
Abstract
Paracoccus spp. are isolated from both terrestrial and aquatic habitats, indicating their ubiquitous existence in the environment. Here we present the first phage isolated from this genus, vB_PmaS-R3, and its complete genome sequence. Paracoccus phage vB_PmaS-R3 is a siphophage isolated from the South China Sea. The genome sequence is 42,093 bp, with a G + C content of 56.36 %. Fifty-two open reading frames were predicted from the genome. The genome can mainly be divided into three regions: genes for DNA metabolism, regulatory genes and structure forming genes. Genes encoding DNA metabolism and structural proteins showed high sequence homology to corresponding genes of Burkholderia phage KL1 and Pseudomonas phage PA73. In addition, four gene transfer agent-like genes were found in the vB_PmaS-R3 genome. A putative L-alanoyl-D-glutamate peptidase was predicted as the endolysin. A MazG gene was found in the vB_PmaS-R3 genome, which indicates genomic adaption to the nutrient-limited marine environment.
Similar content being viewed by others

Introduction
Viruses are the most abundant biological entities in the marine environment [1]. They mediate approximately 20 % mortality of the biomass in the sea per day, accelerate nutrient cycling rates and regulate the host community composition through host-specific infection and lysis of cells [1, 2]. Viruses also play an important role in host evolution through viral infection-mediated lateral gene transfer [3]. Metagenomic studies of several marine viral communities revealed vast genetic diversity and contained a large fraction of novel sequences [2, 4]. Studies on virus isolation, virus-host interaction and genomic analysis at the individual level can provide detailed information on the biological features of a virus and give a better understanding of its role in biogeochemical and evolutionary processes [5]. Paracoccus is an important genus in the Rhodobacteraceae family. Paracoccus spp. are widely distributed in both terrestrial and aquatic environments and frequently isolated from marine environments [6, 7]. Although there are many species identified in the Paracoccus genus, so far there is no report on Paracoccus phage or a phage genome. Here we describe the first phage isolated from this genus and its genome characteristics. According to the methods of nomenclature of viruses of Kropinski et al. [8] the phage was named vB_PmaS-R3.
Organism information
Classification and features
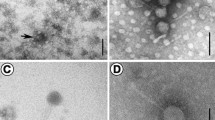
The host of phage vB_PmaS-R3 Paracoccus marcusii strain JL-65 was isolated from the North Pacific Ocean by Du, et al. [6, 9]. Phage vB_PmaS-R3 was a lytic dsDNA phage isolated from the surface seawater of the South China Sea (22.06° N, 118.44° E) (Table 1). According to the transmission electron microscopy image (Fig. 1), vB_PmaS-R3 is a siphophage with an isometric head (ca. 50 nm in diameter) and a long, flexible, non-contractile tail (ca. 122 nm long and ca. 10 nm wide). Phylogenetic analysis based on DNA polymerase gene sequences revealed that vB_PmaS-R3 clustered with Vibrio phage VpKK5, and was also closely related to Pseudomonas phages MP1412, M6, YuA and phage phiJL001 (Fig. 2).

Transmission electron microscopy image of Paracoccus phage vB_PmaS-R3

Neighbor-joining phylogenetic tree based on the DNA polymerase amino acid sequences of vB_PmaS-R3 and 14 siphophages. Enterobacteria phage T5 was used to root the tree. Bootstrap percentage analyses were based on 1000 replications. Bar, 0.2 substitutions per amino acid position
Genome sequencing information
Genome project history
Phage vB_PmaS-R3 is the first phage isolated from the genus Paracoccus , and is a member of the few known siphophage isolates from the marine environment [10]. Genome sequencing of vB_PmaS-R3 would provide a better understanding of the gene diversity of siphophages in the ocean. The genome project is deposited in GOLD and IMG system. The genome sequence and annotation are available in GenBank (KP162168). A summary of the project information is presented in Table 2.
