
Abstract
Background
The discovery of new lead compounds with desired properties and biological activity is an excellent challenge in pesticide chemistry. Chloroacetamide are an important class of synthetic herbicides.
Results
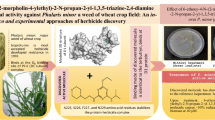
To explore the herbicidal activity of chloroacetamides, several new chloroacetamide derivatives have been designed, and synthesized. The compounds have been described by forming Schiff bases followed by chloroacetylation of imines. The herbicidal activity as a chlorophyll inhibition was evaluated against two broadleaf weeds (Chenopodium album and Anagallis arvensis) and two grass weeds (Lolium temulentum and Echinochloa crus-galli) in comparison with acetochlor as a standard herbicide. 1H-NMR, 13C-NMR and mass spectroscopic analyses confirmed the chemical structures of the synthesized compounds. Several compounds have demonstrated highly potent herbicidal activity compared to the standard herbicide acetochlor. Some of them have been described as the most effective against weeds tested, such as compounds 5b and 18b. Molecular docking to the active sites of Very Long Chain Fatty Acid Synthase (VLCFAS) has indicated that most compounds are low-energy binding agents and show high affinity for the active pocket.
Conclusion
Novel herbicides may be discovered by combining chloroacetamide derivatives with these existing lead structures.
Graphical Abstract

Similar content being viewed by others
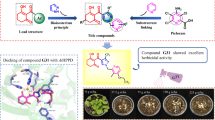
Introduction
Weeds continuously threaten the farming industry by competing with cultivated plants for nutrition resources and dramatically decreasing crop productivity [1, 2]. Despite recent technological advances, there are still diverse challenges for developing effective targeted herbicides. A significant challenge is to create herbicides that are selective to the crops [3]. The significant symmetry between crops and weeds, especially for sites of herbicide action, complicates the discovery process. Moreover, resistance issues need extensive research because there are no herbicides with a new mode of action that have been on the market for decades [4,5,6]. Most studies of herbicide resistance have focused on the biochemical mechanisms of target and non-target proteins. Little attention has been paid to the effects of the chemical structure properties of the inhibitor molecule on the development of resistance [7, 8].
The successful design of new herbicides depends on the careful consideration of several factors, including the choice of the target enzyme, the design of the inhibitor, the delivery of the inhibitor to the target, and its metabolic fate [9,10,11]. In herbicide development, synthetic chemistry plays an important role in the chemical modification of active products already known as herbicides [12]. Amide products are deterministic organic compounds with many biological activities. The amide bond stability derives from synthetic chemicals to prepare such compounds based on this function. Some derivatives of amides are known to illustrate specific biological properties, including herbicidal, antimicrobial, anticancer, and antihistamine activities [13, 14]. Reacting chloroacetyl chloride with various amines produces chloroacetamide derivatives with potential herbicidal properties. The substitution of the aromatic ring of an aromatic amine or aldehyde affects the yield and nature of the final product [15]. Overall, binding and affinity, physical and chemical properties, and synthetic costs for designing novel active compounds based on the binding sites of the target are the primary concerns in the discovery of a target-based herbicide [6].
A group of successful herbicides, classified as group-specific reagents, is the chloroacetamide herbicides. These herbicides contain reactive chlorine, which is a common feature of many known protein modification agents. The main target of such compounds is the inhibition elongation of Very Long Chain Fatty Acid Synthesis (VLCFAs) located in the endoplasmic reticulum. The absence of this protein in the cell and lack of the cuticle waxes consequently loss of membrane stability and leakage, leading to the death of the herbicide-treated plants [16]. Before emergence, chloroacetamide compounds typically affect susceptible weeds (annual grasses and some small-seeded broadleaf weeds) but do not prevent seed germination. The primary absorption and action site of these herbicides on broadleaf species is the roots, while that on grass species is the emerging shoot [17]. In the last few years, the introduction of new chloroacetamide and oxyacetamide herbicides, such as dimethenamid, defense, and flufenacet, has shown that this herbicide class is still going strong in agricultural applications in maize and rice [18,19,20,21]. Therefore, challenges remain for the development of new chloroacetamide herbicides.
In silico studies include pharmacophore mapping, virtual screening, and docking, which are the rational methods for identifying novel hits or leads with diverse chemical scaffolds [22, 23]. As mentioned before, the pharmacophore combines steric and electrostatic characteristics of different compounds that are necessary to ensure optimal supramolecular interactions with a specific structure and trigger or block its biological effects. Molecular docking is widely used to suggest the binding modes of protein inhibitors.
Therefore, the present study is based on synthesizing new chloroacetamide derivatives containing important biological moieties and screening them to evaluate their potential activity against some economic weeds. Herein, the skeleton structure was obtained by replacing the aliphatic oxygenated bridge in chloroacetamide herbicides such as acetochlor, alachlor, and s-metolachlor with aromatic part of commercial herbicides such as phenol moiety of bromoxynil, methoxyphenyl of anisuron, and 2,4-dichlorophenyl of 2,4-D (Fig. 1). The chloroacetamides are done by forming the Schiff base mechanism followed by chloroacetylation. The spectroscopic characterizations of the synthesized derivatives were examined. The synthetic products were compared to acetochlor as a standard herbicide for evaluating their herbicidal activity against two broadleaf weeds and two grass weeds. The computational studies included pharmacophore modeling, and molecular docking. The achieved pharmacophore model can deliver a rational default hypothetical of the primary chemical properties accountable for biological activity and is expected to afford practical information to develop potential new candidates. The results obtained were further supported by molecular docking studies using enzyme VLCFAs to explore the potential binding methods.

The design strategy of novel chloroacetamide derivatives (1b-22b) containing different aromatic moieties
Materials and methods
General methods
Melting points were determined in open glass capillaries using a Griffin melting point apparatus. All spectra of the synthetic compounds were identified and confirmed by 1H-NMR and 13C-NMR spectra using a Bruker NMR 400 MHz (Bruker Biospin, Germany). Deuterated DMSO was used as a solvent. The data were reported as chemical shifts (δ, ppm) relative to tetramethylsilane (TMS) as an internal standard. Molecular weight was determined using an electron impact mass spectrometer (EIMS) at Al-Azhar University, Cairo, Egypt. The relative intensity (%) corresponding to the most characteristic fragments was recorded.
Synthesis of chloroacetamide derivatives
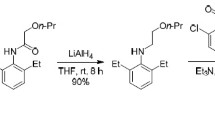
The same strategy for the synthetic method of the typical commercial herbicides (acetochlor, metolachlor, and s-metolachlor) was used for the synthesis of chloroacetamide derivatives based on the formation of Schiff bases followed by chloroacetylation of imines (Fig. 2) [24]. Twenty two Schiff bases (1a–22a, Fig. 2) were synthesized according to Zhu et al. [25] To each amine product dissolved in 20 mL ethyl or methyl alcohol in the 50 mL dry flask, 0.01 mol of the corresponding aldehyde (salicylaldehyde, anisaldehyde, 2,4-dichlorobenzaldehyde) was added dropwise. After the complete addition of aldehyde, 1 mL of glacial acetic acid was added to the reaction mixture and stirred using a magnetic stirrer at room temperature (25 °C) for 10–20 min. The solvent was removed under reduced pressure. The crude product was washed by ether, affording the Schiff base derivative. Afterwards, the imine product (0.01 mol) was dissolved in dichloromethane and cooled at 0–5 °C using ice-water bath. 0.02 mol of chloroacetyl chloride prepared in dichloromethane was added dropwise to the above mixture. The mixtures were stirred for 6 h in water ice mixture and for a further 3 h at room temperature. The solvent was evaporated under reduced pressure to obtain the product. The products were washed with water and crystallized in ethanol or methanol [26].

Synthetic route of synthesized chloroacetamide derivatives (1–22)
2-chloro-N-(2-hydroxybenzyl)-N-(2-hydroxyphenyl)acetamide (1b)
A yellow powder; yield 30%; mp 235–236 °C. 1H-NMR (DMSO, δ/ppm): 4.33 (s, 2H, chloroacetyl), 4.71 (s, 2H, NCH2), 7.25 (m, 2H, hydroxybenzyl, and 2H, hydroxyphenyl), 7.28–7.32 (d, 1H, hydroxyphenyl and 1H, hydroxybenzyl), 7.86–7.88 (d, 1H, hydroxyphenyl and 1H, hydroxybenzyl), and 9.85 (s, 2H, OH of phenyl and OH of benzyl). 13C-NMR (DMSO, δ/ppm): 42.2 (NCH2), 43.7 (CH2, chloroacetyl), 119.42 (C3, hydroxybenzyl), 122.1 (C3, hydroxyphenyl), 123.5 (C5, hydroxybenzyl), 124.5 (C5, hydroxyphenyl), 125.9 (C4, hydroxyphenyl), 126.1 (C4 and C6, hydroxybenzyl), 126.9 (C6, hydroxyphenyl), 130.1 (C1, hydroxyphenyl), 141.9 (C2, hydroxyphenyl), 148.1 (C2, hydroxybenzyl) and 165.5 (C=O). EIMS, m\z (relative abundance, %): 45.30 (22.47); 48.73 (100.00); 162.35 (80.95); 163.06 (59.62); 185.36 (67.33); 275.54 (39.60); 286.14 (13.08); 289.54 (32.28); 291.75. (M+.) (22.22); Anal. Calc. for C15H14ClNO3 was 291.73 and found 291.75.
