
Abstract
Iron-gall inks have been described as complexes of iron ions with gallic or tannic acids, available in gall extracts. To assess this working hypothesis, we have prepared medieval inks using ingredients and methods appropriate to the fifteenth to seventeenth centuries. The five historical inks studied were selected based upon research into Iberian written sources of medieval techniques. Results are supported by comparison with iron complexes with a well-characterized phenol counterpart: gallic, ellagic, and tannic acids as well as digalloyl and pentagalloyl glucose; as either precipitates or prepared as inks by adding gum arabic. Raman and infrared spectroscopies show that medieval writing inks could not have been represented solely by iron complexes with gallic acid. Overall, writing inks display the infrared signature of gallotannins, indicating that complexes of Fe3+-polygalloyl esters of glucose are also formed. Our results also show that the commercial tannic acid solution is far more complex than the gall extracts, and cannot be used to represent a gall extract (as described in historic written sources). High-performance liquid chromatography–electrospray ionisation, HPLC–ESI–MS, reveals that the concentration of gallic acid varies in the gall extracts, depending on the extraction method and ink recipe. Importantly, in certain recipes, gallic acid is found as a minor compound, when compared with the galloyl esters of glucose.
Similar content being viewed by others


Introduction
Degradation of manuscripts catalysed by iron-gall inks is a major conservation issue in heritage collections, posing a serious threat to world written heritage. In Europe, iron-gall ink recipes are profusely described in medieval treatises that mention the use of plant extracts such as Quercus infectoria that were combined with iron salts [1,2,3], Fig. 1. The result of this mixture was an iron-polyphenol complex, to which a polysaccharide such as gum arabic was usually added [4,5,6,7,8]. This writing ink recipe replaced, in part, carbon black inks that were more prone to detachment [9]. Iron-gall inks were rendered obsolete in the twentieth century [9].

Main steps and ingredients in the production of iron-gall ink
The chromophore in iron-gall inks
Iron-polyphenol complex: the metal ion
Although the black colour is central to its use as a writing ink, we know little of the origin of its colour as well as the way it turned brown over time. The fact that iron ions absorb in any region of the visible spectrum, depending on the iron-coordination, only makes this subject more complex [4, 5]. In the phenolic compounds, the catechol ring with 2 hydroxyl or galloyl with 3 hydroxyl groups, provide binding sites for metal ions to chelate [4, 6, 7], Fig. 2. Because polyphenol ligands strongly stabilize Fe3+ over Fe2+, catecholate and gallate complexes of Fe2+ rapidly oxidize in the presence of O2 to give Fe3+-polyphenol complexes [4, 8]. This oxidation of Fe2+ is well documented in food & health research, although the chemistry of iron binding and redox processes is still not completely known [8]. Recently, Dangles et al. found evidence for the mechanism depicted in Scheme 1 [8]. In this mechanism (at neutral pH), the fast first step, the binding of Fe2+ to the catechol, leads to its deprotonation. The large metal-binding stability constants displayed by Fe3+ are the driving force in the second step, where Fe2+ is converted into Fe3+ [4]. In turn, Fe3+ may be reduced, forming quinone or semiquinone species [4]. Based on this model, from these redox reactions no ester hydrolysis is expected, and consequently, no release of gallic acid to the solution is foreseen.

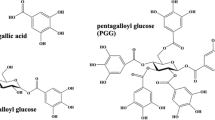
Molecular structures for gallic and ellagic acids; monogalloyl glucose and pentagalloyl glucose

Electron transfer from Fe2+ leads to the formation of Fe3+-phenol complexes, adapted from Dangles et al. [8]
Polyphenol metal ion complex: the ligands
From polyphenol research, it has been shown that “tannins” are found in galls largely as polygalloyl esters of glucose depicted in Fig. 2 [10], however the models used by the scientific community to describe the iron complexes assume that gallic or tannic acids are available in the gall extracts to bind iron [6, 11,12,13,14,15] (Fig. 2 and Additional file 1: Fig. S1). In fact, until 2016, the heritage community was using a structure for the IGI complex that was proved incorrect by Ponce and co-workers [6], Fig. 3. This group, using a reference prepared with gallic acid and iron (II) sulphate, proposed a new Fe3+ gallate structure [Fe(C7O5H3)·xH2O, x = ~ 1.5−3.2], in which Fe3+ binds to the carboxylic acid and three OH groups in the ligand (i.e., the acid and two of the three OH groups are deprotonated) [6]. More recently, relevant contributions into Fe3+ coordination were made by Lerf and Wagner using Mössbauer spectroscopy [15]; these authors proved that Fe3+-gallate complexes, binding through the carboxylate group cannot be formed at the pH found in ink preparation which is between 2 and 3. They propose that the iron center is best represented by iron oxyhydroxides and that these nanoparticles are “covered by a shell of polymerized oxidation products of the phenols” [15]. Iron (II) sulphate was also detected in the precipitates, using both “balanced” and “unbalanced” inks,Footnote 1 as described by Neevel [16].

Basic unit of the structure for the Fe3+-gallate complex, proposed by Ponce et al. [6]
In this work, we measure the concentration of gallic acid in gall extracts prepared from medieval recipes from selected written sources described in the next section, through high-performance liquid chromatography with a diode-array detector (UV–VIS) and with electrospray ionisation-mass spectrometry, HPLC–DAD and HPLC–ESI–MS, respectively. This will allow us to verify if gallic or tannic acids can be used as standards for gall extracts.
Iron-gall inks in technical written sources and references for the colour center
Medieval recipes typically contained the three basic ingredients described in Fig. 1: Fe2+ obtained from an iron sulphate salt, a phenolic extract (tannins), and gum arabic [2, 9]. Additives, such as other metal ions and pigments, and different extraction conditions are described in medieval technical texts as exemplified in Table 1 and Additional file 1: Table S1 [2, 9, 17,18,19,20,21]. A rationale for the different procedures and ingredients that are called for the making of the inks will be discussed in this work.
