
Abstract
The rapid development of sequencing methods over the past decades has accelerated both the potential scope and depth of microbiota and microbiome studies. Recent developments in the field have been marked by an expansion away from purely categorical studies towards a greater investigation of community functionality. As in-depth genomic and environmental coverage is often distributed unequally across major taxa and ecosystems, it can be difficult to identify or substantiate relationships within microbial communities. Generic databases containing datasets from diverse ecosystems have opened a new era of data accessibility despite costs in terms of data quality and heterogeneity. This challenge is readily embodied in the integration of meta-omics data alongside habitat-specific standards which help contextualise datasets both in terms of sample processing and background within the ecosystem. A special case of large genomic repositories, ecosystem-specific databases (ES-DB’s), have emerged to consolidate and better standardise sample processing and analysis protocols around individual ecosystems under study, allowing independent studies to produce comparable datasets. Here, we provide a comprehensive review of this emerging tool for microbial community analysis in relation to current trends in the field. We focus on the factors leading to the formation of ES-DB’s, their comparison to traditional microbial databases, the potential for ES-DB integration with meta-omics platforms, as well as inherent limitations in the applicability of ES-DB’s.
Similar content being viewed by others

Introduction
Interest in categorizing microbial communities across accessible habitats has exposed the vast complexity of microbial life [1,2,3]. What started with the laboratory isolation of microbial species from habitats of interest has expanded both in scope and depth following the advent of meta-omics (metabarcoding, metagenomics, metatranscriptomics, metaproteomics, metabolomics). Metabarcoding, for example, is now commonplace, allowing for an unprecedented systematic cataloguing of microorganisms using identifying biomarkers [4,5,6]. Technological developments over the past couple of decades have greatly expanded microbial community ecology analyses to include, albeit still at great cost and effort, the sequencing of all genomes within a sample (metagenomics). These deep dives into the microbial community allow a higher level of taxonomic precision as well as further opportunities to assess the functional capacity of the system [7,8,9,10]. Coupled to this has been an expansion of gene expression studies across community constituents within a sample (metatranscriptomics). The widening scope of meta-omics has led to the integration of diverse analytical tools into community ecology studies, such as metaproteomics and metabolomics, providing information on the underlying functional activity and metabolic state of the community, respectively [11, 12]. Measurements of physicochemical parameters (i.e., pH, EC, Eh, temperature, nutrients) provide an environmental context for taxonomic and functional fluctuations within the microbial community. Integrating measures of microbial functionality with taxonomic identification and these contextual parameters is essential for a better understanding of inter-microbial relationships and their roles in a particular environment. Nonetheless, databases have largely catalogued their constituent datasets around data type (e.g., sequence data, physiological data) and not the environments from which organisms are being sampled. This practice results in less standardisation across studies utilizing different investigative strategies (i.e., different meta-omics approaches) on the same habitat, ultimately hindering the integration of multiple data types in microbial community ecology assessments.
Several studies have highlighted concerns over the validity of sequencing data accruing from the ever-expanding body of microbial surveys and microbiome studies [13,14,15,16]. One group of reviews has addressed this issue by proposing standards for studies to follow. These reviews target standardisation in the collection and processing of data for microbiome studies with respect to general guidelines [14, 17,18,19] and to specific environmental situations [20,21,22,23,24,25]. Another group of reviews has focused on the efforts to integrate other data types (e.g., mass-spectroscopy spectra, environmental physicochemical data) into sequencing studies [26,27,28,29,30,31]. These efforts notwithstanding, the evolution of microbial database collections from a data type orientation to an environment-specific one has received less attention. A recent commentary in Nature Microbiology addressed the topic of data type integration from the perspective of “microbiome centres”—institutions or consortia designed to accelerate microbiome research by facilitating collaborations between personnel and infrastructure resources [32]. While the inception of the Microbiome Centers Consortium (MCC; http://microbiomecenters.org/) in 2019 marks a milestone for more coordinated standardisation across microbiome studies, database resources are still developed largely independent of one another despite greater connectivity between laboratories around the world. Widespread use of diverse meta-omics techniques over the recent decades drives current efforts to streamline and integrate data types. Better database management achieves multiple aims: expanded access at an assured quality level, a repository for data, as well as more consistent and aligned standardisation for generating data. In this article, we review the development of ecosystem-specific databases (ES-DB’s) to address the unique challenges that arise when working with heterogenous data types inherent to microbial community ecology.
Meta-omics tools to unravel microbial community diversity and function
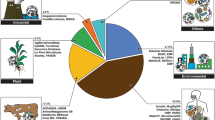
To study microbial community ecology, approaches used may be either DNA-based to study taxonomic diversity (metabarcoding) and gene diversity (metagenomics), or RNA-based to study gene-expression in the active microbial community (metatranscriptomics), or protein- and metabolite-based to study the production and secretion of various molecules (metaproteomics, metabolomics) (Fig. 1). Viruses, while not the primary focus of this review, are studied using both metagenomic and metatranscriptomics techniques, depending on the virus type targeted. Meta-omics tools are often used independently or a couple of them together in an effort to unravel complex interspecies relationships and functions in microbial communities. However, the lack of homogeneity among isolated studies limits their usefulness for making correlations and deriving meaningful hypotheses from similar studies.

Interrelationships between multiple depths of biome characterisation, all which can be unified through microbial database collections
Taxonomic identification and diversity of the microbial community
Metabarcoding studies have largely succeeded in surveying microbial diversity in all major Earth habitats [33,34,35,36,37]. Several synonymous terms in widespread circulation include metataxonomics, community profiling, and amplicon sequencing. While metataxonomics and community profiling emphasise the putative categorical endpoint, amplicon sequencing highlights the methodological contrast to metagenomics. Metabarcoding may be done alone (sequences compared to reference databases) or against a metagenomic sample (sequences compared to dataset of the community genomes in a sample) [38,39,40]. The ability of high-throughput sequencing platforms to rapidly and accurately sequence gene regions has made metabarcoding data the most common data type in microbial sequence database collections [41,42,43]. Practical considerations such as a lower associated cost, lower DNA quantities and lighter bioinformatic analyses required per analysis further lend to the attractiveness of the method [44, 45]. The selection of universal gene sequences for each taxonomic rank of interest has been essential for yielding more exhaustive descriptions of microbial taxonomic diversity [46,47,48,49]. The increasing fidelity of third generation technologies such as the Pacific Bioscience and Nanopore long-read sequencing technologies and accompanying genome assembly infrastructure able to correct misreads represents an important milestone, allowing studies to rely on metabarcoding for species-rank taxonomic assignment [50,51,52].
Far from being a complete story, major biases persist around the metabarcoding approach as reviewed elsewhere [53,54,55,56]. Here we will emphasise the issue of unspecific amplification as a pernicious problem regardless of the focus on DNA- or RNA-based studies. For example, the use of the 16S rRNA gene to target bacteria also amplifies the 16S rRNA gene of plastids (e.g., chloroplasts, mitochondria) especially in host-associated microbial studies [57]. Similarly, investigations into host-associated microbial eukaryote interactions—typically targeting the 18S rRNA gene—simultaneously amplify host 18 s rRNA sequences. The use of excluding or blocking primers may mitigate these issues [58], however the challenges they pose have not been fully resolved.
The requirement for amplification of a known sequence sets metabarcoding apart from other genetic investigation methods and is a significant limitation in exploratory research. For taxonomic studies, targeted genes must be variable enough to distinguish species or strains yet have sufficiently conserved sequences flanking the gene of interest in order to design primers. Common standards include the 16S rRNA gene for prokaryotes [59,60,61,62], the 18S rRNA gene (microbial eukaryotes) [63], or the ITS region (fungi) [64, 65], albeit other sequences such as heat shock proteins have promising perspectives [66, 67]. Additionally, while metabarcoding typically involves sequencing the small subunit of the rRNA gene, it may also be applied to other genes of interest [68, 69]. By identifying the presence of a gene, these procedures are able to provide a clue into the metabolism exhibited by the sampled organism.
For studies aiming only to describe community composition, metabarcoding remains a cost-effective tool compared to culture-independent techniques- especially for well-studied microbiomes [33, 61, 70]. The most significant shortcoming in metabarcoding is the limited flexibility of a single gene sequence to represent total diversity. In contrast to the sequencing of a specific amplicon, targeted metagenomics is a culture-independent technique which limits the scope to a subset of total sample diversity (e.g., prokaryotes or eukaryotes) but targeting all genes within the sample [71, 72]. Ultimately, while sequence data is effective at mapping taxonomic relationships within a sample, investigative work into the mechanisms driving observed shifts in physicochemical parameters require a broader set of tools. Meta-omics addresses this goal by exploring community ecology from different perspectives: what microorganisms may potentially create (metagenomics), what they are in the process of creating (metatranscriptomics), and what they have created (metaproteomics, metabolomics).
Function and metabolic potential of the microbial community
Discerning function within the context of microbial ecosystems is a major challenge for community ecology studies. Metagenomic studies reveal functional potential—DNA sequences that have the potential to be expressed. Conversely, metatranscriptomics can be used to study the pool of expressed sequences. The subset of expressed sequences may then be studied within the context of translated proteins (metaproteomics) or as the biproducts of cellular metabolism (metabolomics). The following section will describe the types of data produced in these studies as well as the unique challenges they pose in terms of database integration.
