biodegradation. 2018;29:429–42.
Daniels CA, Baumgarten S, Yum LK, Michell CT, Bayer T, Arif C, et al. Metatranscriptome analysis of the reef-building coral Orbicella faveolata indicates holobiont response to coral disease. Front Mar Sci. 2015;2:62.
Ziegler M, Seneca FO, Yum LK, Palumbi SR, Voolstra CR. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat Commun. 2017;8(1):14213.
Pootakham W, Mhuantong W, Putchim L, Yoocha T, Sonthirod C, Kongkachana W, et al. Dynamics of coral-associated microbiomes during a thermal bleaching event. MicrobiologyOpen. 2018;7(5):e00604.
Ling J, Zhou W, Yang Q, Yin J, Zhang J, Peng Q, et al. Spatial and species variations of bacterial community structure and putative function in seagrass rhizosphere sediment. Life. 2021;11(8):852.
Szitenberg A, Beca-Carretero P, Azcárate-García T, Yergaliyev T, Alexander-Shani R, Winters G. Teasing apart the host-related, nutrient-related and temperature-related effects shaping the phenology and microbiome of the tropical seagrass Halophila Stipulacea. Environ Microbiome. 2022;17:18.
Aziz RK, Breitbart M, Edwards RA. Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 2010;38(13):4207–17.
Mihara H, Esaki N. Bacterial cysteine desulfurases: their function and mechanisms. Appl Microbiol Biotechnol. 2002;60:12–23.
Hidese R, Mihara H, Esaki N. Bacterial cysteine desulfurases: versatile key players in biosynthetic pathways of sulfur-containing biofactors. Appl Microbiol Biotechnol. 2011;91:47–61.
Alamuri P, Mehta N, Burk A, Maier RJ. Regulation of the Helicobacter pylori Fe-S cluster synthesis protein NifS by iron, oxidative stress conditions, and fur. J Bacterial. 2006;188(14):5325–30.
Dai Y, Outten FW. The E. Coli SufS–SufE sulfur transfer system is more resistant to oxidative stress than IscS–IscU. FEBS Lett. 2012;586(22):4016–22.
Rybniker J, Pojer F, Marienhagen J, Kolly GS, Chen JM, Van Gumpel E, et al. The cysteine desulfurase IscS of Mycobacterium tuberculosis is involved in iron–sulfur cluster biogenesis and oxidative stress defence. Biochem J. 2014;459(3):467–78.
Saragosti E, Tchernov D, Katsir A, Shaked Y. Extracellular production and degradation of superoxide in the coral Stylophora pistillata and cultured Symbiodinium. PLoS ONE. 2010;5(9):e12508.
Storz G, Tartaglia LA, Farr SB, Ames BN. Bacterial defenses against oxidative stress. Trends Genet. 1990;6:363–8.
Zeinali F, Homaei A, Kamrani E. Sources of marine superoxide dismutases: characteristics and applications. Int J Biol Macromol. 2015;79:627–37.
Downs CA, Mueller E, Phillips S, Fauth JE, Woodley CM. A molecular biomarker system for assessing the health of coral (Montastraea faveolata) during heat stress. Mar Biotechnol. 2000;2:533–44.
Downs CA, Fauth JE, Halas JC, Dustan P, Bemiss J, Woodley CM. Oxidative stress and seasonal coral bleaching. Free Radic Biol Med. 2002;33(4):533–43.
Yamada S, Sugimoto H, Kobayashi M, Ohno A, Nakamura H, Shiro Y. Structure of PAS-linked histidine kinase and the response regulator complex. Structure. 2009;17(10):1333–44.
West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem Sci. 2001;26(6):369–76.
McLoon AL, Kolodkin-Gal I, Rubinstein SM, Kolter R, Losick R. Spatial regulation of histidine kinases governing biofilm formation in Bacillus subtilis. J Bacterial. 2011;193(3):679–85.
Acknowledgements
We are grateful to members of the Molecular Invertebrate Systematics and Ecology laboratory, University of the Ryukyus, especially Ms. Marilyn Carletti, Ms. Chloé Loïs Fourreau, Ms. Iori Mizukami, Mr. Yusuke Iwaki, and Mr. Hiroki Nakajima for their help in sampling. Finally, Kohei Hamamoto dedicates this paper to the memory of his late beloved dog Mimimi, who supported him throughout this work.
Funding
This work was supported by a MEXT/JSPS KAKENHI Grants (20H00653 and 24H00784), and the on Environmentally-conscious Developments and Technologies (E-code) research laboratory at the National Institute of Advanced Industrial Science and Technology (AIST).
Author information
Authors and Affiliations
Contributions
The research was designed by KH, MM, AI, and JDR. Sampling was done by KH and AP. Laboratory experimentation was directed by MN, and performed by KH, AM, and KG. Sequence processing, data curation and data analyses were done by KH, MM, and AI. The original draft was written by KH, then reviewed and edited by all authors. AI and JDR supervised entire research. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hamamoto, K., Mizuyama, M., Nishijima, M. et al. Diversity, composition and potential roles of sedimentary microbial communities in different coastal substrates around subtropical Okinawa Island, Japan. Environmental Microbiome 19, 54 (2024). https://doi.org/10.1186/s40793-024-00594-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-024-00594-1