Following a methodology already developed in our critical edition of “The book on how to make all the colour paints for illuminating book” [22], we prepared medieval inks using the recipes that provide the ingredients and methods appropriate to their period [23]. The choice of writing inks is the result of research into selected Iberian written sources of medieval techniques [17,18,19,20,21]. In this work, these medieval inks were analysed, in situ, by colorimetry, Fourier transform infrared spectroscopy (FTIR) and Raman microscopy. Selected historical ink reproductions are compared with standards prepared complexing iron with gallic, ellagic and tannic acids as well as with digalloyl and pentagalloyl glucose, with and without gum arabic, Fig. 1 and Table 2. In addition, the simple model commonly used in literature for “balanced” iron-gall systems is also included: “The ink can be described as a suspension of an Fe(III) gallate precipitate in an aqueous solution containing iron sulphate, gallic acid and gum Arabic (9 g/L gallic acid monohydrate; 40 g/L FeSO4·7H2O; 80 g/L gum Arabic: 1: 4,4:8,9).” [16, 24].
Methods/experimental
Except for the galls and gum arabic, all reagents used were of analytical grade. Spectroscopic or equivalent grade solvents and Millipore water were used for all the chromatographic and spectroscopic studies. Gallnuts (“oak apples from Quercus infectoria”) and gum arabic in grains from A. senegal were purchased from Kremer. The phenolic compounds used to prepare references were: gallic acid (Aldrich), tannic acid (Aldrich), pentagalloyl glucose (Sigma), digalloyl glucose (Extrasynthese). Except for tannic acid, all compounds were pure. As discussed in detail in “Quantification of gallic acid by HPLC–DAD and HPLC–ESI–MS”, tannic acid proved to be a very complex mixture of phenols, in which gallic acid was also present.
Pomegranate was purchased in a local supermarket and a branch was taken from a fig tree in the Caparica Campus. White wine vinegar and organic white wine were acquired in a local organic supermarket.
Preparation of historic ink reconstructions
The inks were prepared following the medieval treatises mentioned previously, Table 1 and Additional file 1: Table S1. The translations may be found as Additional file 1: Table S1b. Unit conversion was based on “Equivalencias entre las pesas y medidas usadas antiguamente en las diversas provincias de España y las Legales del Sistema Métrico-Decimal”, 1886 and on “Memória sobre os pesos e medidas de Portugal, Espanha, Inglaterra e França”, 1838 [25, 26]. This directory describes the units as used in the different cities, Additional file 1: Table S2.
Preparation of the references (iron-polyphenol complexes)
The iron-polyphenol complexes were prepared according to Ponce et al. [6], using the quantities described in Table 2. The polyphenols were completely dissolved in Millipore water, in an ultrasound bath, at 40 °C. Subsequently, Fe(II) sulphate heptahydrate was added to the stirred phenol solution. The latter was then divided in two: (a) to one half, gum arabic was added to prepare an ink that was continuously stirred for a week as indicated in Ponce et al. [6], until the pH dropped to ca. 1.8, indicating the gallic acid was deprotonated as it reacted with the iron ion; (b) the other half, was used to obtain the precipitate, hence no gum arabic was added. After a week, the precipitate was separated by centrifugation at a speed of 12,000 rpm, for 15 min, at 4 °C. The precipitate was collected and washed with water and centrifuged to remove unreacted starting materials and impurities. The precipitate was left to dry at room temperature, ground in an agate mortar, and then dried under vacuum.
Precipitates were analysed as powders. The historic inks were applied on filter paper, to be analysed by Raman spectroscopy and colorimetry, and glass slides, to be analysed by infrared spectroscopy, with the aid of a Pasteur pipette (ca. 50 μL per 1 cm2). The historic inks were prepared and measured five times each to assess reproducibility.
Colourimetry
L*a*b* coordinates were measured using a Microflash mobile colorimeter DataColor International with a Xenon lamp, over an 8 mm-diameter measuring area. CIELAB system was used defining the D65 illuminant and the 10º observer. The instrument was calibrated with a white tile and a black trap, and the measurements were performed on top of filter paper. The described values are the average value of three measurements, which proved to be sufficient to guarantee reproducibility.
In the Lab* cartesian system, L*, relative brightness, is represented in the z-axis. Variations in relative brightness range from white (L* = 100) to black (L* = 0). In the red-green y-axis, a* is usually found between − 60 (green) and + 60 (red). In the yellow-blue x-axis, b* ranges from − 60 (blue) to + 60 (yellow). The (a*, b*) pair represents the hue and chroma of the object.
Micro-Fourier transform infrared spectroscopy
Infrared analyses were performed using a Nicolet Nexus spectrophotometer coupled to a Continuμm microscope (15 × objective) with an MCT-A detector. The spectra were collected in transmission mode, in 50 µm2 areas, resolution setting 4 or 8 cm−1 and 128 scans, using a Thermo diamond anvil compression cell. CO2 absorption at ca 2400–2300 cm−1 was removed from the acquired spectra (4000–650 cm−1). To improve the robustness of the results, at least two spectra were acquired from different sample spots.
Raman microscopy
Raman microscopy was carried out using a Horiba Jobin–Yvon LabRAM 300 spectrometer, equipped with a diode laser with an excitation wavelength of 785 nm and a maximum laser power of 37 mW measured at the sample. Spectra were recorded as an extended scan. The laser beam was focused with a 50x Olympus objective lens and the spot size is of 4 μm. The laser power at the sample surface was between 9.5 and 0.37 mW. No evidence of ink degradation was observed during spectra acquisition. More than three spectra were collected from the same sample. A silicon reference was used to calibrate the instrument.