Metagenomics
The metagenomic approach indiscriminately sequences all DNA fragments from a sample. The goal of this approach is to preserve the vast majority of genomes, with the important caveat that sampling the true total diversity is not physically realistic [73, 74]. Metagenomics is not subject to the same limitations in terms of primer selection and specificity as metabarcoding [45, 75], although data quality may be diminished through pre-processing steps as with other methods [53, 54, 76]. Given the significantly larger datasets than found in metabarcoding studies, metagenomics data is more laborious to process with requirements for sequence preparation and assembly that must be weighed against the potential for greater resolution in discerning metabolic pathways [77]. In assembly, short sequencing reads are sorted to link extracted genomes with the original mixed microbial community constituents. The resulting pool of genomes can be screened for the presence of genes associated with metabolic pathways of interest; however it does not confirm their expression (requiring transcriptomics).
The assembly of DNA-based viruses was initially beset with unique challenges compared to other sources of DNA, however the isolation and sequencing of DNA-based viruses has become considerably more developed and reproducible, as reviewed elsewhere [78,79,80]. Similarly, RNA-based viruses have also been studied through metagenomics, with protocols now achieving a high degree of accuracy and recovery efficiency [81].
Indeed, some pathways may be shared among several genomes, suggesting potential cross-feeding between microbes in the community [82]. Screening genomes for specific genes associated with particular metabolic profiles can be a powerful tool in discerning (i) evolutionary acquisition of genes and (ii) putative biochemical transformations within the ecosystem. Such information may help substantiate observed physicochemical shifts in the ecosystem. Crucially, however, metagenomic sequence data from reference databases cannot linearly translate into functional assignments for homologous sequences [15, 83], in contrast to metatranscriptomics which measures gene expression (84, 85).
Metatranscriptomics
The application of metatranscriptomics to analyse the sum of genes expressed in a same sample, is essential for assigning functionality to members of a microbial community. The metatranscriptomics approach explores the metabolically active fraction of a sample via sequencing RNA transcripts. Here, total RNA or messenger RNA (mRNA) in a sample is converted to complementary DNA (cDNA), allowing it to be further pre-processed as needed in a stable form [86, 87]. The cDNA strands are then sequenced, creating a map of active gene expression and regulation [88, 89]. Analogous to DNA-based approaches, metatranscriptomics can be PCR-mediated or PCR-free, furthermore, reads may be assembled de novo or with the help of a reference database [90,91,92]. The most challenging aspect of metatranscriptomics is the isolation and storage of microbial mRNA as mRNA must be separated from rRNA, since the latter comprises the majority of RNA present, as well as from eukaryotic mRNA [87, 93, 94]. Furthermore, the inherent instability of mRNA reduces the amount of sample available for sequencing [94]. Strategies whereby RNA is stabilised (e.g., poly(A) tailing) limit the need for sequence knowledge prior to cDNA synthesis [95,96,97]. While this improves on previous primer-based sequencing methods, it nonetheless presents new biases and challenges [88, 98,99,100]. RNA- viruses have been characterized through metatranscriptomics [101, 102]. Limitations in the bioinformatics infrastructure for reference databases as well as the quality of submissions nonetheless hampers the ability to work with viral strains [93], albeit the issue is being addressed by organisations such as the World Society for Virology [103]. As the case with virtually all metagenome reference datasets, transcriptome reference databases suffer from significant coverage gaps [92]. Furthermore, the task of relating transcriptomic data to DNA-based taxonomy presents its own set of challenges [104].
Metaproteomics
Instead of focusing exclusively on protein diversity within a sample, metaproteomics provides a temporo-spatial snapshot of the proteins expressed by the metabolically active community [105, 106]. Metaproteomics includes all analyses that isolate or characterise proteins, such as two-dimensional gel electrophoresis, liquid chromatography, mass spectrometry, as well as antibody and protein microarray techniques [107]. Limitations to metaproteomic approaches are typically associated with the complexity of the sample, especially in dynamic environments with multiple trophic levels [108, 109]. The current state of the field and the challenges within have been reviewed elsewhere [110,111,112,113].
Proteins from animals, plants, or otherwise non-target organisms often contribute to samples, and that further complicates already sensitive protein extraction methods [114]. Finally, researchers must contend with computational challenges in the identification and assignment of peptides and proteins [115,116,117] as well as their functional annotation [118].
Metabolomics
Metabolomics seeks to identify and quantify metabolites (compounds ≤ 1500 Da) produced by the metabolically active fraction of the microbial community [105]. Generally, metabolomics is most effective when investigating how a known set of metabolites produced by a particular community may change under experimental conditions. It is particularly effective as a tool to follow the response of organisms to changes in stimuli (nutrition, biotic and abiotic stressors) [119,120,121]. Since changes in the metabolome occur simultaneously with changes in the transcriptome and proteome, mapping of biochemical pathways can theoretically link metabolomics with both -omics results. However, there are practical challenges around handling large amounts of metabolomics data as well amplicon and protein data when there can also be insufficient reference data and difficulties in profiling metabolites. As such, metabolomics is typically split between metabolite profiling (labor-intensive but specific) and fingerprinting (rapid but only partially correlatable snapshot) [122]. Compared to metaproteomics, metabolomics faces greater challenges owing to the colossal quantity of metabolites present in any sample and the large number of uncharacterised metabolites [123]. Furthermore, accurately detecting molecules is limited by detection methods (e.g., interpreting MS peaks) as well as the detection of partially degraded metabolites—both factors contributing to false discoveries [124]. A major asset in unravelling metabolic pathways has been the emergence of constraint-based analyses of metabolic networks, which are able to integrate gathered data with simulated metabolic models. Of these, the most predominant is the flux balance analysis (FBA) which accompanies mechanistic simulations with the stoichiometric matrices for the conservation of mass and biologically relevant objective functions to predict flux distributions. These networks may then be further explored through metabolic pathway analysis, which creates potential pathways between sets of metabolites. A common thread in the analysis of biological data is the excess of variables, lending to a potential over-reliance on reference databases or established models. Developments such as the Metabolomics Standards Initiative support the creation of more reliable protocols to determine whether metabolites of interest are present in samples, while projects such as the Human Metabolome Database create a more specialised reference tool for human studies [125, 126].
Functional assignment
While elucidating function is a major goal of meta-omics, biochemical observations cannot be directly mapped back onto sequence data. Even though several strategies exist to segregate the metabolically active microbial community from the total of detectable genetic sequences [127, 128], a similar strategy does not yet exist to segregate the total exudates predicted by the metatranscriptome from those observed through metabolomic or metaproteomic analyses. Nonetheless, there are several initiatives that incorporate ontological analysis with the large sequence datasets generated from metagenomic studies, which is an area that has been well reviewed [18, 129,130,131]. Ultimately, this is due to a diverse set of challenges: full or partial degradation of exudates before sampling, modification of compounds (e.g. use as reducing equivalents), inadequate sampling resolution, etc. There is a plethora of physiological observations of isolated strains in the laboratory and putative inferences provided by metagenomic analysis, but community ecology aims to describe on a physicochemical level the potential of a microbial community to interact within its ecosystem [127, 132,133,134,135]. For more information on the state of meta-omics for functional community analysis, the reader is referred to recent studies [26, 29, 30, 136,137,138,139,140].
Common microbial databases
The accumulation of data in large taxonomic repositories has opened up new possibilities for research into the organisation and assembly of microbial communities that were previously inaccessible due to sparse coverage. Persistent incomparability of microbiome analyses has been addressed by consolidating studies around the same set of metadata standards [141, 142]. Given the rapid proliferation of heterogenous data generated from different protocols with and without standardisation steps, several prominent institutions have set out to create more internally consistent generic repositories for datasets. A selection of prominent databases are summarised in Table 1, while a more thorough and regularly updated list can be found in the annual issue of Nucleic Acids Research devoted to databases [143].
A fundamental challenge for the collection of microbial community data is the unequal incorporation and treatment of experimental parameters across studies. Biases in data collection, processing, and interpretation are not necessarily controllable or due to human error. Environmental and technological constraints, as well as the inherent need to choose different sampling techniques, hamper reproducibility across studies. The inception of “microbiome centers” as a knowledge sharing network that seeks to promote cross-disciplinary integration and to streamline the collection and analyses of microbial community data is an emergent strategy to respond to this challenge [32].
The microbial database collections described in Table 1 share a fundamental characteristic; they specialise in specific data types and targeted taxa rather than ecosystems. By contrast, ecosystem-specific databases adapt methodologies and analyses to the unique characteristics of the ecosystem under study. Nonetheless, ecosystem-specific databases are still an emerging tool for a better understanding microbial community dynamics. The following sections cover the motivations leading to their emergence, as well as an analysis of their benefits and limitations in describing microbial community ecology.
Ecosystem-specific databases as a platform for standardisation of methodological practices
A decoupled approach, whereby each -omics study presents a subset of the entire picture, addresses some of the inherent practical limitations (cost, expertise, infrastructure requirements) behind multi-omics studies. Common sets of standards are essential for integrating temporally and spatially distinct studies of the same ecosystem into a coherent view of the whole, and in this respect, ecosystem-specific databases provide a useful template [144]. In turn, this likely contributes to the generation of higher quality results in terms of accuracy, precision, and reproducibility. Common standards across studies facilitate the integration of meta-omics tools into community analysis, as well as the ability for multiple studies to be included as additional temporal and spatial snapshots of a sampling region. Furthermore, ES-DB’s are always curated by a research group or consortium with experts of the given ecosystem and/or targeted taxa. While the inclusion of metadata greatly improves the quality of microbial databases [14, 145,146,147,148], reliably identifying errors (mislabeled data, misspelled labels [149]) within large data sets remains a challenge [150,151,152].