2-chloro-N-(2-hydroxybenzyl)-N-(4-hydroxyphenyl)acetamide (2b)
A pale-yellow powder, yield 54%; mp 176–177 °C. 1H-NMR (DMSO, δ/ppm): 4.12 (s, 2H, chloroacetyl), 4.28 (s, 2H, NCH2), 6.855–6.860 (d, 2H, hydroxyphenyl), 6.872–6.877 (d, 3H, hydroxybenzyl), 7.169–7.174 (m, 2H, hydroxybenzyl), 7.186–7.190 (d, 2H, hydroxyphenyl), and 9.67 (s, 2H, OH of phenyl and OH of benzyl). 13C-NMR (DMSO, δ/ppm): 41.9 (NCH2), 43.3 (CH2, chloroacetyl), 116.6 (C3 and C5, hydroxybenzyl), 122.5 (C3 and C5, hydroxyphenyl), 124.8 (C2 and C6 of hydroxyphenyl and C1, C4, and C5 of hydroxybenzyl), 129.1 (C1, hydroxyphenyl), 157.7 (C2 of hydroxyphenyl and C2 of hydroxybenzyl) and 169.2 (C=O). EIMS, m\z (relative abundance, %):211.84 (43.49), 212.96 (100.00), 281.47 (24.80), 286.74 (6.53), 291.42 (M+.) (15.49); Anal. Calc. for C15H14ClNO3 was 291.73 and found 291.42.
2-chloro-N-(2-hydroxybenzyl)-N-(3,5-dichloro-4 hydroxyphenyl)acetamide (3b)
A pale brown crystal, yield 20%; mp 284–285 °C. 1H-NMR (DMSO, δ/ppm): 4.38 (s, 4H, chloroacetyl and NCH2), 6.956–6.976 (d, 1H, hydroxybenzyl), 6.990–7.117 (m, 2H, hydroxybenzyl), 7.23 (s, 2H, hydroxyphenyl), 7.35–7.37 (d, 1H, hydroxybenzyl), and 8.28 (s, 2H, OH of phenyl and OH of benzyl). 13C-NMR (DMSO, δ/ppm): 41.9 (NCH2), 42.18 (CH2, chloroacetyl), 119.4 (C3, hydroxybenzyl), 122.1 (C5, hydroxybenzyl), 123.5 (C2 and C6, hydroxyphenyl), 125.9 (C3 and C5, hydroxybenzyl), 126.1 (C4 and C6, hydroxybenzyl), 126.9 (C1, hydroxybenzyl), 130.1 (C1, hydroxyphenyl), 141.9 (C2, hydroxyphenyl), 148.1 (C2, hydroxybenzyl) and 165.5 (C=O). EIMS, m\z (relative abundance, %): 99.65 (96.52), 164.56 (100.00), 299.71 (64.81), 358.04 (50.35), 360.22 (M+.) (43.43); Anal. Calc. for C15H12Cl3NO3 was 360.62 found 360.22.
2-chloro-N-(2-hydroxybenzyl)-N-(o-tolyl)acetamide (4b)
A dark brownish to red powder, yield 85%; mp 192–193 °C. 1H-NMR (DMSO, δ/ppm): 2.36 (s, 3H, –CH3), 4.47 (s, 2H, chloroacetyl), 5.22 (s, 2H, NCH2), 7.02–7.06 (d, 1H, hydroxybenzyl), 7.35–7.38 (m, 2H, hydroxybenzyl), 7.44–7.47 (d, 1H, phenyl), 7.49–7.55 (m, 2H, phenyl), 7.65–7.68 (d, 1H, hydroxybenzyl), 7.74–7.76 (d, 1H, phenyl), and 10.27 (s, OH). 13C-NMR (DMSO, δ/ppm): 17.2 (CH3), 41.38 (NCH2), 49.1 (CH2, chloroacetyl), 117.4 (C3, hydroxybenzyl), 120 (C6, phenyl), 122.7 (C5, hydroxybenzyl), 123.8 (C5, phenyl), 127.7 (C4, hydroxybenzyl), 129.1 (C6, hydroxybenzyl), 129.8 (C1, hydroxybenzyl), 130.5 (C4, phenyl), 131.9 (C3, phenyl), 132.3 (C2, phenyl), 136.9 (C1, phenyl), 161.2 (C2, hydroxybenzyl) and 164 (C=O). EIMS, m\z (relative abundance, %): 209.55 (56.38), 210.34 (100.00), 211.19 (96.10), 285.96 (2.13), 289.77 (M+.) (12.07); Anal. Calc. for C16H16ClNO2 was 289.76 found 289.77.
2-chloro-N-(2-hydroxybenzyl)-N-(p-tolyl)acetamide (5b)
A yellow crystal, yield 46%; mp 165–166 °C. 1H-NMR (DMSO, δ/ppm): 2.36 (s, 3H, –CH3), 4.22 (s, 4H, CH2 of chloroacetyl and NCH2), 7.12–7.15 (m, 2H, hydroxybenzyl), 7.27–7.29 (d, 2H, hydroxybenzyl), 7.45–7.47 (d, 2H, phenyl), 7.64–7.66 (d, 2H, phenyl), and 10.26 (s, OH). 13C-NMR (DMSO, δ/ppm): 20.8 (CH3), 41.9 (NCH2), 43.9 (CH2, chloroacetyl), 120 (C3 and C5, hydroxybenzyl), 129.7 (C1, C4, and C6 of hydroxybenzyl and C3 and C5 of hydroxyphenyl), 133.6 (C1, C2, C4, and C6 of hydroxyphenyl), 136.2 (C2, hydroxybenzyl) and 165.1 (C=O). EIMS, m\z (relative abundance, %): 209.55 (56.38), 210.34 (100.00), 211.19 (96.10), 285.96 (2.13), 289.77 (93.68), 65.90 (86.95), 255.92 (100.00), 289.92 (M+.) (49.47), 292.08 (M+2.) (33.81); Anal. Calc. for C16H16ClNO2 was 289.76 found 289.92.
2-chloro-N-(4-chlorophenyl)-N-(2-hydroxybenzyl)acetamide (6b)
A white crystal, yield 45%; mp 294–295 °C. 1H-NMR (DMSO, δ/ppm): 4.63 (s, 4H, CH2 of chloroacetyl and NCH2), 7.39–7.51 (m, 2H, hydroxybenzyl), 7.51–7.64 (d, 4H, phenyl), 7.64–7.66 (d, 2H, hydroxybenzyl), and 9.01 (s, OH). 13C-NMR (DMSO, δ/ppm): 41.9 (NCH2), 43.3 (CH2, chloroacetyl), 119.7 (C3 and C5, hydroxybenzyl), 128.4 (C2 and C6 of hydroxyphenyl and C1, C4, and C6 of hydroxybenzyl), 133.1 (C1, C3, C4, and C5 of hydroxyphenyl), 134.2 (C2, hydroxybenzyl) and 164.2 (C=O). EIMS, m\z (relative abundance, %): 80.17 (70.77), 92.31 (62.48), 120.07 (100.00), 226.72 (59.94), 270.15 (28.24), 295.01 (70.75), 305.13 (50.10), 310.80 (M+.) (26.50); Anal. Calc. for C15H13Cl2NO2 was 310.17 found 310.80.
2-chloro-N-(2-chloro-4-methylphenyl)-N-(2-hydroxybenzyl)acetamide (7b)
A white crystal, yield 55%; mp 118–119 °C. 1H-NMR (DMSO, δ/ppm): 2.30 (s, 3H, CH3), 4.22 (s, 2H, chloroacetyl), 4.36 (s, 2H, NCH2), 7.16–7.18 (m, 2H, hydroxybenzyl), 7.36 (s, 1H, phenyl), 7.56–7.58 (d, 4H, hydroxybenzyl and phenyl), and 9.83 (s, OH). 13C-NMR (DMSO, δ/ppm): 20.6 (CH3), 41.9 (NCH2), 43.4 (CH2, chloroacetyl), 126.5 (C3 and C5, hydroxybenzyl), 127.1 (C5 and C6, phenyl), 128.6 (C1, C4, and C6, hydroxybenzyl), 130.2 (C2 and C3, phenyl), 132 (C1 and C4, phenyl), 137.6 (C2, hydroxybenzyl) and 165.6 (C=O). EIMS, m\z (relative abundance, %): 77.19 (100.00), 191.30 (90.04), 317.26 (81.49), 319.24 (88.99), 321.05 (49.40), 324.20 (M+.) (88.99); Anal. Calc. for C16H15Cl2NO2 was 324.20 found 324.20.
