
Abstract
Thermoanaerobacter thermohydrosulfuricus BSB-33 is a thermophilic gram positive obligate anaerobe isolated from a hot spring in West Bengal, India. Unlike other T. thermohydrosulfuricus strains, BSB-33 is able to anaerobically reduce Fe(III) and Cr(VI) optimally at 60 °C. BSB-33 is the first Cr(VI) reducing T. thermohydrosulfuricus genome sequenced and of particular interest for bioremediation of environmental chromium contaminations. Here we discuss features of T. thermohydrosulfuricus BSB-33 and the unique genetic elements that may account for the peculiar metal reducing properties of this organism. The T. thermohydrosulfuricus BSB-33 genome comprises 2597606 bp encoding 2581 protein genes, 12 rRNA, 193 pseudogenes and has a G + C content of 34.20 %. Putative chromate reductases were identified by comparative analyses with other Thermoanaerobacter and chromate-reducing bacteria.
Similar content being viewed by others

Introduction
Thermoanaerobacter thermohydrosulfuricus strain BSB-33 (ATCCBAA-2171 = DSM 25103) is a Gram-positive anaerobic rod-shaped thermophilic bacterium isolated from sediment samples collected at a shallow hot spring in Bakreshwar India in the state of West Bengal [1]. The hot spring sediment was found to be basic (pH 9.2 +/− 0.1) with a temperature range of 66–70 °C supporting a diverse microbial community including Gammaproteobacteria, Cyanobacteria, green nonsulfur and low-GC Gram-positive bacteria [2]. Strain BSB-33 was found to reduce both Cr(VI) and Fe(III) anaerobically at 60 °C while utilizing peptone or pyruvate [1]. In contrast, Thermoanaerobacter thermohydrosulfuricus strain DSM 567T isolated from sugar beet extraction juice, and strain WC1-12 isolated from wood compost are not reported to reduce metals but reduce sulfite and thiosulfate to H2S while fermenting a wide range of carbohydrates [3, 4]. The Fe(III) and Cr(VI) reducing trait of BSB-33 has not been reported for the closely related species Thermoanaerobacter wiegelii strain Rt8.B1 isolated in New Zealand [5] while Thermoanaerobacter siderophilus SR4 isolated from hydrothermal vents in Kamchatka peninsula is capable of reducing Fe(III) only [6]. The related species Thermoanaerobacter ethanolicus JW200 and Thermoanaerobacter pseudethanolicus 39E isolated from Yellowstone National Park [7, 8] also reduce iron but not chromium [9]. Thermoanaerobacter species reported to anaerobically reduce Fe(III) and Cr(VI) (sp X514, sp X513, sp X561) were all collected from the geologically and hydrologically isolated deep subsurface environments in the Piceance Basin in Colorado [10]. Notably however, 16S rRNA and chaperonin-60 universal target (cpn60 UT) region sequence comparison reveals that the chromate-reducing Thermoanaerobacter species (BSB-33, and sp X513-14) are only distantly related. Thus the mechanism(s) of chromium reduction in these two species is likely quite divergent.
Owing to its strong oxidizing nature, soluble Cr(VI) can be toxic, mutagenic, and carcinogenic in various biological systems. Reduction of hexavalent chromium produces the water-insoluble and less mobile Cr(III), which has diminished toxicity due to decreased bioavailability [11]. Chromium (VI) is widely used in many industrial applications and improper disposal of waste has contaminated soil, vadose zones, and groundwater at sites throughout the industrialized world. High levels of Cr(VI) contaminated ground water from leather industries has been reported in South Australia and India [12, 13] and at chromium chemical production sites in Corvalis, Oregon, and Tamil Nadu in India [14, 15]. In addition, the U.S. Department of Energy uranium production and enrichment facilities operating during World War II resulted in high levels of chromium contamination in the groundwater at some U.S. sites [16–18]. Microbe-mediated reduction of soluble Cr(VI) in groundwater into insoluble forms to reduce contamination has been a research pursuit for decades and has been implemented in the U.S. and elsewhere. Pseudomonas strains isolated from chromate containing sewage sludge were among the first microbes reported as capable of biological reduction of chromate [19, 20]. In addition, chromium reducing species like Streptomyces sp MC1 isolated from sugarcane, and specific microbial soil communities have been used to remediate Cr(VI) contaminated soils [21–23]. Thereafter, several anaerobic chromium reducing bacteria were isolated from chromium-contaminated and non-contaminated soil and ground water and identified as candidates for bioremediation [24]. Despite many advances, there remains much foundational knowledge to gain about the mechanism of bacterial resistance to chromate that will improve our fundamental understanding of microbial metal reduction and improve microbial bioremediation strategies.
Our analyses here reveal that the distinctive chromium-reducing capabilities of BSB-33 appear divergent from other members of this species and may be particularly well-suited for bioremediation of chromium contamination. Here we present a summary classification and a set of features for Thermoanaerobacter thermohydrosulfuricus BSB-33 together with the description of the completed genome sequence and annotation. We use this genome sequence for comparative analyses with other chromium reducing bacteria both in and outside of the genus Thermoanaerobacter . We report novel insights into the divergent mechanism(s) of chromate reduction and resistance in this highly divergent species.