HPLC–DAD and HPLC–ESI–MS
HPLC–DAD: Merck-Hitachi Elite Lachrom with DAD L-2455; HLPC–ESI–MS: Finnigan Surveyor Plus HPLC fitted with a PDA Plus detector, an auto-sampler Plus and an LC quaternary pump plus coupled to a Finnigan LCQ Deca XP Plus mass detector equipped with an ESI source and an ion trap quadrupole. The mass spectrometer was operated in the negative-ion mode with the source, with a capillary temperature of 275 °C and capillary voltages of 4.5 kV. The mass spectra were recorded between 250 and 2000 m/z.
The stationary phase was a 150 × 4.6 mm i.d., 5 μm pore size reversed-phase C18 column (Merck) thermostated at 25 °C. The mobile phase was composed by solvent A, 1% (v/v) formic acid, and solvent B, 100% (v/v) acetonitrile. The flow rate was 0.50 mL/min and the gradient method started with a linear gradient ranging from 90% A to 65% A in 50 min, then reaching 100% B in 5 min, a final isocratic gradient of 100% B during 7 min and a final re-equilibration isocratic gradient of 90% A for 5 min.
The historic inks, reproduced five times each, were analysed three times each. All gall extracts and full ink samples were filtered through a syringe filter of 0.45 µm pore size prior to HPLC/DAD–ESI/MS analysis. Reference phenols were also injected, dissolved in water, and the detection was carried out at 280 nm. The compound quantification was achieved through a calibration curve. This calibration curve was obtained by injecting a standard gallic acid (Sigma-Aldrich) with a concentration range of 0.253 to 0.00253 mg/mL and linear calibration curve (r2 = 0.9985). Each sample was prepared in duplicate and injected in triplicate. Unknown concentrations were determined from the regression equation and the results were expressed as mean ± standard deviation and presented as mg mL−1 equivalents of gallic acid.
A study of the repeatability of this method from extraction to HPLC analysis, used commonly in our laboratory, gave a coefficient of variation of less than 5%. The detection (LOD) and quantification (LOQ) limits were 0.0000891 and 0,000267 mg mL−1 respectively, and they were determined as reported elsewhere [27, 28].
Results and discussion
Selection criteria for Iberian medieval recipes dated from fifteenth to seventeenth centuries
Four Spanish and one Portuguese ink recipes, dated between fifteenth and seventeenth centuries, were chosen and are shown in Table 1. These recipes are representative of different institutions where the use of the writing ink was essential, such as universities, notaries, chanceries and the monastic world. The information found in these texts is critical for advancing the history of technique and the study of manuscript cultures. The first of the recipes, in chronological terms (1464), is described in Chancelaria de Braga cx. 31 (Chancellery of the Archiepiscopate of Braga) and stands out for being the oldest found in Portugal [17]. From the manuscript in the Faculty of Medicine of Montpellier, Montpellier H-490, completed between 1469–1480 and studied by Ricardo Córdoba [18], we reproduced the one recipe described for a writing ink (fol. 233v). A single writing ink recipe is also found in the Archivo Histórico Provincial de Córdoba, Sección de Protocolos Notariales de Córdoba (1474; cx. 5, fol. 58v), a text studied by E. Rodríguez [19], and directly related to the world of writing. On the other hand, in “Libro de los Oficios” of the Monastery of Santa Maria de Guadalupe, Cáceres, three iron-gall ink recipes are presented (fol. 201r-v). Studied by Kroustallis [20], this manual from the end of the fifteenth century contains scriptural knowledge linked to the monastic world. The fifth recipe is collected in the Recipe Book Ms. 9226 (fol. 192r-v) composed by Juan Vázquez del Mármol, a priest and book corrector of the Hispanic monarchy, between the sixteenth and the seventeenth centuries [21]. This manuscript covers a wide range of subjects, including woodworking, medicine, candle manufacture, and others. It also assembles a great number of recipes dedicated to writing inks [21]. Henceforth, the manuscripts and respective recipes will be designated as Braga, Montpellier, Córdoba, Guadalupe, and Madrid, respectively.
Preparation of iron-gall inks and reference compounds
The ingredients and the steps to prepare the iron-gall inks and references are summarized in Tables 1 and 2, together with the pH of the dispersion. Colour coordinates are presented in Table 3.
All the recipes share three common raw materials: gall nuts, iron sulphate (in historical recipes named azije and/or caparrosa), and gum arabic. The galls and the metal ion produce dark inks, perceived as black, while the agglutinant facilitates its use as a written vehicle (by maintaining the complex dispersed in solution), Table 3. For all the ink recipes, a decrease in the pH was observed following the addition of iron sulphate, likewise in the references, Tables 1 and 2. This was expected considering that, in a very fast first step, Fe2+ binds to the catechol ring; this binding implies a deprotonation and with it an increase in the concentration of H+ [8].
In the fifteenth century written sources, the term caparrosa has been used both for iron or copper sulphate, indistinctly. Caparrosa is mentioned in two recipes; in Oficios it is used together with azije, and in the sixteenth to seventeenth centuries Madrid as the only source for metal ions. Our experimental results proved that copper sulphate alone cannot produce a dark ink.Footnote 2 So, in the seventeenth century recipe caparrosa possibly indicates a mineral containing iron sulphate or admixed with copper sulphate [29]. In all the recipes, the galls are broken into pieces (partidas) or crushed (quebrantadas) and not ground (moídas). According to the “Diccionario de la lengua Española from the Real Academia Española” [30], partir means to open or divide something into two or more parts, which differs from quebrar, which means to reduce to smaller fragments without grinding. Two cases mention the purification of caparrosa: one by placing it in solution to separate it from dirt (Guadalupe: no echar la tierra) and in Braga, where the iron sulphate is to be sifted to separate it from dirt (peneirado). Usually, following the formation of the black colour, the solution is filtered; only two recipes do not mention a final filtration step, Braga and Madrid. The ingredients ratio in Table 1 show that the Braga recipe uses the highest quantity of azije; the amount of gum arabic is constant in all the recipes (ca. half of the galls in weight) except for Madrid, which presents a similar polyphenol:metal ion:gum arabic ratio, Table 1.