2-chloro-N-(2-hydroxybenzyl)-N-(3,5-dimethylphenyl)acetamide (8b)
A pale brown crystal, yield 60%; mp 144–145 °C. 1H-NMR (DMSO, δ/ppm): 2.25 (s, 6H, –CH3, –CH3), 4.23 (s, 4H, CH2 of chloroacetyl and NCH2), 6.42–6.46 (m, 2H, hydroxybenzyl), 6.48–6.50 (d, 2H, hydroxybenzyl), 6.74 (s, 1H, phenyl), 7.21 (s, 2H, phenyl), and 10.16 (s, OH). 13C-NMR (DMSO, δ/ppm): 21.5 (CH3 and CH3); 44.1 (NCH2), 62.29 (CH2, chloroacetyl), 117.5 (C3 and C5 of hydroxybenzyl and C2 and C6 of phenyl), 117.7 (C4, phenyl), 125.42 (C4 and C6, hydroxybenzyl), 125.8 (C1, hydroxybenzyl), 138.1 (C3 and C5, phenyl), 138.3 (C1, phenyl), 138.8 (C2, hydroxybenzyl) and 164.9 (C=O). EIMS, m\z (relative abundance, %): 67.93 (84.76), 101.17 (100.00), 263.62 (87.56), 301.31 (27.76), 302.23 (77.11), 303.50 (M+.) (38.47); Anal. Calc. for C17H18ClNO2 was 303.79 found 303.50.
2-chloro-N-(2,6-dimethylphenyl)-N-(2-hydroxybenzyl)acetamide (9b)
A golden to black crystal, yield 85%; mp 104–105 °C. 1H-NMR (DMSO, δ/ppm): 2.15 (s, 6H, –CH3, –CH3), 4.31 (s, 4H, CH2 of chloroacetyl and NCH2), 7.08–7.10 (m, 2H, hydroxybenzyl), 7.15–7.16 (d, 2H, hydroxybenzyl), 7.47–7.54 (m, 1H, phenyl), 7.65–7.67 (d, 1H, phenyl), 8.68–8.70 (d, 1H, phenyl), and 9.79 (s, OH). 13C-NMR (DMSO, δ/ppm): 16.2 (CH3 and CH3), 42.4 (NCH2), 44.8 (CH2, chloroacetyl), 114.1 (C3, hydroxybenzyl), 123.5 (C5 of hydroxybenzyl and C4 of phenyl), 124.5 (C3 and C5, phenyl), 125.9 (C1, hydroxybenzyl), 126.9 (C4 and C6, hydroxybenzyl), 130.1 (C2 and C6, phenyl), 136.1 (C1, phenyl), 147.2(C2, hydroxybenzyl) and 166 (C=O). EIMS, m\z (relative abundance, %): 55.23 (43.44), 56.25 (100.00), 77.31 (67.17), 132.24 (26.86), 160.19 (34.39), 303.06 (M+.) (11.36); Anal. Calc. for C17H18ClNO2 was 303.79 found 303.06.
2-chloro-N-(2,5-dichlorophenyl)-N-(2-hydroxybenzyl)acetamide (10b)
An off-white crystal, yield 40%; mp 111–112 °C. 1H-NMR (DMSO, δ/ppm): 4.42 (s, 4H, CH2 of chloroacetyl and NCH2), 7.31–7.33 (m, 2H, hydroxybenzyl), 7.57–7.59 (d, 2H, hydroxybenzyl), 7.70–7.72 (d, 2H, phenyl), 7.90 (s, 1H, phenyl), and 10.01 (s, OH). 13C-NMR (DMSO, δ/ppm): 42 (NCH2), 55.9 (CH2, chloroacetyl), 115.2 (C3, hydroxybenzyl), 120.2 (C5, hydroxybenzyl), 122.8 (C6, phenyl), 123.2 (C4, phenyl), 125 (C4 and C6, hydroxybenzyl), 129 (C1, hydroxybenzyl), 132.9 (C5, phenyl), 135.6 (C2, phenyl), 136.8 (C1, phenyl), 160 (C2, hydroxybenzyl) and 166 (C=O). EIMS, m\z (relative abundance, %): 90.94 (85.99), 184.55 (100.00), 264.16 (58.75), 300.92 (76.29), 324.03 (38.93), 344.62 (M+.) (55.59); Anal. Calc. for C15H12Cl3NO2 was 344.62 found 344.20.
2-chloro-N-(2-hydroxybenzyl)-N-(4-methoxyphenyl)acetamide (11b)
A darkish brown to black powder, yield 80%; mp 129–130 °C. 1H-NMR (DMSO, δ/ppm): 3.75 (s, 3H, OCH3), 4.40 (s, 4H, CH2 of chloroacetyl and NCH2), 7.01–7.03 (d, 2H, methoxyphenyl), 7.33–7.36 (m, 2H, hydroxybenzyl), 7.45–7.47 (d, 2H, hydroxybenzyl), 7.58–7.62 (d, 2H, methoxyphenyl), and 10.27 (s, OH). 13C-NMR (DMSO, δ/ppm): 42 (NCH2), 55.9 (CH2, chloroacetyl), 56.1 (–OCH3), 115.3 (C3 and C5, methoxyphenyl), 117.8 (C3, hydroxybenzyl), 119.8 (C5, hydroxybenzyl), 123 (C2, methoxyphenyl), 123.4 (C6, methoxyphenyl), 124.7 (C4, hydroxybenzyl), 125 (C1 and C6, hydroxybenzyl), 129.2 (C1, methoxyphenyl), 159.2 (C2, hydroxybenzyl), 161.9 (C4, methoxyphenyl) and 165.8 (C=O). EIMS, m\z (relative abundance, %): 71.76 (58.47), 153.45 (100.00), 221.66 (51.53), 302.27 (11.81), 305.95 (M+.) (24.93); Anal. Calc. for C16H16ClNO3 was 305.76 found 305.95.
2-chloro-N-(2-hydroxybenzyl)-N-(naphthalen-1-yl)acetamide (12b)
White to grey flakes, yield 80%; mp 160–161 °C. 1H-NMR (DMSO, δ/ppm): 4.47 (s, 4H, CH2 of chloroacetyl and NCH2), 7.51–7.55 (m, 1H, hydroxybenzyl), 7.57–7.60 (m, 3H, hydroxybenzyl and naphthyl), 7.69–7.70 (d, 2H, hydroxybenzyl), 7.82–7.84 (d, 2H, naphthyl), 7.97–7.99 (m, 1H, naphthyl), 8.06–8.08 (d, 2H, naphthyl), and 10.31 (s, OH). 13C-NMR (DMSO, δ/ppm): 43.8 (NCH2 and CH2 of chloroacetyl), 122.4 (C2, naphthyl), 123 (C3, hydroxybenzyl), 126.1 (C4, naphthyl), 126.4 (C5 of hydroxybenzyl and C8 of naphthyl), 125.5 (C8a, naphthyl), 126.6 (C3, C6, and C7, naphthyl), 128.2 (C4 and C6, hydroxybenzyl), 128.7 (C5 of naphthyl and C1 of hydroxybenzyl), 133.3 (C4a, naphthyl), 134.2 (C1 of naphthyl and C2 of hydroxybenzyl) and 166.1 (C=O). EIMS, m\z (relative abundance, %):146.31 (87.02), 210.0 (100.00), 239.16 (86.37), 324.66 (40.51), 325.87 (M+.) (2.27); Anal. Calc. for C19H16ClNO2 was 325.79 found 325.87.
2-chloro-N-(2-hydroxyphenyl)-N-(4-methoxybenzyl)acetamide (13b)
A pale yellow powder, yield 60%; mp 172–174 °C. 1H-NMR (DMSO, δ/ppm): 1.76 (s, 3H, –OCH3), 1.91 (s, 2H, chloroacetyl), 3.87 (s, 2H, NCH2), 6.73–6.75 (d, 1H, hydroxyphenyl)), 7.27–7.40 (d, 5H, methoxyphenyl and hydroxyphenyl), 7.40–7.52 (m, 2H, hydroxyphenyl), and 10.76 (s, 1H, OH). 13C-NMR (DMSO, δ/ppm): 44.1 (CH2 of chloroacetyl and NCH2), 62.2 (–OCH3), 117.5 (C3 and C5, methoxyphenyl), 117.7 (C3, C5, and C6, hydroxyphenyl), 125.4 (C4, hydroxyphenyl), 125.8 (C1 of methoxyphenyl and C1 of hydroxyphenyl), 138.1 (C2 and C6, methoxyphenyl), 138.3 (C2, hydroxyphenyl), 138.8 (C4, methoxyphenyl) and 165.1 (C=O). EIMS, m\z (relative abundance, %): 51.37 (65.16), 134.14 (68.26), 139.66 (52.51), 153.43 (77.16), 175.13 (100.00), 218.82 (64.77), 301.73 (38.45), 305.87 (M+.) (5.43); Anal. Calc. for C16H16ClNO3 was 305.76 found 305.87.