Organism information
Classification and features
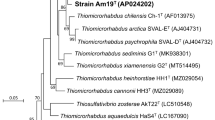
Strain BSB-33 has been previously described as ‘ Thermoanaerobacter -like bacterium’ [1] on the basis of BLAST [25] analysis of a 968 bp 16S rRNA sequence (EU368841) obtained using a single S-D-Bact-0027-a-S-18 primer [26]. Later, BSB-33 16S rRNA gene was re-sequenced by the American Type Culture Collection to generate a 1614 bp long sequence that has been deposited in GenBank (EU368841.2). The BSB-33 strain is deposited in the ATCC as ‘ Thermoanaerobacter indiensis BSB-33’. However, comparison of the 16S rRNA sequence using NCBI BLAST shows that BSB-33 has >99 % 16S rRNA sequence identity with Thermoanaerobacter thermohydrosulfuricus DSM 567, Thermoanaerobacter thermohydrosulfuricus WC1, Thermoanaerobacter wiegelii Rt8.B1, Thermoanaerobacter pseudethanolicus ATCC 33223, and Thermoanaerobacter siderophilus SR4. Thus ‘ Thermoanaerobacter indiensis BSB-33’ cannot be identified as a unique species on the basis of 16S rRNA sequence. Notably however, taxonomic assignment of Thermoanaerobacter species using 16S rRNA sequence is known to be particularly problematic due to highly conserved intervening sequences (IVS) in 16S rRNA of T. thermohydrosulfuricus and T. pseudethanolicus [4]. Phylogenetic analysis for identification of Thermoananerobacter species based on chaperonin-60 universal target region proved more accurate than 16S rRNA sequence and a more accurate predictor of whole genome relatedness and DNA-DNA hybridization values [27]. Therefore, phylogenetic trees have been constructed for BSB-33 using the maximum likelihood (ML) method within MEGA v5.1 software [28]. We used both chaperonin-60 universal target region and 16S rDNA derived from sequenced Thermoanaerobacter genomes to compare the trees and Clostridium thermocellum was used to root the trees (Figs. 1 and 2). Despite apparent phenotypic divergence, BSB-33 exhibited 99.82 %, 99.64 %, 99.64 %, 98.19 % chaperonin-60 UT sequence identity with T. thermohydrosulfuricus WC1, T. wiegelii Rt8.B1, T. siderophilus SR4, and T. thermohydrosulfuricus DSM 567 respectively. The 16S rRNA sequence identity of BSB-33 with T. thermohydrosulfuricus WC1, T. wiegelii Rt8.B1, T. siderophilus SR4 and T. thermohydrosulfuricus DSM 567 is further supported by sequence identity of cpn60 UT region; hence, it is phylogenetically closer with these species than with other Thermoanaerobacter species represented in these analyses. Despite significant genotypic and phenotypic diversity across Thermoanaerobacter sub-species, genetic homology in standard marker genes results in classification of diverse strains as a common species [4]. Because of this difficulty with Thermoanaerobacter species, and to infer whole genome relatedness, digital DNA-DNA hybridization by means of genome-to-genome distances (GGD) were calculated using the GGDC 2 software [29]. The BSB-33 genome (KB910517.1) shares 95.2 %, 84.8 % and 76 % DDH identity with T. thermohydrosulfuricus WC1 (KB731262.1 – KB731323.1), T. siderophilus SR4 (CM001486.1) and T. wiegelii Rt8.B1 (CP002991.1) genomes respectively. The typical cutoff for a unique species is a genome relatedness of less than 70 % [30]. Since BSB-33 shares the greatest sequence identity with WC1, it is therefore most likely that ‘ Thermoanaerobacter indiensis BSB-33’ is a divergent subspecies of T. thermohydrosulfuricus and hereafter referred to as strain BSB-33.

Phylogenetic tree highlighting the position of Thermoanaerobacter thermohydrosulfuricus BSB-33 relative to other type strains within the Thermoanaerobacteraceae. The tree was inferred from aligned characters of the cpn60 UT sequences using maximum likelihood method within the software MEGA v5.1 (bootstrap: calculated with the Kimura 2-parameter model distance correction and 1000 replicates). The strains and their corresponding GenBank accession numbers for cpn60 UT genes are: T. wiegelii Rt8.B1, JGI; T. thermohydrosulfuricus WC1, HM623896; T. thermohydrosulfuricus BSB-33, JGI; T. thermohydrosulfuricus DSM567, HM623910; T. siderophilus SR4, JGI; T. mathranii DSM11426, DQ439966; T. italicus DSM9252, NZ_ACVH01000076; T. ethanolicus ATCC31550, NZ_ACXY01000003; T. brockii subsp. finii DSM3389, NZ_ACQZ01000003; T. brockii subsp. brockii DSM1457, HM623909; Thermoanaerobacter sp. X513, ACPF01000040; Thermoanaerobacter sp. X514, NC_010320; T. pseudethanolicus DSM2355, NC_010321; Thermoanaerobacter sp. X561, ACXP01000037; T. tengcongensis MB4, NC_003869; Clostridium thermocellum ATCC27405, NC_009012. Bootstrap values are indicated at nodes when larger than 60 %. Clostridium thermocellum was used as an out group. The branches are scaled in terms of the expected number of substitutions per site (scale bar)

Phylogenetic tree highlighting the position of Thermoanaerobacter thermohydrosulfuricus BSB-33 relative to other type strains within the Thermoanaerobacteraceae. The tree was inferred from aligned characters of the 16S rRNA sequences using maximum likelihood method within the software MEGA v5.1 (bootstrap: calculated with the Kimura 2-parameter model distance correction and 1000 replicates). The strains and their corresponding GenBank accession numbers for 16S rDNA respectively are: T. wiegelii Rt8.B1, NR_075059.1; T. thermohydrosulfuricus WC1, HM585213.1; T. thermohydrosulfuricus BSB-33, EU368841.2; T. thermohydrosulfuricus DSM567, L09161.1; T. siderophilus SR4, AF120479.1; T. mathranii DSM11426, NR_026397.1; T. italicus DSM9252, AJ250846.1; T. ethanolicus ATCC31550, L09162.1; T. brockii subsp. finii DSM3389, L09166.1; T. brockii subsp. brockii DSM1457, L09165.1; Thermoanaerobacter sp. X513, AF542520.1; Thermoanaerobacter sp. X514, AF542517.1; T. pseudethanolicus DSM2355, L09164; Thermoanaerobacter sp. X561, AF542518.1; T. tengcongensis MB4, AF209708.1; Clostridium thermocellum ATCC27405, L09173.1. Bootstrap values are indicated at nodes when larger than 60 %. Clostridium thermocellum was used as an out group. The branches are scaled in terms of the expected number of substitutions per site (scale bar)
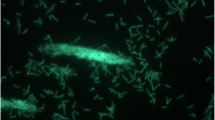
BSB-33 cells are described as straight to curved rods, approximately 0.5 μm in diameter and 2 μm in length occurring singly or in short filaments (Fig. 3) [1]. Strain BSB-33 was determined to be gram-positive at all stages of growth with slight tumbling motility. Colonies were uniformly round, white, opaque and 0.5-0.8 mm in diameter when grown anaerobically for 2–3 days on 2 % agar plates (not shown). The G + C content of the DNA of strain BSB-33 was determined by the thermal denaturation to be 35.70 ± 2 mol% [1], and calculated to be 34.20% from the genome sequence reported here (see Table 3).