Growth conditions and genomic DNA preparation
Phage vB_PmaS-R3 was isolated from surface seawater of the South China Sea. The seawater was filtered through a 0.22-μm polycarbonate membrane filter (Millipore, Bedford, MA, USA) and stored at 4 °C. The host strain was cultured at 28 °C using modified rich organic medium (1 g l−1 yeast extract, 1 g l−1 peptone, 1 l artificial seawater) as described by Yurkov et al. [11]. Two milliliters of seawater filtrate were added to 20 ml exponentially growing bacterial cultures (OD600 = 0.2) and incubated for 48 h. Cultures were then centrifuged at 10,000 × g for 10 min. The supernatants were collected and filtered through 0.22-μm filters to remove bacterial cells. One hundred microliters of the filtrate were added to 1 ml exponentially growing bacterial culture to perform plaque assays according to the method described by Suttle and Chen [12]. The phage isolate was then purified five times using the plaque assay method. For phage DNA preparation, phage suspension was added to 1 l host culture (OD600 = 0.2–0.3) with a multiplicity of infection of 0.01 and incubated overnight at 28 °C, with shaking at 180 rpm. Phage particles in the lysates were harvested and purified using the method described by Chen et al. [13] with some modifications as follows. The lysates were added with 2 mg of both DNase I and RNase A, and incubated at room temperature for 1 h. After the digestion, the NaCl concentration of the lysates was adjusted to 1 M. Then the lysates were incubated on ice for 1 h. The remaining cells and debris were removed by centrifugation at 10,000 × g for 10 min followed by filtration through 0.22-μm filters. Polyethylene glycol 8000 was added to the filtrate to a final concentration of 100 g l−1 and incubated at 4 °C for 2 d. The phage particles were precipitated by centrifugation at 10,000 × g for 80 min, resuspended in 6 ml TM buffer (20 mmol l−1 Tris–HCl, 10 mmol l−1 MgCl2, pH 7.4) and then incubated overnight at 4 °C. CsCl2 was added to the phage suspensions to a final concentration of 0.75 g ml−1 and the mixture was centrifuged at 200,000 × g for 24 h in a S55S-1096 rotor (Hitachi, Tokyo, Japan) using a Himac CS 150 GXL microultracentrifuge (Hitachi, Tokyo, Japan). The visible viral bands were extracted and dialyzed twice in TM buffer overnight at 4 °C. Genomic DNA was extracted according to the method described by Chen et al. [13].
Genome sequencing and assembly
Genome sequencing and assembly were performed at the Chinese National Human Genome Center, Shanghai. The genome of vB_PmaS-R3 was sequenced using massively parallel pyrosequencing technology (454 GS FLX) [14]. Library construction and sequencing were performed following the manufacturer’s instructions. A total of 15,070 reads with average length of 300 bp were obtained, covering the genome 100-fold. Assembly was performed using the GS de novo Assembler software [15] and produced 13 contigs ranging from 975 bp to 14210 bp. Relationships between the contigs were determined by multiplex PCR [16]. Gaps were then filled in by sequencing the PCR products using ABI 3730xl capillary sequencers. Finally, the sequences were assembled using the Phred, Phrap and Consed software packages [17], and low quality regions of the genome were resequenced.
Genome annotation
Open reading frames (ORFs) were identified by combining results from the GeneMark.hmm 2.0 gene prediction program with heuristic models [18], the ORF Finder [19], the RAST server version 2.0 [20], and the Integrated Microbial Genomes-Expert Review platform [21]. Each translated ORF was blasted against the NCBI non-redundant protein database and the KEGG protein database using BLASTP [22]. The protein domains and COG [23] assignment were predicted by RPS-BLAST against the NCBI CDD library [24].
Genome properties
The properties and statistics of the genome are summarized in Tables 3 and 4. Phage vB_PmaS-R3 has a circular double-stranded DNA genome with a length of 42,093 bp and a G + C content of 56.37 % (Table 3). Fifty-two ORFs were predicted from the genome with a coding efficiency of 92.79 %. Among the 52 ORFs, 11 had no matches in the NCBI non-redundant protein database, 33 were homologous to genes identified in phages, while seven were only found in bacteria. Strikingly, one (ORF30) was found to be closely related to a gene identified from an algal virus, Dunaliella viridis virus SI2 (Additional file 1: Table S1). Of the 44 ORFs that have matches in the NCBI database, 28 were assigned to putative functions (Fig. 3, Additional file 1: Table S1). The distribution of genes into COG categories is presented in Table 4.

