
Abstract
Traumatic brain injury (TBI) causes acute and subacute tissue damage, but is also associated with chronic inflammation and progressive loss of brain tissue months and years after the initial event. The trigger and the subsequent molecular mechanisms causing chronic brain injury after TBI are not well understood. The aim of the current study was therefore to investigate the hypothesis that necroptosis, a form a programmed cell death mediated by the interaction of Receptor Interacting Protein Kinases (RIPK) 1 and 3, is involved in this process. Neuron-specific RIPK1- or RIPK3-deficient mice and their wild-type littermates were subjected to experimental TBI by controlled cortical impact. Posttraumatic brain damage and functional outcome were assessed longitudinally by repetitive magnetic resonance imaging (MRI) and behavioral tests (beam walk, Barnes maze, and tail suspension), respectively, for up to three months after injury. Thereafter, brains were investigated by immunohistochemistry for the necroptotic marker phosphorylated mixed lineage kinase like protein(pMLKL) and activation of astrocytes and microglia. WT mice showed progressive chronic brain damage in cortex and hippocampus and increased levels of pMLKL after TBI. Chronic brain damage occurred almost exclusively in areas with iron deposits and was significantly reduced in RIPK1- or RIPK3-deficient mice by up to 80%. Neuroprotection was accompanied by a reduction of astrocyte and microglia activation and improved memory function. The data of the current study suggest that progressive chronic brain damage and cognitive decline after TBI depend on the expression of RIPK1/3 in neurons. Hence, inhibition of necroptosis signaling may represent a novel therapeutic target for the prevention of chronic post-traumatic brain damage.
Similar content being viewed by others


Introduction
With an estimated case load of 69 million per year [1], traumatic brain injury (TBI) represents a leading cause of death and disability in all age groups worldwide, especially in children and young adults. The incidence of TBI is expected to increase in the coming decades, as the number of the two main etiologies—motor vehicle accidents and falls—are expected to rise due to an increase in motorization and an aging population, respectively [2]. The socio-economic impact of TBI is vast, with estimated costs of approximately 400 billion US$ annually [3]. This number does not only include the direct costs due to acute primary care, but also long-term follow-up costs since many TBI survivors suffer from mood changes, memory deficits, and loss of fine motor skills, have difficulties returning to their previous occupation, and therefore require lifelong support [4,5,6]. Consequently, TBI is increasingly recognized as a chronic neurological disorder with socio-economic implications comparable to conditions like Alzheimer’s disease or other neurodegenerative disorders [7].
While the pathophysiology of acute brain damage has been investigated in detail in experimental animals and in humans over the past decades, relatively little is known about the mechanisms determining long-term outcome after TBI. Chronic functional deficits in TBI patients may be caused by progressive brain atrophy in cortex and hippocampus [8, 9] and hydrocephalus formation [10,11,12,13]. So far, clinical and experimental studies suggest that inflammation plays an important role for the development of chronic posttraumatic brain damage [14,15,16,17,18], however, the cellular and molecular mechanisms downstream of this process are not fully understood. Particularly, the trigger and the intracellular signaling cascades causing neuronal cell death weeks and months after TBI are still unknown.
Necroptosis is a form of necrotic regulated cell death, which involves the upstream assembly of the necroptosome complex formed by the interaction of receptor interacting protein kinase 1 and 3 (RIPK1 and 3) [19] and downstream RIPK3-mediated phosphorylation of mixed lineage kinase like protein (MLKL). Necroptosis can be initiated by activation of Toll-like receptors (TLR) 3 and 4, TNF-alpha receptor 1 or, as more recently shown by us and others, by cylindromatosis (CYLD)-mediated deubiquitination of RIPK1 [20]. CYLD is prone to activation by reactive oxygen species (ROS) [21]. Hence, necroptosis may be activated by several events, TNF release, TLR activation, inflammation and ROS production, which are all believed to occur in the brain following TBI [16, 22, 23]. Based on these findings, we hypothesize, that necroptosis may be a relevant intracellular mechanism which triggers chronic neurodegeneration after TBI. To address this issue, we used mice deficient for RIPK1 in neurons or RIPK3 and investigated lesion progression by longitudinal magnetic resonance imaging (MRI), behavioral outcome, and necroptotic signaling up to three months after TBI in a clinically relevant mouse model of TBI. Our results demonstrate that necroptosis is an important novel mediator of chronic neurodegeneration after TBI.