Scanning electron micrograph of T. thermohydrosulfuricus BSB-33 grown anaerobically in nitrogen-purged Luria-Bertani (LB) broth at 60 °C to mid exponential phase
BSB-33 cells are strict anaerobes that can couple the oxidation of peptone to the reduction of Fe(III) oxyhydroxide and Fe(III)-citrate with maximal growth and iron and chromium reduction observed at 60 °C [1]. In basal medium BSB-33 can also couple the oxidation of peptone to the reduction of 0.2 mM K2CrO4, 30 mM MnO2 and 20 mM AQDS [1]. When Fe (III)-citrate is used as the electron acceptor either peptone or pyruvate (organic acid) can be utilized as a substrate (electron donor) while lactate, acetate, citrate, or 100 % hydrogen gas are not utilized [1]. Both the soluble and membrane subcellular fractions of BSB-33 contain anaerobic Fe(III) and Cr(VI) reduction activities at 60 °C using NADH as the electron donor [1]. A summary of the classification and general features of T. thermohydrosulfuricus BSB-33 are presented in Table 1.
Genome sequencing information
Genome project history
BSB-33 was selected for sequencing by the DOE Joint Genome Institute Community Sequencing Program 2012 because of its phylogenetic position and its metal reducing capabilities that are potentially suitable for DOE missions involving bioremediation and microbial fuel cell development. The genome project is deposited in Integrated Microbial Genome (IMG ER) online database in the Genome Online Database GOLD [31] as project Gi14051 and the complete genome sequence is available in GenBank (KB910517.1) under the name ‘ Thermoanaerobacter indiensis BSB-33’. Sequencing, finishing, and annotation of T. thermohydrosulfuricus BSB-33 were performed by DOE Joint Genome Institute. A summary of the project information is shown in Table 2.
Growth conditions and genomic DNA preparation
T. thermohydrosulfuricus BSB-33 was grown anaerobically in nitrogen (N2) purged Luria-Bertani (LB) broth at 60 °C. Extraction of chromosomal DNA was performed on 500 ml of culture grown into late exponential phase (OD600nm = 0.4). Cells were collected by centrifugation at 4 °C and 10,000x g for 10 min. High molecular weight DNA was prepared from these cells using the standard methods as recommended by the DOE Joint Genome Institute (JGI, Walnut Creek, CA, USA). Briefly, cells were suspended in TE buffer (10 mM Tris–HCl, 1.0 mM EDTA, pH 8.0) to a final OD600nm = 1. The sample was treated with lysozyme (1.33 mg/ml) and incubated 5 min at room temperature. Sodium dodecyl sulfate (0.5 %) and Proteinase K (0.1 mg/ml) were added and cells incubated for 1 hr at 37 °C. NaCl (0.5 M) and hexadecyltrimethyl ammonium bromide/NaCl mixture (0.66 %/46 mM) were added and incubated at 65 °C for 10 min and then extracted with chloroform/isoamyl alcohol mixture. Cell lysates were extracted with phenol/chloroform/isoamylalcohol (25:24:1) followed by precipitation with 0.6x volumes of isopropanol. The nucleic acid pellet was washed with 70 % ethanol, dissolved in TE buffer and then treated with DNase-free RNase A at 37 °C for 20 min. DNA concentration and purity was determined by UV/Vis absorbance and DNA quality further assessed by visualizationon 1 % agarose gels stained with ethidium bromide and no plasmid or viral DNA was evident in preparations (not shown).
Genome sequencing and assembly
The draft genome of Thermoanaerobacter thermohydrosulfuricus BSB-33 (‘T. indiensis’ BSB-33 in JGI documents) was generated at the DOE Joint Genome Institute (JGI) using Illumina data [32]. For this genome, a short-insert paired-end library was constructed and sequenced with Illumina giving average insert sizes of 270 +/− 70 bp which generated 26,606,974 reads and an Illumina long-insert paired-end library gave an average insert size of 10871 +/− 1786 bp which generated 40,206,744 reads totalling 8,012 Mbp of Illumina data (unpublished, Feng Chen-JGI). All general aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov/. The initial draft assembly contained 41 contigs in ten scaffold(s). The initial draft data was assembled with Allpaths, version r41554 [33], and the consensus was computationally shredded into 10 Kbp overlapping fake reads (shreds). The Illumina draft data was also assembled with Velvet, version 1.1.05 [34] and the consensus sequences were computationally shredded into 1.5 Kbp overlapping fake reads (shreds). The Illumina draft data was assembled again with Velvet using the shreds from the first Velvet assembly to guide the next assembly. The consensus from the second Velvet assembly was shredded into 1.5 Kbp overlapping fake reads. The fake reads from the Allpaths assembly and both Velvet assemblies and a subset of the Illumina CLIP paired-end reads were assembled using parallel phrap, version 4.24 (High Performance Software, LLC). Possible mis-assemblies were corrected with manual editing in Consed [35–37]. Gap closure was accomplished using repeat resolution software (unpublished, Wei Gu-JGI), and sequencing of bridging PCR fragments with Sanger and/or PacBio (unpublished, Cliff Han-JGI) technologies. For improved high quality draft and noncontiguous finished projects, one round of manual/wet lab finishing may have been completed. Primer walks, shatter libraries, and/or subsequent PCR reads may also be included for a finished project. A total of zero additional sequencing reactions, 11 PCR PacBio consensus sequences, and 0 shatter libraries were completed to close gaps and to raise the quality of the final sequence. The total size of the genome is 2.6 Mb and the final assembly is based on 8,012 Mbp of Illumina draft data, which provides an average 3,082X coverage of the genome.
Genome annotation
Genes were identified using Prodigal [38] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using GenePRIMP gene prediction software [39]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGRFam, Pfam, PRIAM, KEGG, COG, and InterPro databases. Additional gene prediction analysis and functional annotation was performed within the Integrated Microbial Genomes (IMG-ER) platform (http://img.jgi.doe.gov) developed by the Joint Genome Institute, Walnut Creek, CA, USA [40].