In the selected inks, the polyphenols could be extracted using water, vinegar mixed with water, white wine, or white wine mixed with water; the latter two resulted in the darkest inks, Table 1. The Madrid recipe clearly indicates the use of white wine to prepare the ink, while the selected recipe in Guadalupe only specifies the use of wine. Since more recipes of the same manuscript clearly indicate white wine, the latter was chosen for both the Madrid and the Guadalupe recipes.
Regarding additives, two recipes mention their use: Córdoba (fifteenth century), where pomegranate peel is added to the galls extraction, and Madrid (fifteenth to seventeenth centuries), with añil (indigo), refined sugar and alum. The latter was the easiest to apply on the support, the ink being more fluid, darker and the shiniest, when compared with the other selected inks which do not have the presence of additives.
Art technological source research carried out as part of the InkCor project shows that, in most of the 253 recipes analysed (written or printed between 1000 and 1900), the iron (II) sulphate: gall nuts ratio varies between 1:4 and 4:1 in weight, with “an emphasis on proportions 6:6, 6:4, 6:3, 6:2” [2]. In this project, two historically representative model inks were proposed, based on “Aleppo” gall nuts with a low and a high copper content [2]. The first model ink displays the following ratio 1:1:0.5 for gall: vitriol: gum arabic in weight; in the vitriol, the copper sulphate, admixed with iron (II) sulphate is present in 0.01% in weight [2]. The Iberian recipes proportions are in agreement with this data, and the closest to the model historical ink with lower copper content is, possibly, Braga’s.
Multi-analytical characterization of iron-gall inks
Colorimetry
Colour coordinates L*a*b*, in Table 3 show that the Iberian reconstructions are perceived as darker inks when compared to the standards (lower L* values). Iberian inks are characterized by negative b* values (blue) and close to zero, positive a* values (red axis), being perceived as a very dark bluish colour. Overall, these inks display similar Lab* values, with Braga and Guadalupe being the darker ones, with Lab* values of (19.52; 0.84; 3.94) and (19.97; 1.03; − 4.80), respectively, Table 3.
Iron-pentagallate and iron-tannate inks display the same hue, but are not as dark as the Iberian inks. Iron-gallate is characterized by very low a* and b* values and may be described as an achromatic colour grey, with L* = 50. Inks prepared with ellagic acid and digalloyl glucose differentiate from all the others because b* is found in the yellow axis. The higher L* values observed in the references may be related to a lower polyphenol concentration, compared with the reconstructions; this will be investigated in future work.
Raman microscopy
The spectra obtained for the ink reconstructions and for the iron-gallate, tannate, di- and penta-gallate based inks are represented in Figs. 4 and 5, respectively. The single iron-gallate and tannate precipitates were also analysed and their spectra are available in Additional file 1: Figures S2. Due to its fluorescence, it was not possible to obtain good S/N spectra for the iron-tannate precipitate and for this reason the bands are unresolved.

Raman spectra for the ink reconstructions, applied on filter paper, λ = 785 nm

Raman spectra for the iron-gallate, di-, penta-gallate and tannate based inks, applied on filter paper, λ = 785 nm
The first conclusion is that all spectra for the inks produced display the fundamental pattern of an iron-gall ink as described by Lee et al. [31]. These authors considered that the main four Raman bands used for a positive identification of iron-gall inks, in historic documents, are found around 1470 cm−1, between 1315 and 1350 cm−1, 490–640 cm−1 (broad) and at 400 cm−1, Table 4. A closer examination of the region between 1315 and 1580 cm−1, shows a well-resolved envelope characterized by two intense bands (ca 1325–36 and 1464–70 cm−1) and two medium–low intensity peaks at (1422–1430 cm−1 and 1569–77 cm−1), the latter is found in many spectra as a shoulder. We do not have theoretical predictions for vibrational spectra of iron-galloyl complexes, and this is the reason why we will support a first discussion of the Raman data on the study of Huguenin et al. on the acid–base equilibria of gallic acid [32]. In this publication, based on theoretical predictions [33], Raman band assignments for gallic acid (LH4) and its deprotonated species (LH3−, LH2 2−, LH3−, L4−) are discussed [32]. The Raman spectrum presented by Huguenin et al. compares well with the measured spectra, in solution, presented by Biles et al. [33]. The spectrum is dominated by two main bands at 1681 cm−1 and 1612 cm−1, the latter being the more intense. Based on Biles et al.Footnote 3 calculated values for this spectrum (presented as supplementary material in the original paper for the neutral, full protonated gallic acid ligand, LH4 [33]), Huguenin et al. attributes the “vibrational band at 1681 cm−1 to the C=O stretch of the carboxylic groups” and “the most intense band located at 1612 cm−1 to the symmetric C–C stretching vibrations of the aromatic compound” [32]; the latter is slightly shifted during deprotonation of the phenol hydroxyl groups to 1604–7 cm−1. As expected, the 1681 cm−1 band is absent in all deprotonated forms, and is substituted by the carboxylate stretching at 1399 cm−1, and by a new broad band with maxima between 1376 and 1383 cm−1, which is a signature band for all deprotonated species; the fully deprotonated form, L4−, also displays a distinctive OH deformation vibrational mode at 1249 cm−1. More recently, Garrido et al. carried out DFTFootnote 4 predictions and described the SERS spectra for the first mono-protonated form LH3−; it displays a different fingerprint when compared with what published Huguenin et al. As we did not use SERS, we will not discuss this work any further [34].
Thus, based on Huguenin, Biles and co-workers’ assignments, and hypothesising that for the gallate complex we expect an LH3− based structure, we would expect to observe the 1612 cm−1 C–C stretching in the aromatic ring and a shift in the broad band at 1376 cm−1. This is not the case; although we still find two main bands at circa 1470 cm−1 and a second in a variable interval 1325–1345 cm−1. Presently, we cannot explain these very large shifts.