An inherent advantage of ES-DB’s is improving interconnectivity of studies around the same ecosystem. Multiple studies describe the usefulness of manual curation to complement automated assignment tools [153,154,155,156]. As a case study for the statistical power of combining studies into aggregate databases through standardised methodologies, the Earth Microbiome Project Consortium (EMP) collected and analysed data from 97 microbiome studies, 59 of which were published in peer-reviewed journals [157]. Owing to standardised protocols, pooled data from the EMP has been used in meta-analyses to contextualise global patterns derived from individual studies [158]. Within the EMP consortium, individual studies are able to tailor collection and analysis practices to their unique environment under study while adhering to a set of core standards. As such, it represents an example of how both standardization and customization may be weaved together.
Curated databases have allowed for development of sampling protocols tailored to the microbial communities under study. One such example is the Actinobacteria genus, Tetrasphaera, that was routinely underestimated in wastewater treatment systems until microbial screening protocols incorporated adaptations to the cell lysis procedure during DNA extraction [159]. These procedural adaptations, concomitant with a push for greater reproducibility across microbial community studies investigating wastewater treatment, have contributed to the formation of the Microbial Database for Activated Sludge (MIDAS 3). MIDAS has since become a detailed ecosystem-specific database for wastewater treatment systems with resolution at the species level [160, 161]. Since then, the MIDAS team has also included a field guide for researchers interested in submitting their own data [162].
A critical aspect of database management is the development of internal quality standards. What has been described as the reproducibility crisis, a phenomenon whereby microbiome studies often produce poorly comparable datasets and interpretations, may be addressed through the standardisation of methodologies and interconnectivity among researchers [53, 163,164,165]. Each step in the analysis of microbiomes will influence the resulting OTU table from sample storage, DNA extraction, sequencing (including as applicable: amplification, primer choice), sequencing platform, to the choice in bioinformatics pipeline used. As such, while standardisation does not remove biases involved in the process, it may reduce variability across studies in the same field. A recent review on the critical knowledge gap around sampling and handling in microbiome studies identified 95% of studies as having used subjective sampling methods or inadequately describing a methodology [76]. Schloss (2018) recently outlined how microbiome studies can improve their integrity and reproducibility through an evaluative rubric [163]. Data transparency has likewise been shown to improve community cross-validation [16, 166, 167]. The standardisation of bioinformatics processes has been facilitated by independent, community-led initiatives such as the Critical Assessment of Metagenome Interpretation (CAMI), a comprehensive comparison of methodologies for microbiome analysis [168]. Other sources provide more general guidelines and educational tools such as the Statistical Diversity Lab (http://statisticaldiversitylab.com/) [169], as well as resources that summarise best practices in sample preparation for microbiome analyses [14, 170]. In contrast to the above protocols that present ways in which standardisation can be done, ES-DBs establish standards in the context of their specific biome. Table 2 summarises current ES-DB’s in operation and their capabilities.
A roadmap for ecosystem-specific databases
Environment-specific databases typically originate around persistent knowledge gaps and are often associated with challenges in the selection of appropriate sampling techniques. This is the case of the proposed Drinking Water Microbiome Project (DWMP) outlining a knowledge gap from a literature comparison that indicated a lack of knowledge within the drinking water microbiome literature compared to other wastewater treatment microbiomes [171]. They propose that a common database allowing diverse data types to be pooled under standardised conditions can address the challenge of characterizing microbiome dynamics for drinking water systems. A recent perspective article by de Vrieze (2020) discussed the creation of a more applied database than currently available within the MIDAS infrastructure to address the challenges in studying the anaerobic digester microbiome [172, 173]. Here, a strategy of identifying and fingerprinting microbial communities within the anaerobic digestion microbiome is proposed as a tool to complement monitored physicochemical parameters. As measurements reveal shifts in the concentration of specific metabolites, this may be related to shifts in the community composition at large.
In all cases, the integration of functional databases with taxonomic collections requires both top-down and bottom-up engagement as proposed for the DWMP [171]. A recent meta-analysis of DNA barcoding databases that cover European aquatic habitats highlighted issues in quality control and assurance when integrating diverse databases; results pointed to an inconsistent image of taxonomic and subsequently phylogenetic diversity [174]. Despite this, interest in greater biome contextualisation as well as cross-biome studies appears to be growing. A consortium of researchers studying water quality in natural and anthropogenic environments, the Alliance for Freshwater Life, demonstrates how properly curated and inclusive databases may communicate with a larger audience and develop policy and educational platforms beyond their fundamental scientific contribution [175]. Importantly, ecosystem-specific datasets are not limited to environmental studies. In their 2018 article, Kapono et al. recreated the “human environment” as a combination of microbial and chemical data for use in forensics studies [176]. Nor has the applicability of identifying microbiome-associated biomarkers or keystone species been ignored in health and medicine [170, 177, 178]. Similarly, the search for novel genes via bioprospecting depends strongly on accurate genetic annotation and thus may also benefit from more robust reference databases [179, 180].
Limitations of ES-DB’s for meta-omics integration
ES-DB’s appear to be well conceived to address some of the contemporary challenges associated with large microbial community datasets: standardisation of sample methods, processing and analysis, data reproducibility, and the integration of meta-omics technologies from independent studies on the same ecosystem, as reviewed previously [53, 165]. In essence, the goal of ES-DB’s is to ensure that anthropogenic biases (sampling strategies, analysis protocols) are kept to a minimum so that (i) temporal and spatial variability may be better studied across independent studies of the same ecosystem and (ii) independent research groups specializing in different meta-omics analytical strategies are all able to contribute towards a common knowledge pool.
Pinning down an explicit definition for ecosystem-specific databases in contrast to multi-omics databases can become blurred, since it depends on how the ecosystem in question is defined. While in some cases the ecosystem under study is physically constrained (e.g., human body microbiome [181,182,183]), in other cases it describes a global system (e.g., the open ocean [184]). Biomes do not have strict boundaries, so ES-DB’s may suffer from arbitrary exclusions of relevant data from neighboring biomes. Adding or subtracting biomes into the scope of a particular ES-DB will necessarily lead to blurring definitional boundaries and a form of the Sorites paradox, which pursued to its logical conclusion can eventually broaden an ES-DB into a generalised microbial collection. A grey area emerges when it comes to describing the boundary between databases examining multiple biomes within a common specialised environment and databases examining them within a global holistic context. Generic databases thus remain an effective catch-all option for any data type.
Another crucial limitation to ES-DB’s relates to their administration. In order to have professional curation of the dataset, there must be a group of specialists in the field willing and able to provide the service. One way in which the initial entry costs could be lowered would be to establish a standardised template (meta-structure), applicable to any microbial database collection, for data that is to be uploaded or pooled from existing datasets. This strategy could accommodate any dataset size that is collected by a single research group up to an international consortium, with curation rights regulated by each database founder. Not only would this allow better integration between ES-DB’s, but it could decrease the barriers to entry by removing the need for extensive bioinformatics expertise. It would provide a template for decision-making by researchers to follow with respect to sample processing and data organisation. Alongside the emergence of ES-DB’s, several “utilitarian databases” have been proposed that orient themselves around functional analyses, ecosystem services, and/or the organisation of metadata (Table 3). As the scope and depth of these auxiliary tools expands, they will further complement the development of databases and analytical tools catering to unique ecosystems.
Conclusion
The establishment of generic repositories for genetic data marked a milestone for the systematisation of global microbial diversity cataloguing. Having greatly expanded data accessibility, data type specific sequence and omics repositories facilitate novel analyses of data collected from previous studies. However, different standards and practices around data collection and processing reduce data robustness and limit the ability for researchers to compare studies [53, 165]. Although no generalizable model for standardisation can be applied across all ecosystems, standards applied to a restricted ecosystem can be useful. Here we have reviewed how various factors contribute to the emergence of ecosystem-specific databases and what important repercussions for data quality and reproducibility can emerge from well-considered strategies that integrate multiple data types.
Nonetheless, more widespread implementation of ES-DB’s requires more inclusive and accessible bioinformatic infrastructure. While algorithms and methodologies designed to sort and organise existing data are becoming more widespread, only a few resources are available to facilitate spontaneous creation of new ES-DB’s. Concrete standards for data annotation and organisation that permit better synthesis of omics data are necessary to facilitate this development. By consolidating standards for best practices and professionally curating data, higher quality and reproducible datasets will become more commonplace and accessible in the future.
A final point along these lines is that a good database requires good datasets. Standard methods are a representation of best-practices in a world of practical and economic limitations. As technology improves, database curators must decide when and how to update the standard methodology, taking into consideration that each shift damages the reproducibility of the database as a whole. As an ongoing example, significant reductions in the cost of full-length 16S rRNA gene sequencing are making longer reads increasing competitive strategy vs. shorter amplicons—the current recommended sequencing strategy for databases such as the EMP. Currently, the Illumina platform (specializing is short reads) delivers a higher sequencing quality than Pacific Bioscience and Nanopore (long reads)—a crucial decision factor which will also need to be resolved. The entry barrier for new data will need to be set individually across ES-DBs to balance expanding the breadth of incoming datasets against constricting data to only high-quality entries. Nonetheless, curation will only continue to rise in importance as database collections increase in both size and scope.