Genome properties
Detailed genome statistics are provided in Table 3 and Fig. 4. The genome consists of one chromosome with a total length of 2,597,606 bp and a G + C content of 34.20 %. Of the 2,721 predicted genes, 2,581 are protein-coding genes, 140 encode putative RNAs, and 193 pseudogenes were identified. The majority of the protein-coding genes (79.86 %) were assigned a putative function while the remaining genes were annotated as hypothetical proteins. The distribution of genes into functional categories based on clusters of orthologous genes is presented in Table 4.

A graphical overview of the T. thermohydrosulfuricus BSB-33 genome. The nucleotide numbers are shown across the bottom of the figure with a corresponding black and white scale bar. The first row above the scale bar identifies genes on the forward strand of the genome, color coded according to COG categories. The second row above the scale bar is also color coded by COG category and shows genes on the opposite strand (reverse strand) of the genome. The third row of colored tick marks shows the location of identified RNA genes with tRNAs in green, rRNAs in red. The black line above the protein and RNA gene markers shows the GC content across the genome, and the top magenta colored graphical corresponds to the GC skew across the genome
Insights from the genome sequence
Chromate reductases
Chromium contamination is primarily an anthropogenic event and therefore microbial chromium reduction may not have specifically evolved for chromate, it is more likely part of generalized electron transport systems or enzymes with other primary physiological functions [41–45]. A number of both soluble and membrane associated chromate-reducing enzymes have been purified to various degrees, most of which have NAD(P)H dependant oxidoreductase activity [44]. Some of these enzymes have proven to be essential for both Cr(VI) reduction and countering chromate mediated oxidative stress, thereby conferring bacterial chromate resistance [41]. Many Thermoanaerobacter are capable of reducing iron, but strain BSB-33 has both Fe(III) and Cr(VI) reductase activity, suggesting genotypic dissimilarity [1].
To gain insight into the mechanism(s) of chromate reduction and chromate tolerance in BSB-33, comparative genomic analysis was conducted on BSB-33 and its closely related species T. wiegelii Rt8.B1 and T. siderophilus SR4, and the Fe(III) and Cr(VI) reducing Thermoanaerobacter sp X514 isolate. Gene annotations and computational tools available in the Joint Genome Institute Integrated Microbial Genomes database were used for analyses. Independent searches for Cluster of Orthologous Groups [46] and analyzing associated enzyme commission number, KEGG Orthology [47], and TIGRFAMs [48] annotations together with literature and database searches, we identified genes potentially encoding soluble and membrane associated oxidoreductases and dehydrogenases that potentially function in chromate reduction. Genes encoding dihydrolipoamide dehydrogenase, NADH:flavin oxidoreductase (Old yellow enzyme), di-iron [Fe-Fe] hydrogenase, thioredoxin, thioredoxin reductase were found to be potential Cr-reducing proteins common to all four Thermoanaerobacter species in our analysis. Interestingly however, NAD(P)H-nitrite reductase, NADH:ubiquinone oxidoreductase (H(+) translocating) complex, nickel-iron [Ni-Fe] hydrogenase, superoxide dismutase were all limited to BSB-33 and its most closely related species T. wiegelii Rt8.B1 and T. siderophilus SR4 (Table 5). Therefore, differences are expected in the mechanism of chromate reduction by BSB-33 and sp X514. Summary of all features is presented in Fig. 5.

Comparison of candidate chromate reductase genes in Thermoanaerobacter thermohydrosulfuricus BSB-33, T. wiegelii Rt8.B1, T. siderophilus SR4 and T. sp X514 species. The grid is arranged with columns representing Thermoanaerobacter species and rows representing candidate chromate reductase genes. Each grid is colored depending on whether the gene is present (blue) or absent (white)
Candidate genes for soluble metal (chromate) reduction activities
Nitrite reductases
The formate-dependent nitrite reductase (nrfA) in Shewanella oneidensis MR1 is involved in nitrite reduction during anaerobic respiration and also proposed to be responsible for chromate reduction [45]. Recently, the formate-dependent cytochrome c nitrite reductase (nrfA) was shown to catalyze the six electron reduction of nitrite (NO2 −) to ammonium (NH4 +) at the active site heme iron which also catalyzes sulfite reduction [49]. The BSB-33 genome encodes a NAD(P)H-nitrite reductase (nirB) (B044DRAFT_0957; EC:1.7.1.4; COG1251; KO:K00362), which is an iron-sulfur heme flavoprotein containing siroheme which catalyzes the reduction of nitrite to ammonium (N02 − + 6[H∙] = NH4 ++ 20H−). The siroheme prosthetic group bound to nirB gene products are covalently linked to iron-sulfur clusters which facilitate the six electron reduction of nitrite and sulfite [50, 51]. Owing to such electron transfer characteristics, it is possible that the NAD(H)-nitrite reductases of BSB-33 are also involved in Cr(VI) reduction under anaerobic conditions. The genes encoding enzymes involved in the biosynthesis of siroheme, are hemABCDL (B044DRAFT_0544, 0547, 0545, 0546, 0551) to convert glutamyl tRNA to uroporphyrinogen-III, cobA-hemD (B044DRAFT_0546) and MET8 (B044DRAFT_0548) to convert uroporphyrinogen–III to siroheme. These genes are only present in the BSB-33 genome and its most closely related species T. wiegelii Rt8.B1 and T. siderophilus SR4.