Material and methods
Ethical statement
All animal experiments were reviewed and approved by the Ethical Review Board of the Government of Upper Bavaria. The results of the study are reported in accordance with the ARRIVE guidelines [24]. Animal husbandry, health screens, and hygiene management checks were performed in accordance with Federation of European Laboratory Animal Science Associations (FELASA) guidelines and recommendations [25]. Only male mice between 6 and 8 weeks old mice were used. All surgical procedures, behavioral testing, imaging, and data analysis were performed in a randomized fashion by a researcher blinded to the genotype and group allocation of the animals. Group allocation was obtained by drawing lots by a third party not involved in the study or data analysis.
Animals
Inducible neuronal Ripk1 cKO mice were generated as previously described [26] and bred as Ripk1fl/fl (WT) and Ripk1fl/fl::Camk2a-Cre + /Cre (cKO) in our facility. For induction of neuronal RIPK1 deletion, Ripk1 cKO mice (starting at 4 weeks of age) received three intraperitoneal injections of 2 mg Tamoxifen (Sigma-Aldrich, Taufkirchen, Germany, # T5648) in a 100 µl Miglyol suspension (Caelo, Hilden, Germany, #3274) every 48 h (day 1, 3, 5). RIPK3-deficient mice were generated and kindly provided by V. M. Dixit, Genentech Inc., San Francisco, CA, [27] and bred heterozygously to obtain Ripk3−/− (KO) and Ripk3+/+ (WT) cohorts. The genotype of each RIPK1 and RIPK3 deficient mouse was proven by genotyping (Additional file 1: Figure S1a and b). PCR was performed using the AccuStartTM II Mouse Genotyping Kit (Quanta Biosciences, Beverly, MA, #95,135–500) according to the manufacturer’s instructions. Primers were obtained by Metabion (metabion GmbH Planegg, Germany). Neuronal specific conditional knock-out of Ripk1 was further proven by immunohistochemistry for RIPK1 (Additional file 1: Figure S1c). Induction of neuronal specific Cre recombinase resulted in an 80% reduction of RIPK1 expression in cortical neurons (Additional file 1: Figure S1d). Neither neuronal RIPK1 nor global RIPK3 deficient mice had any obvious phenotype and were born at normal Mendelian distributions.
Controlled Cortical Impact model of traumatic brain injury
Animals were subjected to experimental traumatic brain injury using the previously described Controlled Cortical Impact (CCI) method [23, 28,29,30]. CCI induces a highly reproducible focal lesion and causes progressive brain damage and cognitive decline thereby replicating many acute and chronic characteristics of human TBI [23]. In short, after induction of anesthesia with buprenorphine (0.1 mg/kg Bw) and isoflurane (4%, 30 s), animals were sedated with 1.5–2.5% isoflurane in 30% oxygen and 70% nitrogen under continuous monitoring of body temperature and heart rate. After right parietal craniotomy, the impact was directly applied to the intact dura with a pressure-driven steel piston with a diameter of 3 mm (L. Kopacz, University of Mainz, Germany; 8 m/s impact velocity, 1 mm penetration depth, 150 ms contact time). For sham-surgery, the piston was placed on the dura, but no impact was applied. The craniotomy was resealed with tissue glue (VetBond, 3 M animal care products, St. Paul, MN) and animals were kept in an incubator at 34 °C and 60% air humidity until they regained full motor activity in order to prevent hypothermia. Carprofen (4 mg/kg every 24 h) was administered i.p. for the following 72 h for analgesia.
Experimental time-line
Motor function, depression-like behavior, and memory function were evaluated three days before trauma to obtain baseline values and up to three months thereafter. Lesion volume and tissue iron was evaluated by repetitive MRI up to three months after injury and validated by histology. At the end of the observation time brains were removed for immunohistochemical analysis of lesion volume, tissue iron, astrocyte activation, and microglia morphology (Fig. 1).

Experimental groups, methods, and time line
Body weight and general condition
Animals were weighed daily from three days before CCI until day 7 after trauma, then weekly. General condition, surgery wounds, behavior, nutrition, and fluid balance were checked daily in the early postoperative period, then weekly.