Availability of data and materials
Not applicable.
Abbreviations
- CAMI:
-
Critical Assessment of Metagenome Interpretation
- DWMP:
-
Drinking Water Microbiome Project
- EMP:
-
Earth Microbiome Project
- ES-DB:
-
Ecosystem specific database
- MCC:
-
Microbiome Centers Consortium
- MIDAS:
-
Microbial Database for Activated Sludge
References
Mora C, Tittensor DP, Adl S, Simpson AG, Worm B. How many species are there on Earth and in the ocean? PLoS Biol. 2011;9(8):e1001127.
Locey KJ, Lennon JT. Scaling laws predict global microbial diversity. Proc Natl Acad Sci U S A. 2016;113(21):5970–5.
Larsen BB, Miller EC, Rhodes MK, Wiens JJ. Inordinate fondness multiplied and redistributed: the number of species on earth and the new pie of life. Q Rev Biol. 2017;92(3):229–65.
Sanschagrin S, Yergeau E. Next-generation sequencing of 16S ribosomal RNA gene amplicons. J Visual Exp JoVE. 2014;90:51709.
Ruppert KM, Kline RJ, Rahman MS. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Global Ecol Conserv. 2019;17:e00547.
Lamb PD, Hunter E, Pinnegar JK, Creer S, Davies RG, Taylor MI. How quantitative is metabarcoding: a meta-analytical approach. Mol Ecol. 2019;28(2):420–30.
Kozińska A, Seweryn P, Sitkiewicz I. A crash course in sequencing for a microbiologist. J Appl Genet. 2019;60(1):103–11.
Liu Y, Qin Y, Guo X-X, Bai Y. Methods and applications for microbiome data analysis. Yi Chuan Hereditas. 2019;41(9):845–62.
Petersen LM, Martin IW, Moschetti WE, Kershaw CM, Tsongalis GJ. Third-generation sequencing in the clinical laboratory: exploring the advantages and challenges of nanopore sequencing. J Clin Microbiol. 2019;58(1):e01315.
Di Bella JM, Bao Y, Gloor GB, Burton JP, Reid G. High throughput sequencing methods and analysis for microbiome research. J Microbiol Methods. 2013;95(3):401–14.
Liu Z, Ma A, Mathé E, Merling M, Ma Q, Liu B. Network analyses in microbiome based on high-throughput multi-omics data. Brief Bioinform. 2021;22(2):1639–55.
Zhou J, He Z, Yang Y, Deng Y, Tringe SG, Alvarez-Cohen L. High-throughput metagenomic technologies for complex microbial community analysis: open and closed formats. MBio. 2015;6(1):e02288.
Ioannidis JP. Why most published research findings are false. PLoS Med. 2005;2(8): e124.
Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 2019.
Gilbert JA, Dupont CL. Microbial metagenomics: beyond the genome. 2010.
Langille MG, Ravel J, Fricke WF. “Available upon request”: not good enough for microbiome data! Berlin: Springer; 2018.
Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 2011;29(5):415–20.
Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP. Bioconductor Workflow for Microbiome Data Analysis: from raw reads to community analyses. F100Res. 2016;5:1492.
Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, et al. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16(7):410–22.
Yuan S, Cohen DB, Ravel J, Abdo Z, Forney LJ. Evaluation of methods for the extraction and purification of DNA from the human microbiome. PLoS ONE. 2012;7(3):e33865.
Albertsen M, Karst SM, Ziegler AS, Kirkegaard RH, Nielsen PH. Back to basics-the influence of DNA extraction and primer choice on phylogenetic analysis of activated sludge communities. PLoS ONE. 2015;10(7):e0132783.
Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069–76.
Greathouse KL, Sinha R, Vogtmann E. DNA extraction for human microbiome studies: the issue of standardization. Genome Biol. 2019;20(1):212.
Nilsson RH, Anslan S, Bahram M, Wurzbacher C, Baldrian P, Tedersoo L. Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat Rev Microbiol. 2019;17(2):95–109.
Dias CK, Starke R, Pylro VS, Morais DK. Database limitations for studying the human gut microbiome. Peerj Comput Sci. 2020;6:e289.
Muller EE, Glaab E, May P, Vlassis N, Wilmes P. Condensing the omics fog of microbial communities. Trends Microbiol. 2013;21(7):325–33.
Kono N, Arakawa K. Nanopore sequencing: review of potential applications in functional genomics. Dev Growth Differ. 2019;61(5):316–26.
Nannipieri P, Ascher-Jenull J, Ceccherini MT, Pietramellara G, Renella G, Schloter M. Beyond microbial diversity for predicting soil functions: a mini review. Pedosphere. 2020;30(1):5–17.
Mallick H, Ma S, Franzosa EA, Vatanen T, Morgan XC, Huttenhower C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017;18(1):228.
Narayanasamy S, Muller EE, Sheik AR, Wilmes P. Integrated omics for the identification of key functionalities in biological wastewater treatment microbial communities. Microb Biotechnol. 2015;8(3):363–8.
Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019;7(1):154.
Martiny JBH, Whiteson KL, Bohannan BJM, David LA, Hynson NA, McFall-Ngai M, et al. The emergence of microbiome centres. Nat Microbiol. 2020;5(1):2–3.
Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12(9):635–45.
Gilbert JA, Jansson JK, Knight R. Earth microbiome project and global systems biology. Am Soc Microbiol; 2018.
Nealson KH, Venter JC. Metagenomics and the global ocean survey: what’s in it for us, and why should we care? ISME J. 2007;1(3):185–7.
Heather JM, Chain B. The sequence of sequencers: the history of sequencing DNA. Genomics. 2016;107(1):1–8.
Steen AD, Crits-Christoph A, Carini P, DeAngelis KM, Fierer N, Lloyd KG, et al. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 2019;13(12):3126–30.
Manoharan L, Kushwaha SK, Hedlund K, Ahrén D. Captured metagenomics: large-scale targeting of genes based on ‘sequence capture’reveals functional diversity in soils. DNA Res. 2015;22(6):451–60.
Monier A, Welsh RM, Gentemann C, Weinstock G, Sodergren E, Armbrust EV, et al. Phosphate transporters in marine phytoplankton and their viruses: cross-domain commonalities in viral-host gene exchanges. Environ Microbiol. 2012;14(1):162–76.
Suenaga H. Targeted metagenomics: a high-resolution metagenomics approach for specific gene clusters in complex microbial communities. Environ Microbiol. 2012;14(1):13–22.
Winand R, Bogaerts B, Hoffman S, Lefevre L, Delvoye M, Van Braekel J, et al. Targeting the 16s rRNA gene for bacterial identification in complex mixed samples: Comparative evaluation of second (illumina) and third (oxford nanopore technologies) generation sequencing technologies. Int J Mol Sci. 2020;21(1):298.
Kumar KR, Cowley MJ, Davis RL, editors. Next-generation sequencing and emerging technologies. Semin Thromb Hemost; 2019: Thieme Medical Publishers.
Leray M, Knowlton N. Censusing marine eukaryotic diversity in the twenty-first century. Philos Trans R Soc B Biol Sci. 2016;371(1702):20150331.
Latz MA, Grujcic V, Brugel S, Lycken J, John U, Karlson B, et al. Short‐and long‐read metabarcoding of the eukaryotic rRNA operon: evaluation of primers and comparison to shotgun metagenomics sequencing. Mol Ecol Resources. 2021.
Semenov M. Metabarcoding and metagenomics in soil ecology research: achievements, challenges, and prospects. Biol Bull Rev. 2021;11(1):40–53.
Tedersoo L, Tooming-Klunderud A, Anslan S. PacBio metabarcoding of Fungi and other eukaryotes: errors, biases and perspectives. New Phytol. 2018;217(3):1370–85.
Badotti F, Fonseca PLC, Tomé LMR, Nunes DT, Góes-Neto A. ITS and secondary biomarkers in fungi: review on the evolution of their use based on scientific publications. Braz J Bot. 2018;41(2):471–9.
Wei S, Cui H, Zhang Y, Su X, Dong H, Chen F, et al. Comparative evaluation of three archaeal primer pairs for exploring archaeal communities in deep-sea sediments and permafrost soils. Extremophiles. 2019;23(6):747–57.
Thijs S, Op De Beeck M, Beckers B, Truyens S, Stevens V, Van Hamme JD, et al. Comparative evaluation of four bacteria-specific primer pairs for 16S rRNA gene surveys. Front Microbiol. 2017;8:494.
Kirsche M, Schatz MC. Democratizing long-read genome assembly. Cell Syst. 2021;12(10):945–7.
Giesselmann P, Hetzel S, Müller F-J, Meissner A, Kretzmer H. Nanopype: a modular and scalable nanopore data processing pipeline. Bioinformatics. 2019;35(22):4770–2.
Tedersoo L, Albertsen M, Anslan S, Callahan B. Perspectives and benefits of high-throughput long-read sequencing in microbial ecology. Appl Environ Microbiol. 2021;87(17):e00626-e721.
Zinger L, Bonin A, Alsos IG, Balint M, Bik H, Boyer F, et al. DNA metabarcoding-Need for robust experimental designs to draw sound ecological conclusions. Mol Ecol. 2019;28(8):1857–62.