Hydrogenases
Di-iron (Fe-Fe) hydrogenase that couple oxidation of NADH and ferrodoxins simultaneously to produce hydrogen gas (H2) are conserved within the Thermoanaerobacter genus [52]. A six-gene cluster encoding a membrane bound nickel-iron energy conserving hydrogenase (Ech) has been identified in the Thermoanaerobacter strains T. italicus Ab9, T. mathrani subsp mathrani A3, T. wiegelii Rt8.B1, T. siderophilus SR4, and T. thermohydrosulfuricus WC1 [52]. The Ech complex uses reduced ferredoxins generated by pyruvate catabolism for the evolution of H2 [52]. BSB-33 genome contains genes encoding two di-iron (Fe-Fe) hydrogenases (B044DRAFT_1055,1062,) and a nickel-iron (Ni-Fe) hydrogenase (B044DRAFT_0240) in addition to other genes of the Ech complex (B044DRAFT_0236-B044DRAFT_0241). By KEGG orthology (KO) annotation the hydrogenase genes are designated as NADH-quinone oxidoreductase subunit G (KO:K00336) and NADH-quinone oxidoreductase subunit D (KO:K00333) respectively. Based on Enzyme Commission (EC) number, the hydrogenases are assigned (EC:1.6.5.3), which is NADH:ubiquinone reductase (H(+)-translocating), a respiratory chain flavoprotein (FMN) containing iron sulfur clusters and involved in electron transfer from NADH to ubiquinone coupled to transmembrane proton translocation. Soluble bacterial quinone oxidoreductases are thought to reduce Fe(III) and Cr(VI) and counter oxidative stress [53]. The quinone oxidoreductases prevent formation of potentially toxic semiquinone radicals and reactive oxygen species [42]. One of the two di-iron hydrogenase genes in BSB-33 is disrupted by insertion of repetitive elements (B044DRAFT_1062). Therefore, the remaining intact di-iron hydrogenase and the Ni-Fe hydrogenase, which is in the cytoplasmic part of Ech complex, remain candidates for Cr(VI) reduction and oxidative stress responses.
Superoxide dismutases
In contrast to Thermoanaerobacter sp X514 genome, BSB-33, Rt8.B1 and SR4 genomes encode a superoxide dismutase SOD, an antioxidant protein involved in dismutation of superoxide into oxygen and hydrogen peroxide [53]. Reduction of Cr(VI) compounds can give rise to reactive oxygen species which elicit bacterial cell responses that include inducing antioxidant proteins like superoxide dismutases [54]. It has been proposed that improving the efficacy of enzymes to minimize oxidative stress during chromate reduction is one possible way to increase the effectiveness of bioremediation by a bacterial species [54]. Unlike sp X514, the BSB-33 genome contains an additional enzyme (SOD2, B044DRAFT_1026) to counter oxidative stress produced during chromate reduction.
NADH: flavin oxidoreductase (Old Yellow Enzyme)
The NADH:flavin oxidoreductase from Thermus scotoductus SA-01 is related to the Old Yellow Enzyme family and exhibits chromate reductase activity [55]. This chromate-reducing OYE is highly conserved in Bacillus subtilis where it is functional in xenobiotic degradation and oxidative stress responses [55]. Likewise, oxidative stress responses in Azotobacter vinelandii are linked to flavin-containing oxidoreductase enzymes [56]. OYE-related NADH: flavin oxidoreductases are present in all four Thermoanaerobacter species in our analysis. BSB-33 genome encodes three different OYE-related NADH: flavin oxidoreductases (B044DRAFT_0449, 0057, 1188). The NADH: flavin oxidoreductases of BSB-33 (B044DRAFT_0057) and sp X514 (Teth514_0011) share 48 % amino acid sequence identity with the T. scotoductus SA-01 OYE homologue. Hence it is possible that one or more of the BSB-33 NADH:flavin oxidoreductases are also involved in chromate reduction and oxidative stress responses.
Dihydrolipoamide dehydrogenase
Dihydrolipoamide dehydrogenase is part of a multisubunit pyruvate dehydrogenase complex. This complex catalyzes the conversion of pyruvate to acetyl-CoA, and also exhibits chromate reductase activity in Thermus Scotoductus SA-01 [57]. LPD (EC:1.8.1.4) is a flavoprotein belonging to family of pyridine nucleotide-disulfide oxidoreductases (class I active site) [57, 58] and genes encoding LPD have been identified in BSB-33, Rt8.B1, SR4 and sp X514. BLAST analysis reveals that LPD of BSB-33 (B044DRAFT_0196, 0424) shares 36 % and 37 % amino acid identity with T. scotoductus SA-01 LPD respectively. LPD protein sequences from sp X514 (Teth514_0234, 2038) share 93 % and 43 % sequence identity with BSB-33 LPD (B044DRAFT_0196). Hence, the LPD of BSB-33 and sp X514 may also be involved in the observed chromate reduction.
Thioredoxin and thioredoxin reductase
A metal reduction operon (mre) identified in Desulfovibrio desulfuricans G20 encoding thioredoxin, thioredoxin reductase and an additional metal oxidoreductase exhibited Cr(VI) and U(VI) reduction [59]. Cr(VI)-exposed cultures of Caulobacter crescentus and Shewanella oneidensis MR1 showed upregulation of genes encoding thioredoxin and glutaredoxin [44, 60, 61]. BSB-33, Rt8.B1, SR4 and sp X514 genomes encode thioredoxin and thioredoxin reductases. Thus, thioredoxin and thioredoxin reductases in BSB-33 and sp X514 must be formally considered as candidate soluble proteins for chromate reduction by BSB-33.
Candidate genes for membrane associated chromate reduction activities
Mbx and Rnf complex
A 13 gene-cluster similar in genomic structure to membrane bound oxidoreductase (mbx) genes of Pyrococcus furiosus was identified in three Thermoanaerobacter strains, T. wiegelii Rt8.B1, T. siderophilus SR4 and T. thermohydrosulfuricus WC1 [52]. The gene cluster encodes a complex with Fdred:NAD(P)+ oxidoreductase activity where energy is conserved by translocation of cations with oxidation of ferredoxin (Fdred) [52]. BSB-33 is closely related to T. wiegelii Rt8.B1 and T. siderophilus SR4 and also encodes a similar mbx gene cluster (B044DRAFT_0131-B044DRAFT_0143). Six of the mbx cluster genes in BSB-33 encode the formate hydrogenlyases subunits 3, 4, 6, and a NADH-quinone oxidoreductases subunit B, and the 27 kDa, and 49 kDa subunits (B044DRAFT_0138-B044DRAFT_0143) that are annotated as NADH-quinone oxidoreductases, flavoproteins (FMNs) containing iron sulfur clusters. Among these formate hydrogenlyase subunit 3,4,6 (B044DRAFT_0138, 0139, 0143) and NADH-quinone oxidoreductases 49KD subunit (B044DRAFT_0142) have transmembrane helices. In Shewanella putrefaciens MR1, inhibitory studies suggested that cytoplasmic membrane bound multi-component electron transport chains including cytochromes, quinones, flavoproteins and proteins with iron sulfur centres are involved in Cr(VI) reduction [62]. Therefore, the formate hydrogenlyases and NADH-quinone oxidoreductases 49 kDa subunit of the mbx complex in BSB-33 might play an essential role in transferring reducing equivalents to extracellular Cr(VI) ions. In sp X514 all of the 13 genes required for a functional mbx complex could not be identified [52]. Instead a functionally analogous Na+ ion translocating Rnf complex is present [52]. The Rnf gene cluster encodes for NADH:ubiquinone oxidoreductases (COG4656, TIGR01945) which are possibly involved in Cr(VI) reduction in sp X514.