Genome map of Paracoccus phage vB_PmaS-R3 and comparative genomic analysis with Burkholderia phage KL1 and Pseudomonas phage PA73
Insights from the genome sequence
Comparative genomics
According to the BLASTP results, many ORFs in vB_PmaS-R3 genome are homologous to those predicted from the genomes of two siphophages Burkholderia phage KL1 (KL1) and Pseudomonas phage PA73 (PA73) [25, 26]. Phage vB_PmaS-R3 is genomically similar to these two phages with respect to the genome length (42,832 bp and 42,999 bp for KL1 and PA73, respectively), G + C content (54.6 and 53.6 % for KL1 and PA73) and number of predicted ORFs (55 and 52 for KL1 and PA73). Of the 52 ORFs of the vB_PmaS-R3 genome, 22 are found to be homologous to mutual genes in KL1 and PA73, with one additional gene homologous to a KL1 gene (Fig. 3). These genes are mainly distributed in the DNA metabolism and morphogenesis encoding regions, which are deemed the core parts of the genome. The vB_PmaS-R3 genome could be divided into three modules: genes related to DNA replication and metabolism, regulatory genes, and structure forming genes. In the DNA replication and metabolic module, of the seven ORFs with putative functions, six are homologues to genes of KL1 and PA73. However, the DNA polymerase (ORF1) in the vB_PmaS-R3 genome is phylogenetically distant from those in the KL1 and PA73 genomes. Phage vB_PmaS-R3 has DNA polymerase type I, which belongs to DNA polymerase A family, whereas the DNA polymerases in KL1 and PA73 are members of the polymerase B family. Phage vB_PmaS-R3 shares similar structural genes with KL1 and PA73 (Fig. 3). However, the tail tape-measure gene in vB_PmaS-R3 is very different from those in KL1 and PA73. Furthermore, KL1 and PA73 have three tail assembly genes following the tail tape- measure gene, while in the vB_PmaS-R3 genome, the tail tape-measure gene is followed by four gene transfer-agent like genes instead (Fig. 3). The lysis gene of vB_PmaS-R3 is a single L-alanoyl-D-glutamate peptidase-like gene [27], while in KL1 and PA73, a combination of four and three genes function as the lysis gene, respectively.
Auxiliary metabolic genes
AMGs are phage-encoded metabolic genes that could putatively regulate the host metabolism and represent phage genomic adaptations to the host and environment [28]. In the vB_PmaS-R3 genome, ORF17 is predicted to encode a MazG domain-containing protein, which is frequently described as an AMG. Recently, phage-encoded MazG has received much attention. It is widely distributed in marine cyanophages [29, 30] and can also be found in viruses isolated from heterotrophic bacteria, including marine and non-marine bacteria [25, 31–35]. It is suggested that the expression of phage MazG could help to maintain the metabolism of starved infected host cells in nutrient-limited conditions and to optimize the production of progeny phage.
Conclusions
Phage vB_PmaS-R3 represents the first marine phage isolate infecting Paracoccus spp. According to the genome analysis, this phage is most likely a member of the viral family Siphoviridae. The genome is modularly organized, and shows synteny with the genomes of Burkholderia phage KL1 and Pseudomonas phage PA73 in the DNA metabolism and morphogenesis modules. The DNA polymerase, tail tape-measure and lysis genes reveal the difference between phage vB_PmaS-R3 and phages KL1 and PA73. The MazG gene may have the potential to benefit phage propagation of phage vB_PmaS-R3 in its nutrient-limited environment. The genome analysis of phage vB_PmaS-R3 enriches our knowledge of the evolutionary history of marine phages and ecological understanding of phage adaption to the environment.
Abbreviations
- AMG:
-
Auxiliary metabolic gene
- IMG:
-
Integrated microbial genomes
- ORF:
-
Open reading frame
- RAST:
-
Rapid annotation using subsystem technology
References
Suttle CA. Viruses in the sea. Nature. 2005;437:356–61.
Suttle CA. Marine viruses-major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–12.
Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–81.
Edwards RA, Rohwer F. Viral metagenomics. Nat Rev Microbiol. 2005;3:504–10.
Middelboe M, Chan AM, Bertelsen SK. Isolation and life cycle characterization of lytic viruses infecting heterotrophic bacteria and cyanobacteria. In: Wilhelm SW, Weinbauer MG, Suttle CA, editors. Manual of Aquatic Viral Ecology. Waco: American Society of Limnology and Oceanography, Inc.; 2010. p. 118–33.
Du HL, Jiao NZ, Hu YH, Zeng YH. Diversity and distribution of pigmented heterotrophic bacteria in marine environments. FEMS Microbiol Ecol. 2006;57:92–105.