DeSalle R, Goldstein P. Review and interpretation of trends in DNA barcoding. Front Ecol Evol. 2019;7:302.
Elbrecht V, Vamos EE, Meissner K, Aroviita J, Leese F. Assessing strengths and weaknesses of DNA metabarcoding-based macroinvertebrate identification for routine stream monitoring. Methods Ecol Evol. 2017;8(10):1265–75.
Nichols RV, Vollmers C, Newsom LA, Wang Y, Heintzman PD, Leighton M, et al. Minimizing polymerase biases in metabarcoding. Mol Ecol Resour. 2018;18(5):927–39.
Thomas F, Dittami SM, Brunet M, Le Duff N, Tanguy G, Leblanc C, et al. Evaluation of a new primer combination to minimize plastid contamination in 16S rDNA metabarcoding analyses of alga-associated bacterial communities. Environ Microbiol Rep. 2020;12(1):30–7.
Tedersoo L, Drenkhan R, Anslan S, Morales-Rodriguez C, Cleary M. High-throughput identification and diagnostics of pathogens and pests: overview and practical recommendations. Mol Ecol Resour. 2019;19(1):47–76.
Mancabelli L, Milani C, Lugli GA, Fontana F, Turroni F, van Sinderen D, et al. The impact of primer design on amplicon-based metagenomic profiling accuracy: detailed insights into bifidobacterial community structure. Microorganisms. 2020;8(1):131.
Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029.
Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T, et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol. 2016;7:459.
Laursen MF, Dalgaard MD, Bahl MI. Genomic GC-content affects the accuracy of 16S rRNA gene sequencing based microbial profiling due to PCR bias. Front Microbiol. 2017;8:1934.
Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18(5):1403–14.
Tedersoo L, Bahram M, Zinger L, Nilsson H, Kennedy P, Yang T, et al. Best practices in metabarcoding of fungi: from experimental design to results. Authorea Preprints. 2021.
Op De Beeck M, Lievens B, Busschaert P, Declerck S, Vangronsveld J, Colpaert JV. Comparison and validation of some ITS primer pairs useful for fungal metabarcoding studies. PLoS ONE. 2014;9(6):e97629.
Hu Y, Sun F, Liu W. The heat shock protein 70 gene as a new alternative molecular marker for the taxonomic identification of Streptomyces strains. AMB Express. 2018;8(1):1–8.
Bittner L, Gobet A, Audic S, Romac S, Egge ES, Santini S, et al. Diversity patterns of uncultured Haptophytes unravelled by pyrosequencing in Naples Bay. Mol Ecol. 2013;22(1):87–101.
Yamada A, Inoue T, Noda S, Hongoh Y, Ohkuma M. Evolutionary trend of phylogenetic diversity of nitrogen fixation genes in the gut community of wood-feeding termites. Mol Ecol. 2007;16(18):3768–77.
Aigle A, Prosser JI, Gubry-Rangin C. The application of high-throughput sequencing technology to analysis of amoA phylogeny and environmental niche specialisation of terrestrial bacterial ammonia-oxidisers. Environmental Microbiome. 2019;14(1):1–10.
Shah N, Tang H, Doak TG, Ye Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Biocomputing. 2011;165–76.
Grieb A, Bowers RM, Oggerin M, Goudeau D, Lee J, Malmstrom RR, et al. A pipeline for targeted metagenomics of environmental bacteria. Microbiome. 2020;8(1):1–17.
Trindade M, Van Zyl LJ, Navarro-Fernández J, Abd EA. Targeted metagenomics as a tool to tap into marine natural product diversity for the discovery and production of drug candidates. Front Microbiol. 2015;6:890.
Ni J, Yan Q, Yu Y. How much metagenomic sequencing is enough to achieve a given goal? Sci Rep. 2013;3(1):1–7.
Vollmers J, Wiegand S, Kaster A-K. Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective-not only size matters! PLoS ONE. 2017;12(1):e0169662.
Wilson J-J, Brandon-Mong G-J, Gan H-M, Sing K-W. High-throughput terrestrial biodiversity assessments: mitochondrial metabarcoding, metagenomics or metatranscriptomics? Mitochondrial DNA Part A. 2019;30(1):60–7.
Dickie IA, Boyer S, Buckley HL, Duncan RP, Gardner PP, Hogg ID, et al. Towards robust and repeatable sampling methods in eDNA-based studies. Mol Ecol Resour. 2018;18(5):940–52.
Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol. 2017;35(9):833–44.
Edwards RA, Rohwer F. Viral metagenomics. Nat Rev Microbiol. 2005;3(6):504–10.
Rosario K, Breitbart M. Exploring the viral world through metagenomics. Curr Opin Virol. 2011;1(4):289–97.
Nooij S, Schmitz D, Vennema H, Kroneman A, Koopmans MP. Overview of virus metagenomic classification methods and their biological applications. Front Microbiol. 2018;9:749.
Greninger AL. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018;244:218–29.
Frioux C, Dittami SM, Siegel A. Using automated reasoning to explore the metabolism of unconventional organisms: a first step to explore host–microbial interactions. Biochem Soc Trans. 2020;48(3):901–13.
Santo D, Loncar-Turukalo T, Stres B, Crnojevic V, Brdar S, editors. Clustering and classification of human microbiome data: evaluating the impact of different settings in bioinformatics workflows. In 2019 IEEE 19th international conference on bioinformatics and bioengineering (BIBE); 2019. IEEE.
Ahlgren NA, Fuchsman CA, Rocap G, Fuhrman JA. Discovery of several novel, widespread, and ecologically distinct marine Thaumarchaeota viruses that encode amoC nitrification genes. ISME J. 2019;13(3):618–31.
Altermatt F, Little CJ, Mächler E, Wang S, Zhang X, Blackman RC. Uncovering the complete biodiversity structure in spatial networks: the example of riverine systems. Oikos. 2020;129(5):607–18.
Tiirola M, Mäki A. Construction of metatranscriptomic libraries for 5′ end sequencing of rRNAs for microbiome research. Microbial systems biology. Berlin: Springer; 2022. p. 137–46.
Alberti A, Belser C, Engelen S, Bertrand L, Orvain C, Brinas L, et al. Comparison of library preparation methods reveals their impact on interpretation of metatranscriptomic data. BMC Genomics. 2014;15(1):1–13.
Mäki A, Tiirola M. Directional high-throughput sequencing of RNAs without gene-specific primers. Biotechniques. 2018;65(4):219–23.
Urich T, Lanzén A, Qi J, Huson DH, Schleper C, Schuster SC. Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS ONE. 2008;3(6):e2527.
Petters S, Söllinger A, Bengtsson MM, Urich T. The soil microbial food web revisited with metatranscriptomics-predatory Myxobacteria as keystone taxon? bioRxiv. 2018:373365.
Schoonvaere K, De Smet L, Smagghe G, Vierstraete A, Braeckman BP, de Graaf DC. Unbiased RNA shotgun metagenomics in social and solitary wild bees detects associations with eukaryote parasites and new viruses. PLoS ONE. 2016;11(12):e0168456.
Campanaro S, Treu L, Kougias PG, Zhu X, Angelidaki I. Taxonomy of anaerobic digestion microbiome reveals biases associated with the applied high throughput sequencing strategies. Sci Rep. 2018;8(1):1–12.
Cobbin JC, Charon J, Harvey E, Holmes EC, Mahar JE. Current challenges to virus discovery by meta-transcriptomics. Curr Opin Virol. 2021;51:48–55.
Aguiar-Pulido V, Huang W, Suarez-Ulloa V, Cickovski T, Mathee K, Narasimhan G. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis: supplementary issue: bioinformatics methods and applications for big metagenomics data. Evolutionary Bioinformatics. 2016;12:EBO. S36436.
Karst SM, Dueholm MS, McIlroy SJ, Kirkegaard RH, Nielsen PH, Albertsen M. Retrieval of a million high-quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat Biotechnol. 2018;36(2):190.
Botero LM, D’imperio S, Burr M, McDermott TR, Young M, Hassett DJ. Poly (A) polymerase modification and reverse transcriptase PCR amplification of environmental RNA. Appl Environ Microbiol. 2005;71(3):1267–75.
Hassa J, Maus I, Off S, Pühler A, Scherer P, Klocke M, et al. Metagenome, metatranscriptome, and metaproteome approaches unraveled compositions and functional relationships of microbial communities residing in biogas plants. Appl Microbiol Biotechnol. 2018;102(12):5045–63.
Balázs Z, Tombácz D, Csabai Z, Moldován N, Snyder M, Boldogkői Z. Template-switching artifacts resemble alternative polyadenylation. BMC Genomics. 2019;20(1):824.
Roy KR, Chanfreau GF. Robust mapping of polyadenylated and non-polyadenylated RNA 3′ ends at nucleotide resolution by 3′-end sequencing. Methods. 2020;176:4–13.
Nam DK, Lee S, Zhou G, Cao X, Wang C, Clark T, et al. Oligo (dT) primer generates a high frequency of truncated cDNAs through internal poly (A) priming during reverse transcription. Proc Natl Acad Sci. 2002;99(9):6152–6.
Shi M, Zhang Y-Z, Holmes EC. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018;243:83–90.
Batovska J, Mee PT, Lynch SE, Sawbridge TI, Rodoni BC. Sensitivity and specificity of metatranscriptomics as an arbovirus surveillance tool. Sci Rep. 2019;9(1):1–13.