Magnetic resonance imaging and analysis
For longitudinal determination of lesion volume, MRI measurements were performed 15 min, 24 h, 7 days, one, two, and three months after TBI. For all animals, T1 weighted, T2 weighted, and diffusion weighted imaging (DWI) sequences were collected as previously described [23] under isoflurane anesthesia (1–1.5% in 30% oxygen/70% nitrogen) and multimodal monitoring of physiological parameters using a 3 T nanoScan® PET/MR (Mediso, Münster Germany). Sequences were collected in the following order: T2-weighted imaging (2D fast-spin echo (FSE), TR/TE = 3000/57.1 ms, averages 14, matrix size = 96 × 96; field of view = 16 mm × 16 mm; slice thickness = 500 µm, interslice gap = 60 µm), T1-weighted imaging (2D fast-spin echo (FSE), TR/TE = 610/28,6 ms, averages 14, matrix size = 96 × 96; field of view = 16 mm × 16 mm; slice thickness = 500 µm, interslice gap = 60 µm). Total imaging time was approximately 35 min per mouse and time-point. Lesion volume was measured using ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2018) in T2 sequences. 14 slices surrounding the lesion were chosen for each dataset and the area segmented using the polygon tool. Volume was then calculated using the following equation:
with d being the distance between slices in mm (slice thickness + interslice gap), and A being the measured area in mm3.
Hippocampal atrophy was assessed in T2-weighted images as previously described [23]. One section located in the center of the lesion containing the hippocampus was chosen at the same position for each animal. Areas of both hippocampi were segmented using the polygon tool and the ipsilesional area of the hippocampus was expressed as % of the area of the uninjured contralateral hippocampus.
Behavioral testing
To exclude age-related factors as a cause for behavioral changes during the observation period, all behavioral tests were performed with an additional control group of non-traumatized (naive) RIPK3 or RIPK1 deficient mice (Fig. 1).
Motor function—Beam Walk
The Beam Walk Test was performed as previously described [23, 28, 30] on a 1 m long and 1 cm wide suspended wooden rod. Time to cross the beam and the number of missteps was recorded. Animals with more than two missteps in pre-trauma testing were excluded from randomization.
Memory and learning behavior—Barnes Maze
The Barnes Maze test, a well-established paradigm for assessing memory function [31, 32], was performed 1, 2, and 3 months after CCI as previously described [23]. In short, the animal was placed on a brightly lit round platform with 20 identical holes along its outer rim and trained to locate a box affixed below one of the apertures (home cage) as fast as possible. Time to reach the home cage (latency) as well as distance travelled and walking speed were recorded and analyzed using a video tracking software (EthoVison XT©, Version 11, 2014 Noldus Information Technology). Animals were trained for four consecutive days and memory function was evaluated on the sixth day.
Tail Suspension test
The Tail Suspension test is a paradigm to assess depression-like behavior in rodents and was performed as previously described [23]. Briefly, animals were suspended by the tail for three minutes and their movements recorded and analyzed using a video tracking software (EthoVison XT©, Version 11, 2014, Noldus Information Technology). The time of inactivity was used as a proxy for depression-like behavior [33].
Histological assessment
Lesion volume/ hippocampus volume
Three months after TBI, animals were fixed with 4% PFA in deep anesthesia by transcardial perfusion. Fourteen sequential 50 µm thick coronal sections were cut at 500 µm intervals on a vibratome (Leica, Germany) in order to match the tissue volume investigated by MRI, stained with cresyl violet according to Nissl, and evaluated by histomorphometry for lesion volume using the following formula as previously described [23]:
The volume of the hippocampus in the traumatized hemisphere was determined in six slices one mm anterior until four mm posterior to bregma and then normalized to the contralesional side.
Iron deposits
One section per animal was stained with Prussian blue (Iron Stain Kit, Sigma-Aldrich, # HT20) at -1.5 mm from bregma to visualize iron deposits using a light microscope (Axioscope, Carl Zeiss Microscopy GmbH, Jena Germany). Tile scans were then processed in ImageJ by using the color deconvolution tool to separate color channels. The blue channel was binarized and the integrated density was measured using the particle analysis plugin. Values of the traumatized hemisphere were expressed as percentage of the contralateral side.