For iron-gallate, Ponce et al. proposed the following assignments: C–C ring vibrations at 1470 cm−1 and COO− and C–O stretching modes between 1315 and 1350 cm−1. The two medium–low intensity bands, at circa 1430 cm−1 and 1579 cm−1, were attributed to the symmetrical and asymmetrical vibrations of a coordinated –COO−, to a metallic ion in their iron-gallate precipitate, stating that “the peak positions and separations (≈ 149 cm−1) are also consistent with the bridging carboxylate functionality observed in the crystal structure” [6]. Considering that Lerf and Wagner were not able to detect these type of bindings (with carboxylate) in their recent publications [15], we think that further research is necessary to extract more complete and consistent information from the iron-gallate Raman spectra.
Piantanida et al. presents band assignments for ink reconstructions [35]. However, in this publication the proposed assignments are largely based on the papers by Biles and Garrido; i.e., on gallic acid and not on galloyl-iron complexes. Considering that we will show that gallic acid is a minor component in most of our gall extracts (“Quantification of gallic acid by HPLC–DAD and HPLC–ESI–MS”) as well the contradictory assignments present in literature, we prefer to not assign vibrational modes to the bands observed in this work, Table 4. At this point, we think it is better to use the signature bands for iron-gall inks as proposed by Lee et al. [31], and to discuss the spectral patterns found. When we compare the historical inks with the iron-gallate reference, we observe that 2 of the 3 strong signature bands for iron-gall inks display shifts: The band at 1327 cm−1 for iron gallate, is found in an interval between 1332 and 1336 cm−1 for the historical ink reconstructions; likewise, the signature broad band at 594 cm−1 for iron-gallate is shifted to higher wavenumbers, displaying maxima in an interval between 601 and 608 cm−1. Considering both references and gall inks, the bands that possibly display the higher variability are the signature band at 1325–1336 cm−1 and the band found between 1577 and 1569 cm−1; the more stable, is the signature band at 1468–1471 cm−1. Overall, the best match between the references prepared and the historical iron-gall inks lies in-between the spectral fingerprint of iron-PGG and iron-DiGG. This means that the spectra are better described by molecular structures that are characterized by an ester bond and not by a carboxylate group.
MicroFTIR spectroscopy
In Fig. 6 a representative historical reconstruction, the Braga ink, is compared with the iron-gallate, digallate, pentagallate and tannate precipitates. The infrared spectra of all ink reproductions are presented in Additional file 1: Figure S3. Care must be taken with iron (II) sulphate, as its presence was observed by Lerf and Wagner in their Mossbaüer studies of the precipitates [15]. On the other hand, we do not expect gum arabic to interfere with the final fingerprint, as it was added in a low 0.5% in weight solution.

Infrared spectrum of Braga ink compared with the spectra of iron-gallate, di-galloate, penta-galloate and tannate precipitates (for the meaning of the colour bars, please see Table 5)
Contrary to the Raman discussion, the discussion of the infrared signature will be straightforward, because we rely on the research published by Falcão and Araújo on the characterization by infrared spectroscopy of “tannins”, extracted from several vegetal sources, used to dye leather [36, 37]. Spectra were acquired in ATR, but this will not be an obstacle to its use (small shifts and higher bands intensity will be expected on the lower wavenumber part of the spectra). In the nomenclature used by these authors [36, 37], our polyphenol extracts are described as hydrolysable tannins based on the galloyl unit. For gallotannins the following infrared pattern is proposedFootnote 5: (i) 4 common strong bands shared with all the other “tannins” (1615–1606 cm−1; 1442–1446 cm−1; 1211–1196 cm−1; 1043–1030 cm−1); (ii) 2 characteristic bands of hydrolysable tannins (1731–1704 cm−1; 1325–1317 cm−1); (iii) 3 distinctive bands for gallotannins, that are described as marker bands (1088–1082 cm−1; 872–870 cm−1; 763–758 cm−1). As clearly evidenced in Fig. 6 and depicted in Table 5, our iron-gall precipitates are characterized by these 9 bands together with the broad O–H stretching around 3340–70 cm−1. Both hydrolysable and gallotannins are characterized by the ester functional group. Although the tentative band positions presented in Table 5 are assigned based on the findings of Falcão and Araújo and not DFT calculations, the sound work and clear reasoning of these authors enable us to prove that the infrared spectra of our iron-gall inks display bound gallotannins as the main molecular fingerprint. This is in perfect agreement with the results on the gall extracts characterization that we will discuss in "Quantification of gallic acid by HPLC–DAD and HPLC–ESI–MS", and wherein we found gallic acid largely as a minor compound. This also agrees with the Raman data previously discussed in this study.
From the analysis of the infrared spectra depicted in Fig. 6, it is possible to distinguish between the four reference complexes. The well-resolved peaks observed in the fingerprint region, although characterized by an overall similar pattern, display distinctive features, which will be explored in a future work. Again, in agreement with Raman conclusions, the best match for the iron-gall ink precipitate is with iron-pentagalloyl glucose; but, in this case, the C=O ester stretching mode is shifted to lower wavenumbers, Table 5.
In agreement with Lerf and Wagner [14, 15], the data gathered show that the main chromophore in the iron-gall inks we reproduced is not based on a carboxylate-iron binding, as suggested in previous literature, including the recent work by Ponce et al. [6, 11, 12]. As an alternative to iron-carboxylate binding, colour may be developed through the catechol ring, with 2 OH or galloyl with 3 OH groups providing binding sites for metal ions to chelate, as has already been proposed in different scientific areas of research [38; also see 4, 7, 8, 13].
Finally, comparing the spectral features of our spectra with the sumac spectrum presented by Falcão and Araújo [36], observing the much higher intensity of the band at 1088–1082 cm−1, we may conclude that we have a relevant amount of iron (II) sulphate in all ink samples.