Söderlund-Venermo M, Varma A, Guo D, Gladue DP, Poole E, Pujol FH, et al. World Society for Virology first international conference: tackling global virus epidemics. Virology. 2022;566:114–21.
Satinsky BM, Gifford SM, Crump BC, Moran MA. Use of internal standards for quantitative metatranscriptome and metagenome analysis. Methods in enzymology. 531. Elsevier; 2013. p. 237–50.
Lukhele T, Selvarajan R, Nyoni H, Mamba BB, Msagati TA. Acid mine drainage as habitats for distinct microbiomes: current knowledge in the era of molecular and omic technologies. Curr Microbiol. 2020;77(4):657–74.
Wilmes P, Bond PL. Metaproteomics: studying functional gene expression in microbial ecosystems. Trends Microbiol. 2006;14(2):92–7.
Hardouin P, Chiron R, Marchandin H, Armengaud J, Grenga L. Metaproteomics to decipher CF host-microbiota interactions: overview, challenges and future perspectives. Genes. 2021;12(6):892.
Petriz BA, Franco OL. Metaproteomics as a complementary approach to gut microbiota in health and disease. Front Chem. 2017;5:4.
Isaac NI, Philippe D, Nicholas A, Raoult D, Eric C. Metaproteomics of the human gut microbiota: challenges and contributions to other OMICS. Clin Mass Spectrometry. 2019;14:18–30.
Schiebenhoefer H, Van Den Bossche T, Fuchs S, Renard BY, Muth T, Martens L. Challenges and promise at the interface of metaproteomics and genomics: an overview of recent progress in metaproteogenomic data analysis. Expert Rev Proteomics. 2019;16(5):375–90.
Muth T, Benndorf D, Reichl U, Rapp E, Martens L. Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol BioSyst. 2013;9(4):578–85.
Saito MA, Bertrand EM, Duffy ME, Gaylord DA, Held NA, Hervey WJ IV, et al. Progress and challenges in ocean metaproteomics and proposed best practices for data sharing. J Proteome Res. 2019;18(4):1461–76.
Lohmann P, Schäpe SS, Haange S-B, Oliphant K, Allen-Vercoe E, Jehmlich N, et al. Function is what counts: how microbial community complexity affects species, proteome and pathway coverage in metaproteomics. Expert Rev Proteomics. 2020;17(2):163–73.
Verberkmoes NC, Russell AL, Shah M, Godzik A, Rosenquist M, Halfvarson J, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3(2):179–89.
Schiebenhoefer H, Schallert K, Renard BY, Trappe K, Schmid E, Benndorf D, et al. A complete and flexible workflow for metaproteomics data analysis based on MetaProteomeAnalyzer and Prophane. Nat Protoc. 2020;15(10):3212–39.
Starr AE, Deeke SA, Li L, Zhang X, Daoud R, Ryan J, et al. Proteomic and metaproteomic approaches to understand host–microbe interactions. Anal Chem. 2018;90(1):86–109.
Chatterjee S, Stupp GS, Park SKR, Ducom J-C, Yates JR, Su AI, et al. A comprehensive and scalable database search system for metaproteomics. BMC Genomics. 2016;17(1):1–11.
Werner J, Géron A, Kerssemakers J, Matallana-Surget S. mPies: a novel metaproteomics tool for the creation of relevant protein databases and automatized protein annotation. Biol Direct. 2019;14(1):1–5.
Davey MP, Horst I, Duong G-H, Tomsett EV, Litvinenko AC, Howe CJ, et al. Triacylglyceride production and autophagous responses in Chlamydomonas reinhardtii depend on resource allocation and carbon source. Eukaryot Cell. 2014;13(3):392–400.
Obata T, Fernie AR. The use of metabolomics to dissect plant responses to abiotic stresses. Cell Mol Life Sci. 2012;69(19):3225–43.
Rivas-Ubach A, Poret-Peterson AT, Peñuelas J, Sardans J, Pérez-Trujillo M, Legido-Quigley C, et al. Coping with iron limitation: a metabolomic study of Synechocystis sp. PCC 6803. Acta Physiol Plant. 2018;40(2):1–13.
Dunn WB, Bailey NJ, Johnson HE. Measuring the metabolome: current analytical technologies. Analyst. 2005;130(5):606–25.
Bundy JG, Davey MP, Viant MR. Environmental metabolomics: a critical review and future perspectives. Metabolomics. 2009;5(1):3–21.
Singh A. Tools for metabolomics. Nat Methods. 2020;17(1):24.
Fiehn O, Robertson D, Griffin J, van der Werf M, Nikolau B, Morrison N, et al. The metabolomics standards initiative (MSI). Metabolomics. 2007;3(3):175–8.
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R, et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(11):D608–17.
Kieft B, Li Z, Bryson S, Crump BC, Hettich R, Pan C, et al. Microbial community structure-function relationships in Yaquina bay estuary reveal spatially distinct carbon and nitrogen cycling capacities. Front Microbiol. 2018;9:1282.
Emerson JB, Adams RI, Roman CMB, Brooks B, Coil DA, Dahlhausen K, et al. Schrodinger’s microbes: tools for distinguishing the living from the dead in microbial ecosystems. Microbiome. 2017;5(1):86.
Jo J, Oh J, Park C. Microbial community analysis using high-throughput sequencing technology: a beginner’s guide for microbiologists. J Microbiol. 2020;58(3):176–92.
Mitchell AL, Almeida A, Beracochea M, Boland M, Burgin J, Cochrane G, et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 2020;48(D1):D570–8.
Hall MW, Rohwer RR, Perrie J, McMahon KD, Beiko RG. Ananke: temporal clustering reveals ecological dynamics of microbial communities. PeerJ. 2017;5:e3812.
Li F, Neves AL, Ghoshal B. Symposium review: mining metagenomic and metatranscriptomic data for clues about microbial metabolic functions in ruminants. J Dairy Sci. 2018;101(6):5605–18.
He Z, Deng Y, Zhou J. Development of functional gene microarrays for microbial community analysis. Curr Opin Biotechnol. 2012;23(1):49–55.
Jones CM, Graf DR, Bru D, Philippot L, Hallin S. The unaccounted yet abundant nitrous oxide-reducing microbial community: a potential nitrous oxide sink. ISME J. 2013;7(2):417–26.
Leibold MA, Holyoak M, Mouquet N, Amarasekare P, Chase JM, Hoopes MF, et al. The metacommunity concept: a framework for multi-scale community ecology. Ecol Lett. 2004;7(7):601–13.
Reel PS, Reel S, Pearson E, Trucco E, Jefferson E. Using machine learning approaches for multi-omics data analysis: a review. Biotechnol Adv. 2021:107739.
Swift CL, Podolsky IA, Lankiewicz TS, Seppälä S, O’Malley MA. Linking ‘omics’ to function unlocks the biotech potential of non-model fungi. Curr Opin Syst Biol. 2019;14:9–17.
Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019;7(1):1–12.
Gubelit YI, Grossart H-P. New methods, new concepts: what can be applied to freshwater periphyton? Front Microbiol. 2020;11:1275.
Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. Computational meta’omics for microbial community studies. Mol Syst Biol. 2013;9(1):666.
Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res. 2012;40(Database issue):D115–22.
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN, et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature. 2009;462(7276):1056–60.
Rigden DJ, Fernández XM. The 27th annual Nucleic Acids Research database issue and molecular biology database collection. Nucleic Acids Res. 2020;48(D1):D1–8.
Darzi Y, Falony G, Vieira-Silva S, Raes J. Towards biome-specific analysis of meta-omics data. ISME J. 2016;10(5):1025–8.
Spicer RA, Salek R, Steinbeck C. A decade after the metabolomics standards initiative it’s time for a revision. Sci Data. 2017;4(1): 170138.
Schierz AC, Soldatova LN. The metabolomics standards initiative. Nature 200:7.
Orchard S, Montechi-Palazzi L, Deutsch EW, Binz PA, Jones AR, Paton N, et al. Five years of progress in the Standardization of Proteomics Data 4th Annual Spring Workshop of the HUPO-Proteomics Standards Initiative April 23–25, 2007 Ecole Nationale Superieure (ENS), Lyon. France Proteomics. 2007;7(19):3436–40.
Contreras JL. Legal issues for biological research standards. Nat Biotechnol. 2008;26(5):498–9.
Bhattacharjee K, Joshi SR. NEMiD: a web-based curated microbial diversity database with geo-based plotting. PLoS ONE. 2014;9(4): e94088.
Statnikov A, Henaff M, Narendra V, Konganti K, Li Z, Yang L, et al. A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome. 2013;1(1):1–12.
Toronto International Data Release Workshop A, Birney E, Hudson TJ, Green ED, Gunter C, Eddy S, et al. Prepublication data sharing. Nature. 2009;461(7261):168–70.
Trust W, editor Sharing data from large-scale biological research projects: a system of tripartite responsibility. Report of a meeting organized by the Wellcome Trust and held on 14–15 January 2003 at Fort Lauderdale, USA; 2003: Wellcome Trust London.
Keseler IM, Skrzypek M, Weerasinghe D, Chen AY, Fulcher C, Li G-W, et al. Curation accuracy of model organism databases. Database. 2014;2014.
Chandonia J-M, Fox NK, Brenner SE. SCOPe: manual curation and artifact removal in the structural classification of proteins–extended database. J Mol Biol. 2017;429(3):348–55.