2-chloro-N-(4-hydroxyphenyl)-N-(4-methoxybenzyl)acetamide (14b)
A darkish brown powder, yield 75%; mp 202–204 °C. 1H-NMR (DMSO, δ/ppm): 3.69 (s, 2H, chloroacetyl), 3.87 (s, 2H, NCH2), 3.94 (s, 3H, –OCH3), 6.85–6.88 (d, 2H, methoxyphenyl), 6.94–7.97 (d, 2H, hydroxyphenyl), 7.13–7.15 (d, 2H, methoxyphenyl), 7.19–7.21 (d, 2H, hydroxyphenyl), and 9.17 (s, 1H, OH). 13C-NMR (DMSO, δ/ppm): 41.6 (CH2, chloroacetyl), 56.2 (NCH2), 56.4 (–OCH3); 115.4 (C3 and C5, methoxyphenyl), 116.5 (C3 and C5, hydroxyphenyl), 122.8 (C2 and C6, hydroxyphenyl), 123.0–124.8 (C1, methoxyphenyl), 130.1 (C2 and C6, methoxyphenyl), 132.3 (C1, hydroxyphenyl), 157.6 ( C4 of hydroxyphenyl and C4 of methoxyphenyl) and 164.7 (C=O). EIMS, m\z (relative abundance, %): 49.90 (57.88), 101.17 (100.00), 210.19 (51.84), 252.39 (58.07), 272.96 (41.90), 275.72 (31.50), 301.31 (26.56), 303.32(76.13); 305.50 (M+.) (37.43); Anal. Calc. for C16H16ClNO3 was 305.76 found 305.50.
2-chloro-N-(3,5-dichloro-4-hydroxyphenyl)-N-(4 methoxybenzyl)acetamide (15b)
A pale yellow to brown crystal, yield 20%; mp 65–66 °C. 1H-NMR (DMSO, δ/ppm): 1.80 (s, 3H, –OCH3), 4.17 (s, 2H, chloroacetyl), 4.30 (s, 2H, NCH2), 6.92–6.93 (d, 4H, methoxyphenyl), 7.27 (s, 1H, hydroxyphenyl), 7.39 (s, 1H, hydroxyphenyl), and 10.68 (s, 1H, OH). 13C-NMR (DMSO, δ/ppm): 41.8 (CH2, chloroacetyl), 43.6 (NCH2), 56.2 (–OCH3), 115.1 (C3 and C5, methoxyphenyl), 122.8 (C2 and C6, hydroxyphenyl), 126.1 (C3 and C5, hydroxyphenyl), 128.7 (C1, methoxyphenyl), 130.1 (C2 and C6, methoxyphenyl), 132.4 (C1, hydroxyphenyl), 133.1 (C4, hydroxyphenyl), 134.2 (C4, methoxyphenyl) and 169 (C=O). EIMS, m\z (relative abundance, %):146.04 (100.00), 154.18 (68.17), 187.93 (66.11), 336.41 (86.12), 343.44 (56.32), 374.70 (M+.) (64.20), 375.74 (M+1.) (23.09); Anal. Calc. for C16H14Cl3NO3 was 374.64 found 374.70.
2-chloro-N-(4-methoxybenzyl)-N-(p-tolyl)acetamide (16b)
A yellow powder, yield 80%; mp 103–104 °C. 1H-NMR (DMSO, δ/ppm): 2.32 (s, 3H, –OCH3), 3.93 (s, 2H, chloroacetyl), 4.29 (s, 2H, NCH2), 7.12–7.14 (d, 3H, methoxyphenyl and tolyl), 7.51–7.53 (d, 1H, tolyl), 7.76–7.78 (d, 1H, tolyl), 7.87–7.89 (d, 2H, methoxyphenyl), and 8.49–8.51 (d, 1H, tolyl). 13C-NMR (DMSO, δ/ppm): 21 (CH3), 44 (CH2, chloroacetyl), 56.2 (NCH2), 56.6 (–OCH3), 115.7 (C3 and C5, methoxyphenyl), 119.7 (C1, methoxyphenyl), 121.4 (C3 and C5, tolyl), 130.5 (C2 and C6, methoxyphenyl), 135.7 (C2 and C6, tolyl), 136.6 (C4, tolyl), 138.1 (C1, tolyl), 139.1 (C4, methoxyphenyl) and 164.7 (C=O). EIMS, m\z (relative abundance, %): 50.70 (70.29), 156.80 (75.23), 197.02 (100.00), 285.26 (70.99), 301.57 (50.17), 303.87 (M+.) (45.64); Anal. Calc. for C17H18ClNO2 was 303.79 found 303.87.
2-chloro-N-(4-chlorophenyl)-N-(4-methoxybenzyl)acetamide (17b)
A yellow powder, yield 82%; mp 182–183 °C. 1H-NMR (DMSO, δ/ppm): 3.87 (s, 3H, –OCH3), 3.90 (s, 2H, chloroacetyl), 4.29 (s, 2H, NCH2), 7.13–7.15 (d, 2H, methoxyphenyl), 7.39–7.41 (d, 2H, methoxyphenyl), 7.54–7.56 (d, 2H, phenyl), and 7.87–7.89 (d, 2H, methoxyphenyl). 13C-NMR (DMSO, δ/ppm): 26.6 (NCH2), 42 (CH2, chloroacetyl), 43.8 (–OCH3), 120.9 (C3 and C5, methoxyphenyl), 124.4 (C2 and C6, phenyl), 127.3 (C2 and C6, methoxyphenyl), 129 (C3 and C5, phenyl), 130.6 (C1, methoxyphenyl), 136.1 (C4, phenyl), 137 (C1, phenyl), 141.1 (C4, methoxyphenyl) and 165.2 (C=O). EIMS, m\z (relative abundance, %): 62.24 (91.23), 77.29 (98.33), 100.44 (100.00), 233.13 (64.86), 323.88 (20.77), 324.88 (M+.) (21.19); Anal. Calc. for C16H15Cl2NO2 was 324.20 found 324.88.
2-chloro-N-(2-chloro-4-methylphenyl)-N-(4-methoxybenzyl)acetamide (18b)
A white crystal, yield 50%; mp 115–116 °C. 1H-NMR (DMSO, δ/ppm): 2.3 (s, 6H, –CH3, –OCH3), 4.36 (s, 4H, CH2 of chloroacetyl and NCH2), 7.16–7.18 (d, 2H, methoxyphenyl), 7.36–7.38 (d, 2H, methoxyphenyl), 7.56–7.58 (d, 2H, tolyl), and 9.81 (s, 1H, tolyl). 13C-NMR (DMSO, δ/ppm): 20.6 (–CH3 and –OCH3), 43.5 (NCH2 and CH2 of chloroacetyl), 126.5 (C3 and C5, methoxyphenyl), 127.1 (C5 and C6, tolyl), 128.6 (C3 of tolyl and C1, C2, and C6 of methoxyphenyl), 130.1 (C2, tolyl), 130.2 (C4, tolyl), 132 (C1, tolyl), 137.2 (C4, methoxyphenyl) and 165.6 (C=O). EIMS, m\z (relative abundance, %): 118.11 (59.20), 170.74 (73.52), 326.93 (98.63), 335.24 (100.00), 336.91 (19.24), 338.48 (M+.) (37.24); Anal. Calc. for C16H15Cl2NO2 was 338.23 found 338.48.