Quantification of gallic acid by HPLC–DAD and HPLC–ESI–MS
Due to a large variability in the phenolic compound concentration in the first HPLC–DAD analysis, the historic inks were each reproduced five times. This variability is easily explained by the nature of the phenolic and polyphenolic compounds that are naturally produced by plants as a response to external aggressions. In this case, galls are produced in response to the presence of insects. The phenolic composition of each gall, on each tree, may depend on climate, weather, time of the year, irrigation conditions, etc. The main compounds found by HPLC are free gallic acid and derivatives of gallic acid such as galloyl glucose, digalloyl glucose, trigalloyl glucose, tetragalloyl glucose, pentagalloyl glucose, hexagalloyl glucose and heptagalloyl glucose. Gallic acid concentration in the gall extracts is presented in Table 1 and in Fig. 7, a representative chromatogram of the species distribution found in all ink recipes is given using the Braga ink (Additional file 1: Fig. S4). These are characterized by complex high-performance liquid chromatography elution patterns, which include gallic acid but also di- to hepta-polygalloyl esters of glucose. A more detailed analysis of the molecular structure for the polygalloyl esters will be presented in future work. HPLC–DAD and HPLC–ESI–MS data reveals that the percentage of gallic acid varies in the gall extracts, depending on the extraction method and ink recipe, from ca. 0.3 to 1.8 g/L. This data also shows that, except for the Braga ink, iron (II) sulphate is found in very low amounts when compared with the 9 g gallic acid to 40 g iron (II) sulphate proposed for a balanced ink, in literature. Córdoba and Montpellier inks display the highest content of gallic acid; the other three recipes Braga, Madrid and Guadalupe have similarly low values. We still have not found a reason that could explain the differences found in acid gallic concentration.

HPLC–DAD chromatograms of a commercial tannic acid solution and the Braga extract. Gallic acid is the high intensity peak that elutes at ca. tr = 5 min
The gall extracts chromatographic profiles are very interesting, where HPLC–ESI–MS clearly shows the presence of a dominant fraction of polygalloyl esters of glucose, Fig. 7. The results obtained from the aqueous solution of commercial tannic acid surprised us, and for that reason, the chromatogram obtained is presented in Fig. 7. We were expecting a maximum of two peaks, corresponding to some fraction of gallic acid and a broad band for the isomers of decagalloylglucoses [39]. Instead, the chromatographic profile is much more complex than the gall extracts obtained following fifteenth to seventeenth centuries recipes. Definitively, commercial tannic acid cannot be described, as depicted in the commercial catalogues, as penta-m-digalloyl-glucose (meta-depsidic 1,2,3,4,6 pentakis-O-digalloyl-β-d-glucopyranose), Additional file 1: Figure S1, and it is not suitable for preparing a reference medieval iron-gall ink. As described by Quideau et al. [39], commercial tannic acid is in fact “a complex and varying mixture of different gallotannins and simpler galloylglucoses”. This could have been anticipated as tannic acid is described in literature as being extracted from gall-nuts “by boiling ground-up nuts in acidic aqueous solution” [40]. In the future, we plan to study “true” iron-decagalloylglucoses complexes, but we anticipate it will not be an easy task since these molecular structures equilibrate into meta/para-depsidic isomeric mixtures in solution [39].
Conclusions
Based on a multi-analytical research of 5 iron-gall ink reconstructions from 5 historical recipes made on the Iberian Peninsula between fifteenth and seventeenth centuries as well as five different reference precipitates/inks (standards), we show that iron-gallate or tannate complexes do not accurately represent the chromophores present in medieval iron-gall inks. Reference precipitates/inks were made combining iron sulphate with different polyphenols (tannic, gallic and ellagic acid, as well as di- and pentagalloyl glucose). Colourimetry, microRaman spectroscopy, microFTIR and HPLC–DAD/ESI–MS were jointly used to characterize the reconstructed and reference inks.
Quantification of gallic acid both by HPLC–DAD and HPLC–ESI–MS show that it is a minor compound in most of the gall extracts prepared following Iberian recipes; these extracts are characterized by complex high performance liquid chromatography elution patterns, which include gallic acid as well as the derivatives of gallic acid in the form of polygalloyl esters of glucose also named gallotannins (mono- to hepta-galloyl glucose structures were identified). This agrees with the results of Raman and infrared spectroscopies. In fact, based on the study of vegetable tannins used to transform animal skins into leather [36], it was possible to show that the infrared spectra display the fingerprint of an ester-based polyphenol, more precisely a gallotannin. On the other hand, Raman band assignments published in the literature refer largely to gallic acid or to iron-gallate complexes and therefore could not be fully exploited in this work. Raman and infrared spectra, acquired in this work, indicate that iron-pentagalloyl based ink/precipitate best represent medieval inks.
Overall, historically accurate reconstructions of medieval inks and knowledge on the chemistry of polyphenolic compounds used to produce leather, since ancient times, have been crucial to bringing new insights into iron-gall inks complex structure and compounds formed within it, which will advance future preparations of model inks. Quality and representative model ink are critical for studies of iron-gall ink behaviour or when testing new conservation treatments.
Further research characterizing a wider number of medieval recipes, from different geographic provenances, is necessary, but this work and its methodology have already had an impact on our fundamental knowledge on a medieval iron-gall ink molecular composition and its conservation. The paradigm must shift from model compounds acid gallic-based to polygalloy ester-based. In alternative to iron-carboxylate binding, we argue that colour may be developed through the catechol ring, with 2 OH or galloyl with 3 OH groups providing binding sites for metal ions to chelate. This issue will be addressed in future research.
Notes
"It assumes that there is an ideal molar ratio of FeSO4·7H2O and TA 3.6:1, for a good, i.e., balanced ink. Other ratios are called unbalanced inks and supposed to have inferior properties and be more harmful to the paper on which the ink is written" [15].