Conclusions
‘T. indensis BSB-33’ has been classified as T. thermohydrosulfuricus BSB-33 based on 16S and cpn60 UT region sequence identity. Within a given Thermoanaerobacter species there is a notedly broad functional diversity with only genetic microdiversity, even among isolates from a common environment [4]. Here we focus on the divergent metal reducing capabilities among members of the Thermoanaerobacter genus. Despite many years of intensive study, assimilatory and dissimilatory metal reduction processes in microbes remains incompletely understood and particularly difficult to discern from genetic sequence alone [63, 64]. We present genomic analyses between Thermoanaerobacter species to highlight the mechanisms of dissimilatory metal reduction of Cr(IV) and Fe(III) in these microbes. This comparitive genome analysis indicates several oxidoreductases in BSB-33 that are likely to be involved in chromate reduction of which nitrite reductase, dihydrolipoamide dehydrogenase and NADH: flavin oxidoreductase are top candidate genes. These enzymes being redox proteins with flavin and iron sulfur center prosthetic groups which play essential roles in electron transfer have appropriate characteristics to transfer electrons to Cr(VI) [50, 65].
The complete genome sequence of BSB-33 provides the starting point for a detailed analysis of the mechanism of chromate reduction. Novel mechanisms and uncommon dissimilatory metal reduction pathways between Thermoanaerobacter strains can be identified by further comparative genomic analysis and direct redox experimentation. Experimental characterization of these enzymes will provide valuable insight into the variance and mechanisms of chromate reduction by various Thermoanaerobacter strains.
Abbreviations
- Fe(III):
-
Iron in the +3 oxidation state
- Cr(VI):
-
Chromium in the +6 oxidation state
- Cr(III):
-
Chromium in the +3 oxidation state
- U(VI):
-
Uranium in the +6 oxidation state
- rRNA:
-
Ribosomal ribonucleic acid
- H2S:
-
Hydrogen sulfide
- K2CrO4 :
-
Potassium chromate
- MnO2 :
-
Manganese dioxide
- AQDS:
-
9,10-anthraquinone-2,6-disulfonic acid
- NADH:
-
Nicotinamide adenine dinucleotide
- NAD(P)H:
-
Nicotinamide adenine dinucleotide phosphate
References
Bhowmick DC, Bal B, Chatterjee NS, Ghosh AN, Pal S. A low-GC Gram-positive Thermoanaerobacter-like bacterium isolated from an Indian hot spring contains Cr(VI) reduction activity both in the membrane and cytoplasm. J Appl Microbiol. 2009;106:2006–16.
Ghosh D, Bal B, Kashyap VK, Pal S. Molecular phylogenetic exploration of bacterial diversity in a Bakreshwar (India) hot spring and culture of Shewanella-related thermophiles. Appl Environ Microbiol. 2003;69:4332–6.
Lee Y-E, Jain MK, Lee C, Zeikus JG. Taxonomic Distinction of Saccharolytic Thermophilic Anaerobes: Description of Thermoanaerobacterium xylanolyticum gen. nov., sp. nov., and Thermoanaerobacterium saccharolyticum gen. nov., sp. nov.; Reclassification of Thermoanaerobium brockii, Clostridium thermosulfurogenes, and Clostridium thermohydrosulfuricum E100-69 as Thermoanaerobacter brockii comb. nov., Thermoanaerobacterium thermosulfurigenes comb. nov., and Thermoanaerobacter thermohydrosulfuricus comb. nov., Respectively; and Transfer of Clostridium thermohydrosulfuricum 39E to Thermoanaerobacter ethanolicus. Int J Syst Bacteriol. 1993;43:41–51.
Verbeke TJ, Dumonceaux TJ, Wushke S, Cicek N, Levin DB, Sparling R. Isolates of Thermoanaerobacter thermohydrosulfuricus from decaying wood compost display genetic and phenotypic microdiversity. Fems Microbiol Ecol. 2011;78:473–87.
Cook GM, Rainey FA, Patel BK, Morgan HW. Characterization of a new obligately anaerobic thermophile, Thermoanaerobacter wiegelii sp. nov. Int J Syst Bacteriol. 1996;46:123–7.
Slobodkin AI, Tourova TP, Kuznetsov BB, Kostrikina NA, Chernyh NA, Bonch-Osmolovskaya EA. Thermoanaerobacter siderophilus sp. nov., a novel dissimilatory Fe(III)-reducing, anaerobic, thermophilic bacterium. Int J Syst Bacteriol. 1999;49(4):1471–8.
Wiegel J, Ljungdahl LG. Thermoanaerobacter ethanolicus gen. nov., spec. nov., a new, extreme thermophilic, anaerobic bacterium. Arch Microbiol. 1981;128:343–8.
Onyenwoke RU, Kevbrin VV, Lysenko AM, Wiegel J. Thermoanaerobacter pseudethanolicus sp. nov., a thermophilic heterotrophic anaerobe from Yellowstone National Park. Int J Syst Evol Microbiol. 2007;57:2191–3.
Onyenwoke RU, Geyer R, Wiegel J. Characterization of a soluble oxidoreductase from the thermophilic bacterium Carboxydothermus ferrireducens. Extremophiles. 2009;13:687–93.