2-chloro-N-(3,5-dimethylphenyl)-N-(4-methoxybenzyl)acetamide (19b)
A white to pale greenish powder, yield 57%; mp 137–138 °C. 1H-NMR (DMSO, δ/ppm): 2.25 (s, 9H, –CH3, –CH3, –OCH3), 4.23 (s, 4H, CH2 of chloroacetyl and NCH2), 6.74 (s, 1H, tolyl), 7.21 (s, 2H, tolyl), 7.32–7.34 (d, 2H, methoxyphenyl), and 7.36–7.38 (d, 2H, methoxyphenyl). 13C-NMR (DMSO, δ/ppm): 21.6 (CH3, CH3, –OCH3), 44.1 (NCH2, and CH2, chloroacetyl), 117.5 (C3 and C5 of methoxyphenyl and C2 and C6 of tolyl), 125.8 (C4 of tolyl and C1 of methoxyphenyl), 138.3 (C2 and C6 of methoxyphenyl and C3 and C5 of tolyl), 138.8 (C1 of tolyl and C4 of methoxyphenyl) and 165 (C=O). EIMS, m\z (relative abundance, %): 63.24 (91.23), 77.29 (98.33), 100.44 (100.00), 233.13 (64.86), 264.48 (59.89), 308.72 (25.24); 319.08 (M+.) (32.69); 323.88 (M+4.) (20.77); 324.88 (M+5.) (21.19); Anal. Calc. for C18H20ClNO2 was 317.81 found 319.08.
2-chloro-N-(4-methoxybenzyl)-N-(naphthalen-1-yl)acetamide (20b)
A darkish brown powder, yield 30%; mp 110–111 °C. 1H-NMR (DMSO, δ/ppm): 3.88 (s, 2H, chloroacetyl), 4.29 (s, 3H, –OCH3), 4.44 (s, 1H, NCH2), 7.13–7.16 (d, 1H, methoxyphenyl), 7.55–7.57 (d, 1H, methoxyphenyl), 7.58–7.61 (m, 2H, naphthyl), 7.68–7.70 (d, 2H, methoxyphenyl), 7.82–7.84 (d, 1H, naphthyl), 7.88–7.90 (d, 1H, naphthyl), 7.96–7.99 (m, 1H, naphthyl), and 8.08–8.10 (d, 2H, naphthyl). 13C-NMR (DMSO, δ/ppm): 41.9 (CH2, chloroacetyl), 43.7 (NCH2), 55.43 (–OCH3), 113.9 (C2, naphthyl), 115 (C3 and C5, methoxyphenyl), 122.8 (C4 and C8, naphthyl), 122.9 (C7 and C8a, naphthyl), 126.1 (C6, naphthyl), 126.60 (C3, naphthyl), 126.7 (C1 of methoxyphenyl and C5 of naphthyl), 128.4 (C2 and C6, methoxyphenyl), 132.4 (C4a, naphthyl), 133.1 (C1, naphthyl), 134.1 (C4, methoxyphenyl) and 169.2 (C=O). EIMS, m\z (relative abundance, %): 76.52 (38.88), 114.03 (28.36), 115.08 (100.00), 142.40 (40.31), 143.23 (73.45), 338.21 (4.25); 339.79 (M+.) (16.61); Anal. Calc. for C20H18ClNO2 was 339.82 found 339.79.
2-chloro-N-(2,4-dichlorobenzyl)-N-(3,5-dimethylphenyl)acetamide (21b)
A white crystal, yield 77%; mp 141–142 °C. 1H-NMR (DMSO, δ/ppm): 2.25 (s, 6H, –CH3, –CH3), 4.23 (s, 4H, CH2 of chloroacetyl and NCH2), 6.74 (s, 1H, phenyl), 7.21 (s, 2H, phenyl), 7.63–7.66 (d, 1H, benzyl), 7.87–7.90 (d, 1H, benzyl) and 10.16 (s, 1H, benzyl). 13C-NMR (DMSO, δ/ppm): 21.5 (CH3 and CH3), 44.1 (NCH2 and CH2 of chloroacetyl), 117.6 (C2 and C6, phenyl), 125.8 (C3, C5, and C6 of benzyl and C4 of phenyl), 138.3 (C2 and C4 of benzyl and C3 and C5 of phenyl), 138.8 (C1 of benzyl and C1 of phenyl) and 164.9 (C=O). EIMS, m\z (relative abundance, %): 210.58 (72.67), 217.97 (69.54), 227.81 (67.25), 264.53 (100.00), 356.06 (M+.) (28.24), 358.25 (M+2.) (21.98); Anal. Cal. for C20H18Cl2NO2 was 356.67 found 356.06.
2-chloro-N-(2,4-dichlorobenzyl)-N-(2,5-dichlorophenyl)acetamide (22b)
A beige crystal, yield 72%; mp 118–119 °C. 1H-NMR (DMSO, δ/ppm): 4.42 (s, 4H, CH2 of chloroacetyl and NCH2), 7.31–7.33 (d, 2H, benzyl), 7.57–7.59 (d, 2H, phenyl) and 7.90 (s, 2H, phenyl and benzyl). 13C-NMR (DMSO, δ/ppm): 41.1 (CH2, chloroacetyl), 43.6 (NCH2), 120.84 (C6, chlorophenyl), 124.4 (C4, chlorophenyl), 127.3 (C5, chlorobenzyl), 127.8 (C3, chlorophenyl), 128.8 (C6, chlorobenzyl), 129 (C3, chlorobenzyl), 129.3 (C5, chlorophenyl), 130.7 (2C, C2, and C4, chlorobenzyl), 131.5 (C2, chlorophenyl), 136.1 (C1, chlorobenzyl), 141.2 (C1, chlorophenyl) and 167 (C=O). EIMS, m\z (relative abundance, %): 156.42 (45.05), 220.73 (100.00), 247.23 (93.03), 264.23 (88.56), 380.27 (61.44), 397.98 (M+.) (50.97), 399.82 (M+2.) (29.22); Anal. Calc. for C15H10Cl5NO was 397.50 found 397.98.
Herbicidal activity evaluation
An evaluation of herbicidal activity was conducted using the foliar application method for spraying the four weed species; Chenopodium album and Anagallis arvensis as broadleaf weeds, Lolium temulentum and Echinochloa crus-galli as grass weeds. Commercial herbicide acetochlor was obtained from Egyptchem International for Agrochemicals, Egypt, under the trade name of Host Core 90% EC, with an application rate of 2.6 L/hectare. Plants were grown in seedling-growing peat moss trays in (19 × 11 cm2) pots for 7–21 days in a greenhouse with a 15 h at 22–29 °C. Plants were grown to 3 to 5 leaf stages before applications [27]. Stock solutions were prepared by weighing an amount (determined by the highest rate to be tested that calculated as acetochlor rate) of each compound in a 25 mL glass vial and dissolved in 4 mL of acetone/DMSO as a general solution (GS, 97:3, v/v). Each stock solution (5000 mg/L) was diluted with 20 mL of an aqueous mixture composed of containing H2O, GS, isopropanol, Tensiofix D33, and Tween 80 at a ratio of 45:43:11:2.0:0.03 (v/v) to form the spray mixture associated with the highest application rate. Serial dilutions of 12 and 6 mL of the stock solution were used to prepare the low application rates (2500 and 1250 mg/L). Formulated compounds were applied to the weeds with an atomizer nozzle adjusted to deliver 2.6 L/hectare over an application area of 0.5 cm2. The control was sprayed with a mixture of solvent blank. The treated and controlled weeds were kept in a greenhouse and irrigated for 21 days. The chlorophyll content of the treated and the control plants was measured by the chlorophyll meter (SPAD 502). This technique is more suitable than the extraction method, where the total chlorophyll (mg/mg plant tissue) is assessed on the same leaf over time with five replicates [28]. The data were subjected to the probit regression analysis [29] in IBM SPSS software version 25.0 (SPSS, Chicago, IL, USA) [30] for calculation of the EC50 values.
Computational methods
Generation of 3D-pharmacophore models
3D pharmacophore models of the synthesized chloroacetamide derivatives (1b–22b) were generated and compared using Discovery Studio (DS) software [31] after drawing chemical structures of all compounds were drawn by ChemDraw professional 18.2 software and saved in Standard Delay Format (SDF) file to submitted to the DS [32]. Properties such as HBA (hydrogen bond acceptor), HBD (hydrogen bond donor), and AR (aromatic ring) were used to establish reliable pharmacophoric sites. Ten models have been obtained, and the best hypothesis contains a high score of the site points. Finally, the best pharmacophore model was used to map all tested molecules.
Molecular docking
The crystal structure of VLCFAs was obtained from the Protein Data Bank (PDB ID: 1oxh) (http://www.rcsb.org) at 1.54 Å [33] and imported onto the Molecular Operating Environment (MOE) 2014.13 software (Chemical Computing Group Inc, Montreal, Quebec, Canada). The structure of each enzyme was visualized by the MOE [34]. The protein was prepared by removing heteroatoms and crystallographic water molecules. After the protein preparation, the active site was defined based on the volume occupied by the known ligand pose already in an active site [35]. The compounds were converted to the 3D structure and the Merck Molecular Force Field (MMFF94) power was reduced with a 200-iteration limit and the power threshold value of 15 kcal/mol [36]. The triangle-matching algorithm from MOE was used to dock the compounds into the active site of the enzyme. The free energy of binding of the compounds was calculated based on hydrophobic, ionic and hydrogen bond interactions. An enzyme-ligand complex is considered to have an acceptable ligand if its docking score (or interaction energy) is greater than a certain value.