This was easily verified with the naked eye and Lab* coordinates were also obtained but are not shown in this work.
Biles et al present a thorough discussion of the calculated vibrational modes, but we think this discussion falls outside the scope of this work [33]; however, we would like to stress that: (i) their calculated Raman spectra showed an "over expression" of C–H and O–H vibrational modes; (ii) they could "regard the vibrational bands at the 1695 and 1635 cm−1, respectively as vibrational modes characterized by CO stretch coordinates of the carboxylic groups", but "all other internal coordinates are distributed between lots of vibrational modes and cannot be regarded as bases of characteristic vibrational modes".
Density Functional Theory.
In brackets are indicated the spectral intervals where the bands may be found.
References
Quideau S, Deffieux D, Douat-Casassus C, Pouységu L. Plant polyphenols: chemical properties, biological activities, and synthesis. Angew Chem Int Ed. 2011;50:586–621.
Stijnman A. Iron-gall inks in history: ingredients and production. In: Kolar J, Strlič M, editors. Iron–gall Inks: on manufacture, characterization, degradation and stabilization. Ljubljana: National and University Library; 2006. p. 25–167 (and Appendix 4).
Vivas N, Bourgeois G, Vitry C, Glories Y, Freitas V. Determination of the composition of commercial tannin extracts by liquid secondary ion mass spectrometry (LSIMS). J Sci Food Agric. 1996;72:309–17.
Perron NR, Brumaghim JL. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem Biophys. 2009;53:75–100.
Bartecki A, Burgess J. The colour of metal compounds. 1st English ed. Amsterdam: Gordon and Breach Science Publishers; 2000.
Ponce A, Brosto LB, Gibbons SK, Zavalij P, Viragh C, Hooper J, Gaskell KJ, Eichhorn B. Elucidation of the Fe(III) Gallate Structure in Historical Iron-gall Inks. Anal Chem. 2016;88:5152–8.
Andjelković M, Van Camp J, De Meulenaer B, Depaemelaere G, Socaciu C, Verloo M, Verhe R. Iron-chelation properties of phenolic acids bearing catechol and galloyl groups. Food Chem. 2006;98(1):23–31.
Nkhili E, Loonis M, Mihai S, El Hajji H, Dangles O. Reactivity of food phenols with iron and copper ions: binding, dioxygen activation and oxidation mechanisms. Food Funct. 2014;5:1186–202.
Zerdoun-Bat Yehounda M. Les encres noires au Moyen Âge (jusqu’à 1600). 1st ed. Paris: CNRS Éditions; 2003.
Han J, Wanrooij L, van Bommel M, Quye A. Characterisation of chemical components for identifying historical Chinese textile dyes by ultra high performance liquid chromatography–photodiode array–electrospray ionisation mass spectrometer. J Chromatogr A. 2016;1479:87–96.
Wunderlich CH. Geschichte und Chemie der Eisengallustinte. Restauro. 1994;6:414–21.
Krekel C. The chemistry of historical iron-gall inks: understanding the chemistry of writing inks used to prepare historical documents. Int J For Doc Exam. 1999;5:54–8.
Feller RK, Cheetham AK. Fe (III), Mn(II), Co(II), and Ni(II) 3, 4, 5-trihydroxybenzoate (gallate) dihydrates; a new family of hybrid framework materials. Solid State Sci. 2006;8(9):1121–5.
Wagner FE, Lerf A. Mössbauer spectroscopic investigation of FeII and FeIII 3, 4, 5-trihydroxybenzoates (Gallates)–proposed model compounds for iron-gall inks. Z Anorg Allg Chem. 2015;641(14):2384–91.
Lerf A, Wagner FE. Model compounds of iron-gall inks—a Mössbauer study. Hyperfine Interact. 2016;237(1):36.
Neevel JG. Phytate: a potential conservation agent for the treatment of ink corrosion caused by irongall inks. Restaurator. 1995;16(3):143–60.
Azevedo Santos MJ. Da Visigótica à Carolina a escrita em Portugal de 882–1172: Aspectos Técnicos e Culturais. Lisboa: Fundaçao Calouste Gulbenkian: Junta nacional de investigaçao científica e tecnológica; 1994. p. 303–8.
Córdoba de la Llave R. Un recetario técnico castellano del siglo XV: el manuscrito H490 de la Facultad de Medicina de Montpellier. En la España medieval. 2005;28:7–48.
Rodríguez Díaz EE. Técnicas de escritura y del libro manuscrito. In: García Ballester L, editor. Historia de la Ciencia y de la Técnica en la Corona de Castilla, vol. 2. Valladolid: Junta de Castilla y León, Consejería de Educación y Cultura; 2002. p. 590.
Kroustallis S. El oficio de Pergaminería y el Reglamento del Scriptorium del Monasterio de Ntra. Sra. de Guadalupe. In: Brinquis MS, Cabanes ML, editors. Libro de los Oficios del Monasterio de Nuestra Señora de Guadalupe, vol. 2. Badajoz: Secretaría General Técnica del Ministerio de Cultura, Subdirección General de Publicaciones, Información y Documentación, Junta de Extremadura, Consejería de Cultura y Turismo y Monasterio de Guadalupe; 2007. p. 246–7.
Criado Vega MT. Tratados y recetarios de técnica industrial en la España medieval. La Corona de Castilla, siglos XV–XVI. Córdoba: Universidad de Córdoba: Servicio de Publicaciones; 2013.
Melo MJ, Castro R, editors. The “Book on How to Make Colours”, o livro de como se fazem as cores das tintas todas. Medieval Colours for Practitioners; 2016. http://www.dcr.fct.unl.pt/LivComoFazemCores. Accessed 21 Aug 2017.
Vitorino T, Melo MJ, Carlyle L, Otero V. New insights into brazilwood lake pigments manufacture through the use of historically accurate reconstructions. Stud Conserv. 2016;61(5):255–73.