Liu ZP, Wang BJ, Liu XY, Da X, Liu YH, Liu SJ. Paracoccus halophilus sp. nov., isolated from marine sediment of the South China Sea, China, and emended description of genus Paracoccus Davis 1969. Int J Syst Evol Microbiol. 2008;58:257–61.
Kropinski AM, Prangishvili D, Lavigne R. Position paper: the creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ Microbiol. 2009;11:2775–7.
Harker M, Hirschberg J, Oren A. Paracoccus marcusii sp. nov., an orange gram-negative coccus. Int J Syst Bacteriol. 1998;48:543–8.
Hurwitz BL, Sullivan MB. The Pacific Ocean Virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS One. 2013;8:e57355.
Yurkov VV, Krieger S, Stackebrandt E, Beatty JT. Citromicrobium bathyomarinum, a novel aerobic bacterium isolated from deep-sea hydrothermal vent plume waters that contains photosynthetic pigment-protein complexes. J Bacteriol. 1999;181:4517–25.
Suttle CA, Chen F. Mechanisms and rates of decay of marine viruses in seawater. Appl Environ Microbiol. 1992;58:3721–9.
Chen F, Wang K, Stewart J, Belas R. Induction of multiple prophages from a marine bacterium: a genomic approach. Appl Environ Microbiol. 2006;72:4995–5001.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80.
Droege M, Brendon H. The Genome Sequencer FLX™ System—Longer reads, more applications, straight forward bioinformatics and more complete data sets. J Biotechnol. 2008;136:3–10.
Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing[J]. Nucleic Acids Res. 1997;25:2745–51.
Tettelin H, Radune D, Kasif S, Khouri H, Salzberg SL. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 1999;62:500–7.
Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999;27:3911–20.
Rombel IT, Sykes KF, Rayner S, Johnston SA. ORF-FINDER: a vector for high-throughput gene identification. Gene. 2002;282:33–41.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Markowitz VM, Mavromatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6.
Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, et al. CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2007;35 Suppl 1:D237–40.
Kwan T, Liu J, DuBow M, Gros P, Pelletier J. Comparative genomic analysis of 18 Pseudomonas aeruginosa bacteriophages. J Bacteriol. 2006;188:1184–7.
Lynch KH, Stothard P, Dennis JJ. Comparative analysis of two phenotypically-similar but genomically-distinct Burkholderia cenocepacia-specific bacteriophages. BMC Genomics. 2012;13:223.
Mikoulinskaia GV, Odinokova IV, Zimin AA, Lysanskaya VY, Feofanov SA, Stepnaya OA. Identification and characterization of the metal ion-dependent L-alanoyl-D-glutamate peptidase encoded by bacteriophage T5. FEBS J. 2009;276:7329–42.
Breitbart M. Marine Viruses: Truth or Dare. Annu Rev Mar Sci. 2012;4:425–48.
Bryan MJ, Burroughs NJ, Spence EM, Clokie MRJ, Mann NH, Bryan SJ. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One. 2008;3:e2048.
Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010;12:3035–56.
Angly F, Youle M, Nosrat B, Srinagesh S, Rodriguez-Brito B, McNairnie P, et al. Genomic analysis of multiple Roseophage SIO1 strains. Environ Microbiol. 2009;11:2863–73.
Duhaime MB, Wichels A, Waldmann J, Teeling H, Glöckner FO. Ecogenomics and genome landscapes of marine Pseudoalteromonas phage H105/1. ISME J. 2011;5:107–21.
Kang I, Jang H, Oh HM, Cho JC. Complete Genome Sequence of Celeribacter Bacteriophage P12053L. J Virol. 2012;86:8339–40.
Kang I, Oh HM, Kang D, Cho JC. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc Natl Acad Sci USA. 2013;110:12343–8.
Field D, Garrity GM, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9.
Acknowledgements
We thank Jianda Ji, Qiang Zheng and Sijun Huang for valuable suggestions. This work was supported by NSFC (41376132), NSFC (91428308), the SOA Project (GASI-03-01-02-05), and the 973 project (2013CB955700).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YX performed the experiments and data analysis; YX wrote the first draft of the manuscript; YX, RZ and NJ contributed to data interpretation and preparation of the final manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
Paracoccus phage vB_PmaS-R3 gene annotations. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, Y., Zhang, R. & Jiao, N. Complete genome sequence of Paracoccus marcusii phage vB_PmaS-R3 isolated from the South China Sea. Stand in Genomic Sci 10, 94 (2015). https://doi.org/10.1186/s40793-015-0089-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0089-7