Roh Y, Liu SV, Li G, Huang H, Phelps TJ, Zhou J. Isolation and characterization of metal-reducing Thermoanaerobacter strains from deep subsurface environments of the Piceance Basin. Colorado Appl Environ Microbiol. 2002;68:6013–20.
McLean J, Beveridge TJ. Chromate Reduction by a Pseudomonad Isolated from a Site Contaminated with Chromated Copper Arsenate. Appl Environ Microbiol. 2001;67:1076–84.
Kamaludeen SPB, Arunkumar KR, Avudainayagam S, Ramasamy K. Bioremediation of chromium contaminated environments. Indian J Exp Biol. 2003;41:972–85.
Zayed AM, Terry N. Chromium in the environment: factors affecting biological remediation. Plant Soil. 2003;249:139–56.
Krishnamurthy S, Wilkens MM. Environmental chemistry of Cr. Northeast Geol. 1994;16(1):14–7.
Jeyasingh J, Philip L. Bioremediation of chromium contaminated soil: optimization of operating parameters under laboratory conditions. J Hazard Mater. 2005;118:113–20.
Faybishenko B, Hazen TC, Long PE, Brodie EL, Conrad ME, Hubbard SS, et al. In Situ Long-Term Reductive Bioimmobilization of Cr(VI) in Groundwater Using Hydrogen Release Compound. Environ Sci Technol. 2008;42:8478–85.
Osterberg C, Cutshall N, Cronin J. Chromium-51 as a Radioactive Tracer of Columbia River Water at Sea. Science. 1965;150:1585–7.
Wang Y-T, Shen H. Bacterial reduction of hexavalent chromium. J Ind Microbiol. 1995;14:159–63.
Lebedeva EV, Lyalikova NN. Reduction of crocoite by Pseudomonas chromatophilia sp. nov. Mikrobiologiya. 1979;48:517–22.
Romanenko VI, Koren’kov VN. Pure culture of bacteria using chromates and bichromates as hydrogen acceptors during development under anaerobic conditions. Mikrobiologiia. 1977;46:414–7.
Bader JL, Gonzalez G, Goodell PC, Ali A-MS, Pillai SD. Aerobic Reduction of Hexavalent Chromium in Soil by Indigenous Microorganisms. Bioremediation J. 1999;3:201–11.
Oliver DS, Brockman FJ, Bowman RS, Kieft TL. Microbial reduction of hexavalent chromium under vadose zone conditions. J Environ Qual. 2003;32:317–24.
Polti MA, Garcia RO, Amoroso MJ, Abate CM. Bioremediation of chromium (VI) contaminated soil by Streptomyces sp. MC1. J Basic Microbiol. 2009;49:285–92.
Turick CE, Apel WA, Carmiol NS. Isolation of hexavalent chromium-reducing anaerobes from hexavalent-chromium-contaminated and noncontaminated environments. Appl Microbiol Biotechnol. 1996;44:683–8.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Alm EW, Oerther DB, Larsen N, Stahl DA, Raskin L. The oligonucleotide probe database. Appl Environ Microbiol. 1996;62:3557–9.
Verbeke TJ, Sparling R, Hill JE, Links MG, Levin D, Dumonceaux TJ. Predicting relatedness of bacterial genomes using the chaperonin-60 universal target (cpn60 UT): Application to Thermoanaerobacter species. Syst Appl Microbiol. 2011;34:171–9.
Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9.
Auch AF, von Jan M, Klenk HP, Goker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2:117–34.
Wayne LG, ICSB JC. International Committee on Systematic Bacteriology: announcement of the report of the ad hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Zentralblatt fur Bakteriologie, Mikrobiologie, und Hygiene Series A, Medical microbiology, infectious diseases, virology, parasitology. 1988;268:433–4.
Pagani I, Liolios K, Jansson J, Chen IMA, Smirnova T, Nosrat B, et al. The Genomes OnLine Database (GOLD) v. 4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2012;40:D571–9.
Bennett S. Solexa Ltd Pharmacogenomics. 2004;5:433–8.
Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–20.
Zerbino DR, Birney E. Velvet algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Ewing B, Green P. Base-calling of automated sequencer traces using phred. II Error probabilities. Genome Res. 1998;8:186–94.
Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I Accuracy assessment. Genome Res. 1998;8:175–85.
Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202.
Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119.
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods. 2010;7:455–7.
Markowitz VM, Mavromatis K, Ivanova NN, Chen IMA, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinforma Oxf Engl. 2009;25:2271–8.
Clark DP. Chromate reductase activity of Enterobacter aerogenes is induced by nitrite. Fems Microbiol Lett. 1994;122:233–7.
Gonzalez CF. ChrR, a Soluble Quinone Reductase of Pseudomonas putida That Defends against H2O2. J Biol Chem. 2005;280:22590–5.
Ishibashi Y, Cervantes C, Silver S. Chromium reduction in Pseudomonas putida. Appl Environ Microbiol. 1990;56:2268–70.
Ramírez-Díaz MI, Díaz-Pérez C, Vargas E, Riveros-Rosas H, Campos-García J, Cervantes C. Mechanisms of bacterial resistance to chromium compounds. BioMetals. 2007;21:321–32.
Viamajala S, Peyton BM, Apel WA, Petersen JN. Chromate/nitrite interactions in Shewanella oneidensis MR-1: Evidence for multiple hexavalent chromium [Cr(VI)] reduction mechanisms dependent on physiological growth conditions. Biotechnol Bioeng. 2002;78:770–8.
Tatusov RL. The COG, database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–6.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4.
Haft DH. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31:371–3.
Einsle O. Structure and function of formate-dependent cytochrome c nitrite reductase. NrfA Meth Enzymol. 2011;496:399–422.
Crane BR, Siegel LM, Getzoff ED. Structures of the siroheme-and Fe4S4-containing active center of sulfite reductase in different states of oxidation: heme activation via reduction-gated exogenous ligand exchange. Biochemistry. 1997;36:12101–19.
Murphy MJ, Siegel LM, Tove SR, Kamin H. Siroheme: a new prosthetic group participating in six-electron reduction reactions catalyzed by both sulfite and nitrite reductases. Proc Natl Acad Sci. 1974;71:612–6.