Results and discussion
Chemistry
The target compounds (1b–22b) were synthesized via acylation of imine derivatives (1a–22a) (Fig. 2). The effect of substituent patterns on the yields was investigated. It was found that the different aromatic substituents displayed diverse yields (20–85%) (Table 1). The presence of methyl group on the phenyl ring as shown in compounds 4b, 7b, 8b, 9b, 11b, 16b, 18b, 19b, and 21b gave a high yield (50–85%) except compound 5b (46%) but slowly reaction (≈ 6 h) on the other hand. In contrast, the presence of chlorine substituent on the phenyl ring (3b, 6b, 10b, 15b, and 18b) gave a little yield (20–50%) except compounds 17b and 22b (82% and 72% respectively) with rapid reaction (≈ 1 h). All synthesized compounds were compatible with Lipinski rules (MW, ALogP, HBA, HBD, RB, and PSA) except compounds 21b and 22b with an ALogP value higher than 5 as presented in Table 1. These characteristics proved that the synthesized compounds have adequate hydrophobic to penetrate the biological membranes, as determined by the Lipinski rule-of-five [37, 38]. According to Lipinski's rules, most of these compounds should be readily bioavailable, allowing them to be used as herbicides. Moreover, the Log S values for all derivatives are between -3.14 and -6.62, which indicate moderately to highly soluble in water [39].
The chemical structures of the synthesized compounds were confirmed by 1H-NMR (Supplementary data Figures S1a–S22a), 13C-NMR (Supplementary data Figures S1b–S22b), and MS (Supplementary data Figures S1c–S22c). All 1H-NMR spectra of the derivatives (1b–22b) showed peaks at a chemical shift from 4 to 5 ppm, which corresponded to the hydrogen of –NCH2 and –CH2 of chloroacetyl. In addition, the presence of new peaks by 13C-NMR at a chemical shift from 41 to 43 ppm for the carbon of –NCH2 and from 48 to 58 ppm for –CH2 of chloroacetyl also confirmed the chemical structures of these compounds. The peak at 160 to 170 ppm represents the carbonyl group C=O of synthesized compounds.
Explanation of 3D-pharmacophore models
Table 2 shows the ten top-scored 3D hypothetical pharmacophores generated from the synthesized chloroacetamide derivatives (1b–22b) with information on predictive power. The model of Hypo 1 for this group was the highest score (fit value = 4). The features of this model include two RA, two Hyd, and two HBA with differences in composition, orientation, and sectorial directions (Fig. 3A). These features indicate different numbers of pharmacological properties. Table 3 shows the mapping of compounds 1b–22b on this model. The results proved that the fit values of the compounds ranged from 1.16 to 4. Compound 20b exhibited the highest fit value 4, and the 3D orientation on the best model is shown in Fig. 3B.

A The best pharmacophore model for the synthesized chloroacetamide derivatives (1b–22b). B Mapping of the highest fit compound (20b) on this model of synthesized chloroacetamide derivatives
Herbicidal activity
The herbicidal activity of the synthesized compounds against four weed species including two broadleaf weeds (C. album and A. arvensis) and two grass weeds (L. temulentum and E. crus-galli) in comparison with acetochlor as a standard herbicide are shown in Tables 4, 5, 6, 7. The data are presented as EC50 values in mg/L and their statistical parameters. The results revealed that most of the tested compounds exhibited remarkable inhibition against the chlorophyll content of four weeds. In addition, the herbicidal activity differed according to the substituents on the molecular structure. In general, most of the compounds exhibited higher activity against grass weeds than broadleaf weeds.
Table 4 shows the herbicidal activity of the derivatives against broadleaf weed C. album. Eleven compounds exhibited higher herbicidal activity than the standard acetochlor, with EC50 ranging between 2482 to 3564 mg/L. Compounds 5b and 18b were the most active with EC50 = 2903 and 2482 mg/L, respectively, and superior to the standard (acetochlor). Comparing the results of compound 5b with 6b indicates that the presence of the methyl group at position 4 was much more effective than the presence of chlorine at the same position. Similarly, compound 21b versus compound 22b showed that the presence of methyl groups increased the herbicidal activity more than the chlorine atoms. Fortunately, all the results are in parallel with the standard acetochlor, which contains methyl and ethyl groups, reflecting that the effective substituent has to be alkyl groups (methyl or ethyl) rather than hydroxyl or chlorine atoms. Interestingly, compound 16b versus 17b shows that substituting the methyl group at position 4 is more critical than the chlorine atom. Furthermore, compound 1b versus 4b proves that the alkyl group has the most significant influence on herbicidal activity. However, the hydroxyl group at the same position 4 as shown in compound 2b dramatically reduced the herbicidal activity. Additionally, substitution with chlorine atom at position 2 in the presence of methyl group at position 4 significantly increased the herbicidal activity (16b versus 18b). Compounds 9b, 15b, and 22b presented similar herbicidal activity as obtained by acetochlor with EC50 = 3861, 3765, and 3805 mg/L, respectively. On the other hand, compounds 1b–3b, 10b–13b, and 17b showed lower herbicidal activity than acetochlor. Therefore, it could be concluded that compounds with a hydroxyl group at position 2 or 4 on the phenyl ring (compounds 1b and 2b) or methoxy group at position 4 on the phenyl ring (11b) were less active than the standard.
Table 5 shows the herbicidal activity of the tested derivatives against broadleaf weed, A. arvensis. Seventeen compounds showed higher herbicidal activity than the standard herbicide acetochlor, EC50 = 3890 mg/L, with EC50 ranging from 2614 to 3654 mg/L. Most of these derivatives contain one or two methyl groups such as 5b, 16b, 18b, and 19b with EC50 = 2661, 2998, 2614, and 2977 mg/L, respectively. In addition, all these potent compounds contain one or two methyl groups. For example, compounds 1b and 13b exhibited similar herbicidal activity to acetochlor with EC50 = 3975 and 3887 mg/L, respectively. However, compounds 2b and 3b containing hydroxyl group in their chemical structure showed lower herbicidal activity than the standard herbicide (acetochlor) with EC50 = 5173, 4762, and 4380 mg/L, respectively.
The result of the herbicidal activity against grass weeds (L. temulentum) is shown in Table 6, representing that nine compounds are more effective than the standard. The EC50 values of the test compounds were between 2213 and 3909 mg/L, which are lower than acetochlor (EC50 = 4141 mg/L). However, compounds 4b, 5b, and 12b exhibited herbicidal activity similar to the standard acetochlor with EC50 = 4217, 4070, and 4084 mg/L, respectively. Some compounds e.g. 1b–3b, 7b, 10b–11b, and 13b–16b presented lower activity than acetochlor.
Table 7 shows the herbicidal activity of the tested derivatives against grass weeds (E. crus-galli) compared to the acetochlor (standard). Sixteen compounds exhibited higher herbicidal activity than the standard herbicide acetochlor, with EC50 ranging from 1472 to 2592 mg/L. The most potent compounds were 1b, 4b, 5b, 6b, 8b, 17b, 18b, and 20b. Interestingly, all the compounds showing high herbicidal activity contain methyl groups in their chemical structure. However, compounds 3b, 7b, and 10b–13b showed lower herbicidal activity with a range of EC50 2988–3670 mg/L than acetochlor.
Generally, the results indicated that one or two methyl groups on the phenyl ring increased the inhibition of chlorophyll content. However, more than one hydroxyl group or chlorine atom on the phenyl ring decreased the inhibition. Changing the phenyl ring to a naphthyl moiety significantly increased herbicidal activity, as shown by compound 20b, the fourth compound exhibiting herbicidal activity with an EC50 value of 1896 mg/L. Probably, if the naphthyl moiety is substituted with an alkyl group (methyl or ethyl group) will increase the herbicidal activity. According to the post‐emergence herbicidal activity, the main structure‐activity relationships (SARs) of the compounds 1b–22b can be revealed. The categories of substituents such as hydroxyphenyl, methoxyphenyl, and 2,4-dichlorophenyl rings significantly decreased the herbicidal activity.
The mode of action of the chloroacetamide group is the inhibition biosynthesis of Very Long Chain Fatty Acid Synthesis (VLCFAs) leading to the herbicidal action [16, 19, 21]. Chloroacetamide herbicides act systemically through the lipophilic walls of the phloem tubes [40]. Our obtained results showed that the compounds which contain one or two methyl groups at the phenyl ring exhibited high herbicidal action reflecting a similar effect to the well-known 2-chloroacetamide herbicides [41, 42]. Methoxy benzyl derivatives (13b–20b) showed good herbicidal action, suggesting that the hydrophilicity of the methoxy group may have contributed to the herbicidal activity. These results indicate that the steric factor around the nitrogen atom of 2-chloroacetamide may have played an essential role in the activity of this type of compound [43]. Several derivatives of the tested compounds proved to be more or equally effective herbicides based on comparisons with acetochlor. Consequently, finding one or more of the tested candidates possible can offer good herbicidal efficacy against the tested weed.