Rouchon V, Belhadj O, Duranton M, Gimat A, Massian P. Application of Arrhenius law to DP and zero-span tensile strength measurements taken on iron-gall ink impregnated papers: relevance of artificial ageing protocols. Appl Phys A. 2016;122:773.
Equivalencias entre las pesas y medidas usadas antiguamente en las diversas provincias de España y las legales del sistema métrico decimal, publicadas de Real Orden Dirección General del Instituto Geográfico y Estadístico. Madrid: Imp. de la Dirección General del Instituto Geográfico y Estadístico; 1886.
Barreiros JF. Memória sobre os pesos e medidas de Portugal, Espanha, Inglaterra e França: que se empregäo nos trabalhos do corpo de engenheiros e da arma de artilheria. Lisboa: Typog. da Acad. Real das Sciencias; 1838.
FDA. Guidance for industry—studies to evaluate the metabolism and residue, kinetics of veterinary drugs in food-producing animals: validation of analytical methods used in residue depletion studies. Washington: U.S. Department of Health and Human Services; 2015.
García-Estévez I, Escribano-Bailón MT, Rivas-Gonzalo JC, Alcalde-Eon C. Validation of a mass spectrometry method to quantify Oak Ellagitannins in Wine samples. J Agric Food Chem. 2012;60(6):1373–9.
Nabais P, Melo MJ, Castro R, Sousa L, Videira Lopes G. Singing with light: an interdisciplinary study on the medieval Ajuda Songbook. JMIS. 2016;8:283–312.
Real Academia Española. Diccionario de la lengua Española; 2017. http://dle.rae.es/?w=diccionario. Accessed 27 Aug 2018.
Lee AS, Mahon PJ, Creagh DC. Raman analysis of iron-gall inks on parchment. J Vib Spectorsc. 2006;41(2):170–5.
Huguenin J, Saad O, Bourson P. Monitoring deprotonation of gallic acid by Raman spectroscopy. J Raman Spectrosc. 2015;46(11):1062–6.
Billes F, Mohammed-Ziegler I, Bombicz P. Vibrational spectroscopic study on the quantum chemical model and the X-ray structure of gallic acid, solvent effect on the structure and spectra. Vib Spectrosc. 2007;43(1):193–202.
Garrido C, Diaz-Fleming G, Carcamo JJ, Campos-Vallette MM. Surface enhanced Raman scattering and theoretical characterization of the gallic acid anion silver surface interaction. Vib Spectrosc. 2017;93:12–6.
Piantanida G, Menart E, Strlic M. Classification of Iron-based Inks by means of micro-Raman spectroscopy and multivariate data analysis. J Raman Spectrosc. 2013;44(9):1299–305.
Falcão L, Araújo MEM. Tannins characterization in historic leathers by complementary analytical techniques ATR-FTIR, UV-Vis and chemical tests. J Cult Herit. 2013;14:499–508.
Falcão L, Araújo MEM. Vegetable tannins used in the manufacture of historic leathers. Molecules. 2018;23:1081.
Rahim MA, Kempe K, Muellner M, Ejima H, Ju Y, van Koeverden MP, Suma T, Braunger JA, Leeming MG, Abrahams BF, Caruso F. Surface-confined amorphous films from metal-coordinated simple phenolic ligands. Chem Mater. 2015;27(16):5825–32.
Sylla T, Pouységu L, Da Costa G, Deffieux D, Monti JP, Quideau S. Gallotannins and tannic acid: first chemical syntheses and in vitro inhibitory activity on Alzheimer’s amyloid β-peptide aggregation. Angew Chem Int Ed. 2015;54:8217–21.
Giorgi R. Inorganic nanomaterials for the deacidification of paper. In: Baglioni P, Chelazzi D, editors. Nanoscience for the conservation of works of art. Cambridge: The Royal Society of Chemistry; 2013. p. 396–429.
Authors’ contributions
MJM contributed with the conception and design of the research work; acquisition, analysis and interpretation of data. NT, in collaboration with VF, was responsible for the quantification of gallic acid and other relevant polyphenols by HPLC–DAD and HPLC–ESI–MS. PN contributed with the preparation of the reference iron complexes, the acquisition of the spectral data, Raman and infrared, as well as with the colorimetric measurements; and with the writing and revision of the version to be published. RJDH and RC oversaw the research on the Iberian written sources, and the first contributed with the reconstructions of historic iron-gall inks, and the writing and revision of the version to be published. VS, at her master thesis, performed the first ink reconstructions and respective molecular characterization. FP contributed with the interpretation of the chemical origins of colour in iron-complexes. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Most of the data on which the conclusions of the manuscript rely is published in this paper, and the full data is available for consultation on request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Portuguese Science Foundation, FCT-MCTES: project PTDC/QUI-OUT/29925/2017; post-doctoral scholarship FOOD-RL1-PHD-QUINOA-01-02, CORES PhD programme for PD/BD/105895/2014; scientific infrastructures RECI/QEQ-MED/0330/2012, REM2013 and the Associated Laboratory for Sustainable Chemistry—Clean Processes and Technologies—LAQV, which is financed by national funds from FCT/MEC (UID/QUI/50006/2015) and co-financed by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007265). Support was also given by the Calouste Gulbenkian Foundation award ‘Estímulo à Investigação 2016′ (146301). FEDER funds through COMPETE, POPH/FSE, QREN. The Spanish Ministry of Economy and Competitiveness & the European Regional Development Fund for project HAR2015-67619-P.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
Iron-gall inks’ molecular data and recipes.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Díaz Hidalgo, R.J., Córdoba, R., Nabais, P. et al. New insights into iron-gall inks through the use of historically accurate reconstructions. Herit Sci 6, 63 (2018). https://doi.org/10.1186/s40494-018-0228-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40494-018-0228-8