Pfeiffer F, Oesterhelt D. A manual curation strategy to improve genome annotation: application to a set of haloarchael genomes. Life. 2015;5(2):1427–44.
Xavier JS, Nguyen T-B, Karmarkar M, Portelli S, Rezende PM, Velloso JP, et al. ThermoMutDB: a thermodynamic database for missense mutations. Nucleic Acids Res. 2021;49(D1):D475–9.
Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551(7681):457–63.
Gilbert JA, Jansson JK, Knight R. The Earth Microbiome project: successes and aspirations. BMC Biol. 2014;12(1):1–4.
Nielsen PH, McIlroy SJ, Albertsen M, Nierychlo M. Re-evaluating the microbiology of the enhanced biological phosphorus removal process. Curr Opin Biotechnol. 2019;57:111–8.
Nierychlo M, Andersen KS, Xu Y, Green N, Jiang C, Albertsen M, et al. MiDAS 3: An ecosystem-specific reference database, taxonomy and knowledge platform for activated sludge and anaerobic digesters reveals species-level microbiome composition of activated sludge. Water Research. 2020:115955.
Jørgensen VR, Dueholm MS, Knutsson S, Nierychlo MA, Kristensen JM, Petriglieri F, et al., editors. Global reference database of microbes in anaerobic digesters. In: IWC-16th world conference on anaerobic digestion; 2019.
McIlroy SJ, Saunders AM, Albertsen M, Nierychlo M, McIlroy B, Hansen AA, et al. MiDAS: the field guide to the microbes of activated sludge. Database (Oxford). 2015;2015:bav062.
Schloss PD. Identifying and Overcoming Threats to Reproducibility, Replicability, Robustness, and Generalizability in Microbiome Research. mBio. 2018;9(3).
Taberlet P, Bonin A, Zinger L, Coissac E. Environmental DNA: For biodiversity research and monitoring. Oxford: Oxford University Press; 2018.
Zinger L, Gobet A, Pommier T. Two decades of describing the unseen majority of aquatic microbial diversity. Mol Ecol. 2012;21(8):1878–96.
Amann RI, Baichoo S, Blencowe BJ, Bork P, Borodovsky M, Brooksbank C, et al. Toward unrestricted use of public genomic data. Science. 2019;363(6425):350–2.
Baker M. 1,500 scientists lift the lid on reproducibility. 2016.
Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, et al. Critical assessment of metagenome interpretation—a benchmark of metagenomics software. Nat Methods. 2017;14(11):1063–71.
Willis AD. Rigorous Statistical Methods for Rigorous Microbiome Science. mSystems. 2019;4(3).
McDonald D, Hyde E, Debelius J, Morton J, Gonzalez A, Ackermann G, et al. Knight R. 2018. American Gut: an open platform for citizen science microbiome research. mSystems 3: e00031–18. 2018.
Hull NM, Ling F, Pinto AJ, Albertsen M, Jang HG, Hong P-Y, et al. Drinking water microbiome project: is it time? Trends Microbiol. 2019;27(8):670–7.
De Vrieze J. The next frontier of the anaerobic digestion microbiome: from ecology to process control. Environmental Science and Ecotechnology. 2020:100032.
McIlroy SJ, Kirkegaard RH, McIlroy B, Nierychlo M, Kristensen JM, Karst SM, et al. MiDAS 2.0: an ecosystem-specific taxonomy and online database for the organisms of wastewater treatment systems expanded for anaerobic digester groups. Database (Oxford). 2017;2017(1).
Weigand H, Beermann AJ, Ciampor F, Costa FO, Csabai Z, Duarte S, et al. DNA barcode reference libraries for the monitoring of aquatic biota in Europe: gap-analysis and recommendations for future work. Sci Total Environ. 2019;678:499–524.
Darwall W, Bremerich V, De Wever A, Dell AI, Freyhof J, Gessner MO, et al. The Alliance for Freshwater Life: a global call to unite efforts for freshwater biodiversity science and conservation. Aquatic Conserv Mar Freshwater Ecosyst. 2018;28(4):1015–22.
Kapono CA, Morton JT, Bouslimani A, Melnik AV, Orlinsky K, Knaan TL, et al. Creating a 3D microbial and chemical snapshot of a human habitat. Sci Rep. 2018;8(1):3669.
Belk A, Xu ZZ, Carter DO, Lynne A, Bucheli S, Knight R, et al. Microbiome data accurately predicts the postmortem interval using random forest regression models. Genes (Basel). 2018;9(2):104.
Cao Y, Fanning S, Proos S, Jordan K, Srikumar S. A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies. Front Microbiol. 2017;8:1829.
Roumpeka DD, Wallace RJ, Escalettes F, Fotheringham I, Watson M. A review of bioinformatics tools for bio-prospecting from metagenomic sequence data. Front Genet. 2017;8:23.
Cuadrat RRC, Ionescu D, Davila AMR, Grossart HP. Recovering genomics clusters of secondary metabolites from lakes using genome-resolved metagenomics. Front Microbiol. 2018;9:251.
Integrative HMPRNC. The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16(3):276–89.
Sa B. A framework for human microbiome research. Nature. 2012;486(7402):215–21.
Human Microbiome Project C. Structure. function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14.
Klemetsen T, Raknes IA, Fu J, Agafonov A, Balasundaram SV, Tartari G, et al. The MAR databases: development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 2018;46(D1):D692–9.
Guo X, Chen F, Gao F, Li L, Liu K, You L, et al. CNSA: a data repository for archiving omics data. Database. 2020;2020.
Kamburov A, Galicka H, Lehrach H, Herwig R. ConsensusPathDB: assembling a more complete picture of cell biology.
Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc. 2016;11(10):1889.
Stoesser G, Griffith OL, Griffith M. DDBJ (DNA Databank of Japan). Dictionary of Bioinformatics and Computational Biology. 2004.
Tateno Y, Imanishi T, Miyazaki S, Fukami-Kobayashi K, Saitou N, Sugawara H, et al. DNA Data Bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 2002;30(1):27–30.
Ogasawara O, Kodama Y, Mashima J, Kosuge T, Fujisawa T. DDBJ Database updates and computational infrastructure enhancement. Nucleic Acids Res. 2020;48(D1):D45–50.
Athar A, Füllgrabe A, George N, Iqbal H, Huerta L, Ali A, et al. ArrayExpress update–from bulk to single-cell expression data. Nucleic Acids Res. 2019;47(D1):D711–5.
Leinonen R, Akhtar R, Birney E, Bower L, Cerdeno-Tárraga A, Cheng Y, et al. The European nucleotide archive. Nucleic acids research. 2010;39(suppl_1):D28-D31.
Amid C, Alako BT, Balavenkataraman Kadhirvelu V, Burdett T, Burgin J, Fan J, et al. The European nucleotide archive in 2019. Nucleic Acids Res. 2020;48(D1):D70–6.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30(1):38–41.
Perez-Riverol Y, Bai M, da Veiga Leprevost F, Squizzato S, Park M, Haug K, et al. Omics Discovery Index-Discovering and Linking Public ‘Omics’ Datasets. 2016.
Perez-Riverol Y, Bai M, da Veiga LF, Squizzato S, Park YM, Haug K, et al. Discovering and linking public omics data sets using the Omics Discovery Index. Nat Biotechnol. 2017;35(5):406–9.
Sarkans U, Gostev M, Athar A, Behrangi E, Melnichuk O, Ali A, et al. The BioStudies database—one stop shop for all data supporting a life sciences study. Nucleic Acids Res. 2018;46(D1):D1266–70.
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol. 2017;67(5):1613–7.
Silvester N, Alako B, Amid C, Cerdeno-Tarraga A, Clarke L, Cleland I, et al. The European Nucleotide Archive in 2017. Nucleic Acids Res. 2018;46(D1):D36–40.
Karsch-Mizrachi I, Takagi T, Cochrane G, International Nucleotide Sequence Database C. The international nucleotide sequence database collaboration. Nucleic Acids Res. 2018;46(D1):D48-D51.
Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Pillay M, et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2014;42(Database issue):D568–73.
Meyer F, Bagchi S, Chaterji S, Gerlach W, Grama A, Harrison T, et al. MG-RAST version 4-lessons learned from a decade of low-budget ultra-high-throughput metagenome analysis. Brief Bioinform. 2019;20(4):1151–9.
Keegan KP, Glass EM, Meyer F. MG-RAST, a metagenomics service for analysis of microbial community structure and function. Microbial environmental genomics (MEG). Springer; 2016. p. 207-33.
Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank. Nucleic Acids Res. 2005;33(Database issue):D34–8.
Schuler GD, Epstein JA, Ohkawa H, Kans JA. [10] Entrez: Molecular biology database and retrieval system. Methods Enzymol. 1996;266:141–62.
Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33(Database issue):D501–4.
Lobo I. Basic local alignment search tool (BLAST). Nature Education. 2008;1(1).
Guillou L, Bachar D, Audic S, Bass D, Berney C, Bittner L, et al. The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2012;41(D1):D597–604.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590–6.
Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu Y, et al. The UCSC genome browser database. Nucleic Acids Res. 2003;31(1):51–4.
Stoddard SF, Smith BJ, Hein R, Roller BR, Schmidt TM. rrn DB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015;43(D1):D593–8.
Sierra M, Bhattacharya C, Ryon K, Meierovich S, Shaaban H, Westfall D, et al. The Microbe Directory v2. 0: An Expanded Database of Ecological and Phenotypical Features of Microbes. 2019.