Molecular docking
The docking of the synthesized molecules 1b–22b on the main target protein (VLCFAs) was performed using MOE software. The data were analyzed based on the docking score (ΔG, kcal/mol), hydrogen bonds, and van der Waals connections nearby. Table 8 shows the binding scores and binding interactions of the synthesized derivatives. The results showed that the synthesized derivatives have a well binding convergence with the active sites of the target enzyme with docking energy ranging from − 5.57 to − 7.38 kcal/mol compared to -5.11 kcal/mol for acetochlor. Compounds 5b, 6b, 8b, 13b, and 17b showed HBD and hydrophobic interactions together. Compound 20b showed HBA and hydrophobic interactions. However, compounds 19b and 21b showed only HBD and HBA, respectively, without hydrophobic interaction. The other fourteen derivatives exhibited van der Waals and hydrophobic interactions. All compounds have a distance of 2.88- 3.46 between atoms that bind and hydrogens in amino acid residues. However, the distance of the hydrophobic was in the range of 3.61–4.76 Å. No hydrophobic interaction was found in the case of acetochlor. It can be seen that the amino acids Asn 310, Ala 309, and Thr 307 were bound to most of the derivatives.
Figure 4A and B show the optimal 2D and 3D binding interaction diagrams of the most active compound 18b (ΔG = − 6.65 kcal/mol) compared with the standard herbicide acetochlor (Fig. 4C and D). The interactions of compound 18b with the enzyme show that the phenyl ring interacted through a hydrophobic with a gamma-2 carbon atom of the amino acid threonine (CG2-Thr 307- 6-ing, 3.75 Å). In addition, the compound interacted with 15 amino acids of the enzyme by van der Waals. Therefore, according to the molecular docking results, this compound showed the highest herbicidal activity, and it may be considered a promising inhibitor of the VLCFAs enzyme. On the other hand, acetochlor docked into the enzyme through van der Waals (11 amino acids). Also, the oxygen of the carbonyl group has one H-bonding interaction (HBA) with the nitrogen of alanine (N-Ala 309- O5, 3.36 Å) (Fig. 4C).

Docking of the most herbicidal active compound 18b (ΔG = − 6.65 kcal/mol) (A and B) and the most popular standard herbicide acetochlor (ΔG = − 5.11 kcal/mol) (C and D) in the binding site of VLCFAs (PDB ID 1OXH). Left: 2D interaction diagrams of compounds with 1OXH complex and right are the complex structures in 3D
It is well known that molecular docking is a powerful tool to explore the mechanisms of enzyme-herbicide interactions. This technique is often applied to enantioselectivity studies [44,45,46]. Moreover, it was widely applied to screening novel compounds [47]. The bond interactions were helpful for the elucidation of several biological activities of the tested compounds as herbicides [44]. Sartori et al. indicated that histone deacetylase was the target enzyme in plants for the 18 anilide derivatives compared to s-metolachlor [48]. The authors found that through molecular docking, the affinities of the most active anilide compounds for the binding sites of this enzyme were equal to or higher than those calculated for its inhibitors. Filimon et al.[49] added that s-metolachlor as a chloroacetamide herbicide was able to bind to the active sites of dehydrogenase, phosphatase, and protease.
Conclusion
A series of novel chloroacetamide derivatives was synthesized through a facile and practical protocol optimized in the current study. The molecular structures of these compounds were confirmed by spectroscopic methods. A study of pharmacophore modeling revealed that these compounds displayed the highest effect and were able to meet the planned common characteristics locates by aligning them onto the pharmacophore model. The compounds were evaluated for in vitro herbicidal activity against four weed species; C. album and A. arvensis as broadleaf weeds, L. temulentum and E. crus-galli as grass weeds using the determination of chlorophyll content as an indication of the inhibition. A structure–activity relationship (SAR) study identified that the presence of a methyl group on the phenyl ring led to the highest herbicidal activity. Other compounds exhibited moderate activity. The in silico molecular docking of the synthesized compounds proved that the compounds showed an excellent affinity to bind with the active sites of the main target protein (VLCFAs), and the docking energy ranged from − 5.57 to − 7.38 kcal/mol compared to − 5.11 kcal/mol for acetochlor as a standard herbicide. More importantly, this research identified a new class of lead compounds with potential for herbicide development. In addition, the mode of herbicidal action, selectivity, and quantitative structure–activity relationship (QSAR) study for this type of chemical is now being conducted in our laboratory. At present, we are focusing our efforts on optimizing the herbicidal activity of the most promising compounds, and some structure modifications are being investigated that will be described in a future publication.
Availability of data and materials
All data generated or analyzed during this study are included in this article. Also, the related datasets are available from the corresponding author on reasonable request.
References
MacLaren C, Storkey J, Menegat A, Metcalfe H, Dehnen-Schmutz K. An ecological future for weed science to sustain crop production and the environment: a review. Agron Sustain Dev. 2020;40:1–29. https://doi.org/10.1007/s13593-020-00631-6.
Gianessi LP. The increasing importance of herbicides in worldwide crop production. Pest Manag Sci. 2013;69:1099–105. https://doi.org/10.1002/ps.3598.
Lamberth C, Jeanmart S, Luksch T, Plant A. Current challenges and trends in the discovery of agrochemicals. Science. 2013;341:742–6. https://doi.org/10.1126/science.1237227.
Heap I. Global perspective of herbicide-resistant weeds. Pest Manag Sci. 2014;70:1306–15. https://doi.org/10.1002/ps.3696.
Duke SO. Why have no new herbicide modes of action appeared in recent years? Pest Manag Sci. 2012;68:505–12. https://doi.org/10.1002/ps.2333.
Qu RY, He B, Yang JF, Lin HY, Yang WC, Wu QY, et al. Where are the new herbicides? Pest Manag Sci. 2021;77:2620–5. https://doi.org/10.1002/ps.6285.
Powles SB, Yu Q. Evolution in action: plants resistant to herbicides. Annu Rev Plant Biol. 2010;61:317–47. https://doi.org/10.1146/annurev-arplant-042809-112119.
Jugulam M, Shyam C. Non-target-site resistance to herbicides: recent developments. Plants. 2019;8:417. https://doi.org/10.3390/plants8100417.
Abell LM, Schloss JV, Rendina AR. Target-site directed herbicide design. In: Duke SO, Menn JJ, Plimmer JR, editors. Pest control with enhanced environmental safety. Washington, DC: American Chemical Society; 1993. p. 16–37. https://doi.org/10.1021/bk-1993-0524.ix001.
Krähmer H, Walter H, Jeschke P, Haaf K, Baur P, Evans R. What makes a molecule a pre-or a post-herbicide—how valuable are physicochemical parameters for their design? Pest Manag Sci. 2021;77:4863–73. https://doi.org/10.1002/ps.6535.
Yang Z, Li Q, Yin J, Liu R, Tian H, Duan L, et al. Design, synthesis and mode of action of novel 3-chloro-6-pyrazolyl picolinate derivatives as herbicide candidates. Pest Manag Sci. 2021;77:2252–63. https://doi.org/10.1002/ps.6250.
Vyvyan JR. Allelochemicals as leads for new herbicides and agrochemicals. Tetrahedron. 2002;58:1631–46. https://doi.org/10.1016/S0040-4020(02)00052-2.
Nandurbar AD. Schiff’s bases and amides of selected five membered heterocyclic compounds: a review. J Chem Pharm Res. 2013;5:14–25.
Pardeshi VAS, Chundawat NS, Pathan SI, Sukhwal P, Chundawat TPS, Singh GP. A review on synthetic approaches of benzimidazoles. Synth Commun. 2021;51:485–513. https://doi.org/10.1002/slct.201904832.
Gein VL, Nosova NV, Yankin AN, Bazhina AY, Dmitriev MV. Stereoselective synthesis of novel functionalized cyclohexanone derivatives via the condensation of aromatic aldehydes with acetoacetamide and the influence of the ortho-effect and autocondensation. Tetrahedron Lett. 2019;60:1592–6. https://doi.org/10.1016/j.tetlet.2019.05.023.
Götz T, Böger P. The very-long-chain fatty acid synthase is inhibited by chloroacetamides. Zeitschrift für Naturforschung C. 2004;59:549–53. https://doi.org/10.1515/znc-2004-7-818.