Verbeke TJ, Zhang X, Henrissat B, Spicer V, Rydzak T, Krokhin OV, et al. Genomic Evaluation of Thermoanaerobacter spp. for the Construction of Designer Co-Cultures to Improve Lignocellulosic Biofuel Production. PLoS ONE. 2013;8:e59362.
McCord JM, Fridovich I. Superoxide dismutase: the first twenty years (1968–1988). Free Radic Biol Med. 1988;5:363–9.
Ackerley DF, Barak Y, Lynch SV, Curtin J, Matin A. Effect of Chromate Stress on Escherichia coli K-12. J Bacteriol. 2006;188:3371–81.
Opperman DJ, Piater LA, van Heerden E. A Novel Chromate Reductase from Thermus scotoductus SA-01 Related to Old Yellow Enzyme. J Bacteriol. 2008;190:3076–82.
Yannone SM, Burgess BK. Identification of a palindromic sequence that is responsible for the up-regulation of NAPDH-ferredoxin reductase in a ferredoxin I deletion strain of Azotobacter vinelandii. J Biol Chem. 1997;272:14454–8.
Opperman DJ, Van Heerden E. A membrane-associated protein with Cr(VI)-reducing activity from Thermus scotoductus SA-01. Fems Microbiol Lett. 2008;280:210–8.
Argyrou A, Blanchard JS. Flavoprotein disulfide reductases: advances in chemistry and function. Prog Nucleic Acid Res Mol Biol. 2004;78:89–142.
Li X, Krumholz LR. Thioredoxin is involved in U(VI) and Cr(VI) reduction in Desulfovibrio desulfuricans G20. J Bacteriol. 2009;191:4924–33.
Chourey K, Thompson MR, Morrell-Falvey J, Verberkmoes NC, Brown SD, Shah M, et al. Global molecular and morphological effects of 24-hour chromium(VI) exposure on Shewanella oneidensis MR-1. Appl Environ Microbiol. 2006;72:6331–44.
Hu P, Brodie EL, Suzuki Y, McAdams HH, Andersen GL. Whole-genome transcriptional analysis of heavy metal stresses in Caulobacter crescentus. J Bacteriol. 2005;187:8437–49.
Myers CR, Carstens BP, Antholine WE, Myers JM. Chromium (VI) reductase activity is associated with the cytoplasmic membrane of anaerobically grown Shewanella putrefaciens MR-1. J Appl Microbiol. 2000;88:98–106.
Yannone SM, Hartung S, Menon AL, Adams MW, Tainer JA. Metals in biology: defining metalloproteomes. Curr Opin Biotechnol. 2012;23:89–95.
Cvetkovic A, Menon AL, Thorgersen MP, Scott JW, Poole II FL, Jenney Jr FE, et al. Microbial metalloproteomes are largely uncharacterized. Nature. 2010;466:779–82.
Ackerley DF, Gonzalez CF, Keyhan M, Blake R, Matin A. Mechanism of chromate reduction by the Escherichia coli protein, NfsA, and the role of different chromate reductases in minimizing oxidative stress during chromate reduction. Environ Microbiol. 2004;6:851–60.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci. 1990;87:4576–9.
Garrity GM, Holt JG. The Road Map to the Manual. In: Garrity GM, Boone DR, Castenholz RW, editors. Bergey’s Manual of Systematic Bacteriology, vol. 1. 2nd ed. New York: Springer; 2001. p. 119–69.
Gibbons NE, Murray RGE. Proposals concerning the higher taxa of bacteria. Int J Syst Bacteriol. 1978;28:1–6.
Murray RGE. The Higher Taxa, or, a Place for Everything…? In: Holt JG, editor. Bergey’s Manual of Systematic Bacteriology, vol. 1. 1st ed. Baltimore: The Williams and Wilkins Co; 1984. p. 31–4.
List Editor. List of new names and new combinations previously effectively, but not validly, published. List no. 132. Int J Syst Evol Microbiol. 2010;60:469–72.
Rainey FA. Class II. Clostridia class nov. In: De Vos P, Garrity G, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB, editors. Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 3. New York: Springer; 2009. p. 736.
Weigel J. Order III. Thermoanaerobacterales ord. nov. In: P. De Vos, Garrity GM, Jones D, Krieg NR, Ludwig FA, Rainey KH, Schleifer and Whitman W, editors. Bergey’s Manual of Systematic Bacteriology, Second edition, Volume 3 (Firmicutes). Dordrecht, Heidelberg, London, New York: Springer; 2009. p. 1224
Weigel J. Family I. Thermoaaerobacteraceae fam. nov. In: De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman W, editors. Bergey’s Manual of Systematic Bacteriology, second ed, Volume 3. New York: Springer; 2009. p. 1225.
Weigel J, Oyenwoke R. Genus I. Thermoanaerobacter. In: De Vos P, George M, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman W, editors. Bergey’s Manual of Systematic Bacteriology, Second Edition, Volume 3. New York -Heidelberg: Springer Verlag; 2009. p. 1225–39.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontology Consortium Nat Genet. 2000;25:25–9.
Acknowledgements
We express our deep gratitude to Binayak Dutta-Roy, who has been the main inspiration behind this work. We thank Dr. Subrata Pal for his input and isolating BSB-33. Funding was provided by a Fulbright Doctoral and Professional Scholarship to P.B. (15111363) and through the ENIGMA Scientific Focus Area Program at Lawrence Berkeley National Laboratory, supported by the Office of Science, Office of Biological and Environmental Research, of the U. S. Department of Energy under Contract No. DE-AC02-05CH11231. The work conducted by the U.S. Department of Energy Joint Genome Institute was supported under Contract No. DE-AC02-05CH11231 by the Office of Science of the U.S. Department of Energy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PB and AB carried out the molecular genetic studies, participated in the sequence alignments and analyses. MZ and MA prepared, mounted and imaged cells using scanning electron microscopy. LG managed the sequencing and annotation project. SMY directed the research and drafted the manuscript with PB. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bhattacharya, P., Barnebey, A., Zemla, M. et al. Complete genome sequence of the chromate-reducing bacterium Thermoanaerobacter thermohydrosulfuricus strain BSB-33. Stand in Genomic Sci 10, 74 (2015). https://doi.org/10.1186/s40793-015-0028-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-015-0028-7