Immunohistochemistry
Fifty micrometer thick floating coronal sections were prepared as previously described [34]. Blocking and incubation with the primary antibody was performed in 1% bovine serum albumin, 0.1% gelatin from cold water fish skin, 0.5% Triton X-100 in 0.01 M PBS at pH 7.2–7.4 for 72 h at 4 °C. The following primary antibodies were used: IBA-1 (rabbit, Wako, #019–19,741, 1:200), GFAP-Cy3 (mouse, Sigma Aldrich, #2905, 1:200), RIPK1 (rabbit, Novusbio, #NBP1-77077SS, 1:100), NeuN (guinea pig, Synaptic Systems, #266 004), and phosphorylated MLKL (rabbit, Cell signaling technology, # 91689S, 1:100). After incubation, sections were washed in PBS and incubated with the following secondary antibodies: anti-rabbit coupled to Alexa-fluor 594 (goat anti-rabbit, Thermo Fisher Scientific, #A-11012), anti-guinea pig coupled to Alexa-fluor 488 (goat anti-guinea pig, Thermo Fisher Scientific, # A-11073), and anti-mouse coupled to Alexa-fluor 647 (goat anti-mouse, Thermo Fisher Scientific, #A- 32,728) in 0.01 M PBS at pH 7.2–7.4 containing 0.05% Tween 20. Nuclei were stained with 4’,6-Diamidin-2-phenylindol (DAPI, Invitrogen, #D1306) 1:10,000 in 0.01 M PBS.
Imaging was performed using a ZEISS LSM 900 confocal microscope (Carl Zeiss Microscopy GmbH, Jena Germany). GFAP staining was recorded using a 10 × objective (EC Plan-Neofluar 10x/0.30 Pol M27) with an image matrix of 512 × 512 pixel, a pixel scaling of 0.2 × 0.2 μm and a depth of 8 bit. Whole brain images were collected in z-stacks as tile scans with a slice-distance of 2 μm and a total range of 14 μm [35]. For microglia analysis, images were acquired using a 40 × objective (EC Plan-Neofluar 40x/1.30 Oil DIC M27) with an image matrix of 1024 × 1024 pixel, a pixel scaling of 0.2 × 0.2 μm and a depth of 8 bit. Specific regions of interest were collected in Z-stacks to include the entire slice thickness with a slice-distance of 0.4 μm at 100 μm away from the lesion in the hippocampus and at 300 μm in the cortex. For p-MLKL staining, a 5 × objective was used (EC Plan-Neofluar 5x/0.16 Pol M27) with an image matrix of 1434 × 1434 pixel, a pixel scaling of 3.321 × 3.321 μm and a depth of 8 bit. Whole brain images were collected in z-stacks as tile scans with a slice-distance of 5 μm and a total range of 25 μm. To demonstrate the intracellular localization of p-MLKL, a 100 × objective (Epiplan-Neofluar 100x/1.3 Oil Pol M27) was used with an image matrix of 512 × 512 pixel, a pixel scaling of 0.166 × 0.166 μm and a depth of 8 bit. Images were collected in z-stacks with a slice-distance of 0,280 μm and a total range of 5.6 μm. After obtaining a maximum intensity projection, images were imported into ImageJ [36] and intensity of p-MLKL measured in the rim of the lesion and normalized to the signal of DAPI to correct for differences in staining. The corrected signal was then normalized to the same sized region of interest on the contralateral hemisphere.
Analysis of astrocyte coverage
Assessment of astrocyte coverage was performed using ImageJ in sections stained for GFAP (see above). Z -stacks were imported into Fiji and split into individual channels. GFAP intensity in five ROI (250 × 250 μm; at 0, 250, 500, 750, and 1000 μm distance from the lesion in the striatum) was then measured using the mean grey value and normalized to the measurements of the contralesional hemisphere to adjust for possible differences in staining intensity.
Analysis of microglia coverage and morphology
Microglia coverage was manually assessed in maximum intensity projections of iba-1 stained sections. One section per animal was chosen at 1.5 mm from bregma and two ROIs chosen on the ipsilesional hemisphere, one in layer V of the cortex at 300 µm away from the lesion and one in the CA1a region of the hippocampus. The number of microglia was normalized to total DAPI positive cell count and expressed in as percentage of coverage in sham operated animals.
To assess microglia morphology, Sholl and fractal analysis were performed to indicate ramification, cell range, total cell size, and circularity using a modified protocol from Young and Morrison [37]. Z-stack images were converted to a maximum intensity projection and cells were individually cut out using the polygon selection tool in ImageJ [35]. Only cells fully captured within the z-stack were selected. After background subtraction, images were binarized and resized to 600 × 600 pixels keeping the original scale. Speckles or debris around the cells were removed using the paintbrush tool. Sholl analysis was performed using the Sholl analysis plugin in ImageJ [38]. Centered on the soma, concentric circles with an increasing radius of 2 μm were drawn, the number of intersections measured at each radius. After converting binary images to outlines, fractal analysis was performed using the FracLac plugin for ImageJ [39]. As described previously [37], the total number of pixels present in the cell image of either the filled or outlined binary image were calculated and later transformed to μm2 (pixel area = 0.208 μm2). Cell circularity was calculated as Circularity = 4*π*Area/Perimeter2. Maximum span across the convex hull represents the maximum distance between two points in the convex hull.