Sardrood BP, Goltapeh EM. Weeds, herbicides and plant disease management. In: Lichtfouse E, editor. Sustainable agriculture reviews 31. Berlin: Springer; 2018. p. 41–178. https://doi.org/10.1007/978-3-319-94232-2_3.
Böger P. Mode of action for chloroacetamides and functionally related compounds. J Pestic Sci. 2003;28:324–9. https://doi.org/10.1584/jpestics.28.324.
Schmalfuß J, Matthes B, Mayer P, Böger P. Chloroacetamide mode of action, I: inhibition of very long chain fatty acid synthesis in Scenedesmus acutus. Zeitschrift Fur Naturforschung C. 1998;53:995–1003. https://doi.org/10.1515/znc-1998-11-1210.
Mallory-Smith CA, Retzinger EJ. Revised classification of herbicides by site of action for weed resistance management strategies. Weed Technol. 2003;17:605–19. https://doi.org/10.1614/0890-037X(2003)017[0605:RCOHBS]2.0.CO;2.
Matthes B, Schmalfuß J, Böger P. Chloroacetamide mode of action, II: inhibition of very long chain fatty acid synthesis in higher plants. Zeitschrift für Naturforschung C. 1998;53:1004–11. https://doi.org/10.1515/znc-1998-11-1211.
Reddy AS, Pati SP, Kumar PP, Pradeep H, Sastry GN. Virtual screening in drug discovery-a computational perspective. Curr Protein Pept Sci. 2007;8:329–51. https://doi.org/10.2174/138920307781369427.
Fu Y, Liu Y-X, Kang T, Sun Y-N, Li J-Z, Ye F. Identification of novel inhibitors of p-hydroxyphenylpyruvate dioxygenase using receptor-based virtual screening. J Taiwan Inst Chem Eng. 2019;103:33–43. https://doi.org/10.1016/j.jtice.2019.08.005.
Abdel-Latif E, Fahad MM, Ismail MA. Synthesis of N-aryl 2-chloroacetamides and their chemical reactivity towards various types of nucleophiles. Synth Commun. 2020;50:289–314. https://doi.org/10.1080/00397911.2019.1692225.
Zhu S, Xu S, Jing W, Zhao Z, Jiang J. Synthesis and herbicidal activities of p-menth-3-en-1-amine and its Schiff base derivatives. J Agric Food Chem. 2016;64:9702–7. https://doi.org/10.1021/acs.jafc.6b03977.
Murtaza S, Altaf AA, Hamayun M, Iftikhar K, Tahir MN, Tariq J, et al. Synthesis, antibacterial activity and docking studies of chloroacetamide derivatives. Eur J Chem. 2019;10:358–66. https://doi.org/10.5155/eurjchem.10.4.358-366.1859.
Epp JB, Alexander AL, Balko TW, Buysse AM, Brewster WK, Bryan K, et al. The discovery of Arylex™ active and Rinskor™ active: two novel auxin herbicides. Biorg Med Chem. 2016;24:362–71. https://doi.org/10.1016/j.bmc.2015.08.011.
Yamamoto A, Nakamura T, Adu-Gyamfi JJ, Saigusa M. Relationship between chlorophyll content in leaves of sorghum and pigeonpea determined by extraction method and by chlorophyll meter (SPAD-502). J Plant Nutr. 2002;25:2295–301. https://doi.org/10.1081/Pln-120014076.
Finney DJ. Probit analysis. 3rd ed. Cambridge: Cambridge University Press; 1971.
IBM Corp. Released 2017. IBM SPSS statistics for windows, version 25.0. Armonk: IBM Corp.; 2017.
Discovery Studio. 2.1, Accelrys Inc. San Diego, CA, USA. 2008. https://www.3ds.com/products/biovia/discovery-studio.
Badawy MEI, El-Zemity SR. 3D Pharmacophore-based ligand alignment, virtual screening and molecular docking protocols towards the discovery of 2-((1H–1, 2, 4-triazol-1-yl) methyl) derivatives as antifungal inhibitors. Curr Bioact Compd. 2020;16:498–513. https://doi.org/10.2174/1573407215666190131110930.
Moche M, Dehesh K, Edwards P, Lindqvist Y. The crystal structure of β-ketoacyl-acyl carrier protein synthase II from Synechocystis sp. at 1.54 Å resolution and its relationship to other condensing enzymes. J Mol Biol. 2001;305:491–503. https://doi.org/10.1006/jmbi.2000.4272.
MOE (Molecular Operating Environment), Chemical Computing Group. 2008. https://www.chemcomp.com/en/Products.htm.
Fu Y, Sun Y-N, Yi K-H, Li M-Q, Cao H-F, Li J-Z, et al. 3D pharmacophore-based virtual screening and docking approaches toward the discovery of novel HPPD inhibitors. Molecules. 2017;22:959. https://doi.org/10.3390/molecules22060959.
Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–9. https://doi.org/10.1002/(SICI)1096-987X(199905)20:7%3c720::AID-JCC7%3e3.0.CO;2-X.
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev. 1997;23:3–25. https://doi.org/10.1016/S0169-409X(96)00423-1.
Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1:337–41. https://doi.org/10.1016/j.ddtec.2004.11.007.
Romero RB, Romero AL. Inibição de ciclooxigenases 1 (COX-1) e 2 (COX-2) por monoterpenos: um estudo in silico. J Health Sci. 2014;16. https://doi.org/10.17921/2447-8938.2014v16n4p%25p.
Kar S, Roy K, Leszczynski J. On applications of QSARs in food and agricultural sciences: history and critical review of recent developments. Adv QSAR Model. 2017;24:203–302. https://doi.org/10.1007/978-3-319-56850-8_7.
Okamoto H, Kato S, Ogasawara M, Konnai M, Takematsu T. Synthesis and herbicidal activity of N (1-arylethenyl)-2-chloroacetamides. Agric Biol Chem. 1991;55:2733–6. https://doi.org/10.1080/00021369.1991.10871034.
Couderchet M, Schmalfuß J, Böger P. A specific and sensitive assay to quantify the herbicidal activity of chloroacetamides. Pestic Sci. 1998;52:381–7. https://doi.org/10.1002/(SICI)1096-9063(199804)52:4%3c381::AID-PS735%3e3.0.CO;2-8.
Jablonkai I. Alkylating reactivity and herbicidal activity of chloroacetamides. Pest Manag Sci. 2003;59:443–50. https://doi.org/10.1002/ps.634.
Xie J, Zhao L, Liu K, Guo F, Gao L, Liu W. Activity, toxicity, molecular docking, and environmental effects of three imidazolinone herbicides enantiomers. Sci Total Environ. 2018;622:594–602. https://doi.org/10.1016/j.scitotenv.2017.11.333.
Wu Y-P, Wang Y, Li J-H, Li R-H, Wang J, Li S-X, et al. Design, synthesis, herbicidal activity, in vivo enzyme activity evaluation and molecular docking study of acylthiourea derivatives as novel acetohydroxyacid synthase inhibitor. J Mol Struct. 2021;1241:130627. https://doi.org/10.1016/j.molstruc.2021.130627.
Xu C, Sun X, Niu L, Yang W, Tu W, Lu L, et al. Enantioselective thyroid disruption in zebrafish embryo-larvae via exposure to environmental concentrations of the chloroacetamide herbicide acetochlor. Sci Total Environ. 2019;653:1140–8. https://doi.org/10.1016/j.scitotenv.2018.11.037.
Sun X, Ji Z, Wei S, Ji Z. Design, synthesis and herbicidal activity of 5-cyclopropyl-N-phenylisoxazole-4-carboxamides. J Mol Struct. 2020;1220:128628. https://doi.org/10.1016/j.molstruc.2020.128628.
Sartori SK, Alvarenga ES, Franco CA, Ramos DS, Oliveira DF. One-pot synthesis of anilides, herbicidal activity and molecular docking study. Pest Manag Sci. 2018;74:1637–45. https://doi.org/10.1002/ps.4855.
Filimon MN, Roman DL, Caraba IV, Isvoran A. Assessment of the effect of application of the herbicide S-metolachlor on the activity of some enzymes found in soil. Agriculture. 2021;11:469. https://doi.org/10.3390/agriculture11060469.
Acknowledgements
Not applicable.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). This research did not receive any grant or specific funding from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study's conception and design. K.E.E performed synthesis, bioassays, data collection, and spectroscopic analysis. S.R.E and M.E.I.B revised the spectroscopic data, in silico studies, and statistical analysis of the bioassay results. All authors participated in manuscript writing, proofreading, sentence correction, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El-Zemity, S.R., Esmaiel, K.E.E. & Badawy, M.E.I. Design, synthesis, pharmacophore modeling, and molecular docking of some novel chloroacetamide derivatives as herbicidal agents. Chem. Biol. Technol. Agric. 11, 124 (2024). https://doi.org/10.1186/s40538-024-00646-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-024-00646-1