Wilson JL, Nägele T, Linke M, Demel F, Fritsch SD, Mayr HK, et al. Inverse data-driven modeling and multiomics analysis reveals phgdh as a metabolic checkpoint of macrophage polarization and proliferation. Cell Rep. 2020;30(5):1542–52. e7.
Bissett A, Fitzgerald A, Meintjes T, Mele PM, Reith F, Dennis PG, et al. Introducing BASE: the Biomes of Australian Soil Environments soil microbial diversity database. Gigascience. 2016;5(1):s13742-016-0126-5.
Mikaelyan A, Kohler T, Lampert N, Rohland J, Boga H, Meuser K, et al. Classifying the bacterial gut microbiota of termites and cockroaches: a curated phylogenetic reference database (DictDb). Syst Appl Microbiol. 2015;38(7):472–82.
Gilbert JA, Meyer F, Antonopoulos D, Balaji P, Brown CT, Brown CT, et al. Meeting report: the terabase metagenomics workshop and the vision of an Earth microbiome project. Stand Genomic Sci. 2010;3(3):243–8.
Karthikeyan S, Rodriguez-R LM, Heritier-Robbins P, Hatt J, Huettel M, Kostka JE, et al. Genome Repository of Oiled Systems (GROS): an interactive and searchable database that expands the catalogued diversity of crude oil-associated microbes. BioRxiv. 2019:838573.
Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, et al. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol. 2007;5(3):e16.
Goedert JJ, Hua X, Yu G, Shi J. Diversity and composition of the adult fecal microbiome associated with history of cesarean birth or appendectomy: Analysis of the American Gut Project. EBioMedicine. 2014;1(2–3):167–72.
Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford). 2010;2010:baq013.
Öpik M, Vanatoa A, Vanatoa E, Moora M, Davison J, Kalwij J, et al. The online database MaarjAM reveals global and ecosystemic distribution patterns in arbuscular mycorrhizal fungi (Glomeromycota). New Phytol. 2010;188(1):223–41.
Ehrlich SD, Consortium M. MetaHIT: The European Union Project on metagenomics of the human intestinal tract. Metagenomics of the human body. Springer; 2011. p. 307-16.
Stokholm-Bjerregaard M, McIlroy SJ, Nierychlo M, Karst SM, Albertsen M, Nielsen PH. A critical assessment of the microorganisms proposed to be important to enhanced biological phosphorus removal in full-scale wastewater treatment systems. Front Microbiol. 2017;8:718.
Seedorf H, Kittelmann S, Henderson G, Janssen PH. RIM-DB: a taxonomic framework for community structure analysis of methanogenic archaea from the rumen and other intestinal environments. PeerJ. 2014;2:e494.
Caro C, Pinto R, Marques JC. Use and usefulness of open source spatial databases for the assessment and management of European coastal and marine ecosystem services. Ecol Ind. 2018;95:41–52.
de Vargas C, Audic S, Henry N, Decelle J, Mahe F, Logares R, et al. Ocean plankton. Eukaryotic plankton diversity in the sunlit ocean. Science. 2015;348(6237):1261605.
Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. 2020:1–10.
Prestat E, David MM, Hultman J, Taş N, Lamendella R, Dvornik J, et al. FOAM (functional ontology assignments for metagenomes): a hidden Markov model (HMM) database with environmental focus. Nucleic acids research. 2014;42(19):e145-e.
Chien C-H, Chow C-N, Wu N-Y, Chiang-Hsieh Y-F, Hou P-F, Chang W-C, editors. EXPath: a database of comparative expression analysis inferring metabolic pathways for plants. BMC Genomics; 2015.
Christensen V, Walters CJ. Ecopath with Ecosim: methods, capabilities and limitations. Ecol Model. 2004;172(2–4):109–39.
Plantier-Santos C, Carollo C, Yoskowitz DW. Gulf of Mexico Ecosystem Service Valuation Database (GecoServ): Gathering ecosystem services valuation studies to promote their inclusion in the decision-making process. Mar Policy. 2012;36(1):214–7.
Camino F, Ramos E, Acevedo A, Puente A, Losada ÍJ, Juanes JA. OCLE: A European open access database on climate change effects on littoral and oceanic ecosystems. Prog Oceanogr. 2018;168:222–31.
LeBauer D, Kooper R, Mulrooney P, Rohde S, Wang D, Long SP, et al. BETYdb: A yield, trait, and ecosystem service database applied to second-generation bioenergy feedstock production. GCB Bioenergy. 2018;10(1):61–71.
Coggan NV, Hayward MW, Gibb H. A global database and “state of the field” review of research into ecosystem engineering by land animals. J Anim Ecol. 2018;87(4):974–94.
Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Sundaramurthi JC, Lee J, et al. Genomes OnLine Database (GOLD) v. 8: overview and updates. Nucleic Acids Research. 2021;49(D1):D723-D33.
Perez-Riverol Y, Zorin A, Dass G, Vu M-T, Xu P, Glont M, et al. Quantifying the impact of public omics data. Nat Commun. 2019;10(1):1–10.
Guhlin J, Silverstein KA, Zhou P, Tiffin P, Young ND. ODG: Omics database generator-a tool for generating, querying, and analyzing multi-omics comparative databases to facilitate biological understanding. BMC Bioinformatics. 2017;18(1):1–8.
Acknowledgements
We would like to thank Lokeshwaran Manoharan (Department of Laboratory Medicine, National Bioinformatics Infrastructure Sweden (NBIS)) for helpful edits and comments when writing this manuscript. We thank the reviewers for their insightful comments and editorial suggestions.
Funding
Open access funding provided by University of Gothenburg. This work was funded by the Swedish Research Council FORMAS (Joyce 2017–00242) and the European Union ERA-Net Cofund on Food Systems and Climate project (FOSC) project BLUECYCLING (Joyce 2020–03175). A.G. acknowledges support by the Institut Français de Recherche pour l’Exploitation de la Mer (IFREMER).
Author information
Authors and Affiliations
Contributions
VL conceived the idea for the manuscript and wrote it with input from AG and AJ. The figure was prepared with equal contributions from VL and AG. All authors reviewed and approved the final manuscript.
Authors' information
Alyssa Joyce is an Associate Professor and Victor Lobanov a Ph.D. candidate in her group within the Department of Marine Sciences at the University of Gothenburg, Sweden.
Angélique Gobet is a researcher at the IFREMER institute and in the MARine Biodiversity, Exploitation and Conservation research (MARBEC) unit (UMR) in Sète and Palavas-les-flots, France.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to the publication of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Biome
-
Total biotic diversity within a habitat
- Community ecology
-
Identification of taxonomic and phylogenetic relationships between organisms in a community including how they react to their non-living surroundings.
- Database
-
A collection of data arranged around specific characteristics making it easier for retrieval. Sequence databases contain digitalized representation of biological informational units (e.g., nucleic acids, proteins) however databases may include representations or descriptions for other types of biological data
- Ecosystem
-
A biological community of interacting organisms and their physical environment
- Functional analysis
-
Relating expressed genes or metabolites produced to taxonomic identity utilizing meta-omics data
- Genomics
-
A field of study involving all aspects of genomes from their structure, evolution, as well as readability (mapping) and functionality
- Habitat
-
Physical (abiotic) and biotic resources present in a particular area
- Keystone taxon
-
A taxon having a disproportionate influence on community structure, where the influence is due to strong biotic interactions rather than high abundance
- Meta-
-
A prefix indicating that the following term applies to a community sample (see metagenomics vis à vis genomics). The term “metabolomics” has a different root and does not follow this pattern
- Metabarcoding
-
Method allowing for the identification of all the species in the community by targeting a specific gene or gene region
- Metadata
-
All data describing the characteristics of the entry, how it is stored and defined
- Metadatabase
-
A compilation of databases based on their metadata. This centralises screening, comparing, and filtering databases making the data more accessible
- Metagenomics
-
Method targeting the amplification of all genes directly from an environmental sample
- Meta-omics
-
An umbrella term encompassing genomics, transcriptomics, proteomics, and metabolomics. Sometimes referred to as multi-omics
- Metaproteomics
-
A term encompassing all experimental approaches related to the study all proteins in microbial communities, generally their identification and quantification in complex samples
- Metastructure
-
The underlying structure used to organise metadata
- Metatranscriptomics
-
Method allowing for the identification of all expressed genes in an environmental sample
- Microbiome
-
Refers to the total conceptual collection of microorganisms and their genomes within a specific environment
- Microbiota
-
Refers to the total physical collection of microorganisms within a specific environment
- -Omics
-
A suffix referring to a range of biological disciplines, often grouped together as a tool-kit for biological analyses of microbial communities: genomics, proteomics, metabolomics, metagenomics, phenomics and transcriptomics
- Proteomics
-
The isolation and study of proteins from a single organism
- Targeted metagenomics
-
Subset of metagenomics whereby subsequent sequencing analysis constricts the study focus, i.e., to a specific gene cluster or particular group of organisms
- Transcriptomics
-
Analysis of gene expression (entire genome, single gene, or gene cluster) within a single organism
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lobanov, V., Gobet, A. & Joyce, A. Ecosystem-specific microbiota and microbiome databases in the era of big data. Environmental Microbiome 17, 37 (2022). https://doi.org/10.1186/s40793-022-00433-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-022-00433-1