Statistical analysis
Sample size was calculated with the following parameters: alpha error = 0.05, beta error = 0.2, calculated standard deviation ranged from 15 to 20% (depending on the parameter investigated), and biologically relevant difference = 30%. All data is given as mean ± standard deviation (SD) if not indicated otherwise. For comparison between groups, Student t-test was used for normally distributed data and Mann–Whitney Rank Sum test for non-normally distributed data according to the result of Shapiro–Wilk normality test. Measurements over time were tested between groups using One-way or Two-way ANOVA for Repeated Measurements, followed by Tukey’s multiple comparisons test for normally and Holm-Sidak's multiple comparisons test for non-normally distributed data as post hoc test. Calculations were performed with Sigma Plot version 14.0 (Systat Software GmbH, Erkrath, Germany).
Results
A total of 33 male RIP1 deficient mice (naïve RIP1fl/fl Cre group: n = 4, RIP1fl/fl Cre and RIP1fl/fl sham groups = 5 each, CCI RIP1fl/fl group: n = 10, RIP1fl/fl Cre group: n = 9) and 32 male RIP3 deficient mice (naïve RIP3−/−, RIP3+/+, and RIP3−/− sham group: n = 5 each, CCI RIP3+/+ group n = 9, CCI RIP3−/− group n = 8) were operated, assessed, and analyzed for the present study. One RIP3+/+ animal was excluded from randomization and not used for the study due to its performance in the Beam Walk Test before TBI (more than 2 missteps at baseline).
Traumatic brain injury induces long-term necroptotic signaling in neurons
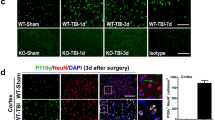
Phosphorylated MLKL (pMLKL) was used as a specific marker for necroptosis [40]. Three months after TBI large amounts of pMLKL were found in the rim of the traumatic cavity, the presumed site of progressive chronic post-trauma brain damage (Fig. 2a, upper panel). pMLKL was found by high resolution confocal imaging in the cytoplasm of selected neurons as small dots, suggesting that pMLKL is part of a protein complex such as the necrosome (Fig. 2a, lower panel, white arrowheads). pMLKL staining was almost absent in neuronal RIPK3 deficient mice suggesting that necroptotic signaling in neurons did essentially not occur in these animals (Fig. 2b). Quantification of pMLKL staining showed a highly significant increase of activated MLKL in wild type mice of both strains, while neuronal Ripk1 or global Ripk3 knock-out completely blunted this response (Fig. 2c and d).

Phosphorylated MLKL, a marker of necroptosis is reduced in RIP knockout animals. a. Exemplary staining of pMLKL and NeuN (upper panel) and a pMLKL positive neuron at higher magnification (lower panel) in the rim of a traumatic contusion in a wild type mouse. b. pMLKL is significantly reduced in a RIPK3 deficient mouse. c and d p-MLKL signal intensity was significantly increased after TBI in wild type animals (white bars) compared to sham animals indicating significant presence of necroptosis three months after TBI; this increase was significantly blunted in RIPK1 (c) as well as in RIPK3 d mice where there was no difference between TBI and sham animals. Data are presented as mean ± SD; n = 9–10 for RIPK1, n = 8–9 for RIPK3. Two-way RM ANOVA with Tukey’s multiple comparisons test was used. ***p < 0.001, n.s. indicates no significant statistical difference between groups
Chronic posttraumatic brain damage is reduced in RIPK1 or RIPK3 deficient mice
After demonstrating neuronal necroptotic signaling three months after TBI in wild type mice and showing that RIPK1 or 3 deficiency prevented this process, we evaluated lesion volume in cortex and hippocampus of wild type and RIPK deficient mice by longitudinal MR imaging. Two animals (one Ripk1 fl/fl::CamK2a Cre and one Ripk3 WT) of the study cohort died for unknown reasons before TBI and were excluded from analysis. All other animals completed the study. Anesthesia and craniotomy did not have any influence on general outcome parameters: sham-operated and CCI animals recovered equally well from surgery in terms of bodyweight (Ripk1 cKO: Additional file 1: Figure S2a, Ripk3 KO: Additional file 1: Figure S2b) and general health score (Ripk1 cKO: Additional file 1: Figure S2c, Ripk3 KO: Additional file 1: Figure S2d). Exemplary three dimensional reconstructions of the brain showed a large lesion and a small hippocampus in the ipsilateral hemisphere of traumatized wild type mice three months after TBI, while the lesion was significantly smaller and the hippocampus significantly larger in neuronal RIPK1 deficient mice (Fig. 3a). Longitudinal investigation of lesion size and hippocampal volume by repetitive MRI showed that the primary damage measured 15 min after trauma was comparable in RIPK1 deficient mice and their respective wild type controls (Fig. 3b, t = 15 min: Ripk1 cKO: 18.4 ± 2.6 mm3, wt: 18.4 ± 1.7 mm3) indicating that the initial trauma was similar in all investigated animals. In agreement with previous results in this model, lesion size peaked 24 h after TBI in both experimental groups (Ripk1 cKO: 27.0 ± 1.8 mm3, + 47% vs. 15 min; wt: 29.8 ± 4.3 mm3, + 61% vs. 15 min) as a representation of acute secondary brain damage. Lesion volume was not different between wild type and neuronal RIPK1 deficient mice at this time point indicating that necroptosis does not play a significant role for acute lesion progression. Within the first month after TBI, removal of necrotic tissue and scar formation resulted in an apparent shrinkage of the lesion. One to two months after TBI progressive loss of brain tissue started to occur, a process we previously demonstrated to continue for at least one year after experimental trauma [23]. Chronic post-trauma tissue injury was significantly reduced in neuronal RIPK1 deficient mice as compared to their wild type littermate controls (t = 1 month: Ripk1 cKO: 5.7 ± 1.6 mm3, wt 8.2 ± 1.9 mm3, p = 0.0355, t = 3 mon Ripk1 cKO: 6.5 ± 1.7 mm3, wt: 11.3 ± 2.0 mm3, p = 0.0002). A similar dynamic was seen in RIPK3 deficient animals. Acute injury was not affected by RIP 3 knock-out (Fig. 3c t = 24 h, Ripk3 KO: 24,8 ± 3,3 mm3, wt: 26,1 ± 5,3 mm3), while chronic lesion progression was significantly attenuated from one to three months after TBI (t = 1 month: Ripk3 KO: 8.8 ± 2.9 mm3, wt: 14.0 ± 1.9 mm3, p = 0.006; t = 3 months: Ripk3 KO: 7.3 ± 1.3 mm3, wt: 11.6 ± 3.6 mm3, p = 0.04). Individual traces for lesion volumes in each animal are given in Additional file 1: Figure S3a (RIP1 KO) and Additional file 1: Figure S3b (RIP3 KO). The MRI findings were corroborated by histopathological evaluations, i.e. acute brain damage 24 h after TBI was similar in RIP deficient and wild type animals (Additional file 1: Figure S4), while chronic brain damage three month after TBI correlated well with the injury assessed by MRI (Additional file 1: Figure S5a and b) and was significantly reduced in both knock-out strains (Additional file 1: Figure S5c and d).

RIPK1 and RIPK3 deficiency significantly reduces posttraumatic brain damage. a 3D reconstruction of lesion volume (blue) in relation to ipsi- and contralateral hippocampus (green) for a RIPK3 wild type and a RIPK3 deficient mouse 3 months after TBI. Scale bar = 5 mm. b and c Lesion volume over time quantified by repetitive T2-weighted MR imaging in RIPK1 (b) and RIPK3 (c) knockout animal. d and e. Hippocampal atrophy over time assessed in longitudinal T2-weighted MRI. RIPK1 (d) and RIPK3 (e) knockout mice show better preservation of hippocampal tissue over time. Mean ± SD; n = 9–10 for RIPK1, n = 8–9 for RIPK3. Two-way RM ANOVA with Sidak's multiple comparisons test was used. *p < 0.05, **p < 0.005, ***p < 0.001
Since memory deficits are a hallmark of chronic posttraumatic brain damage in mice and TBI patients [23, 41, 42], we next investigated long-term hippocampal damage by MRI. In the current TBI model, the hippocampus is only marginally injured acutely after TBI, but severely affected by progressive chronic damage as previously shown [23]. All wild type mice showed significant loss of hippocampal tissue in the traumatized hemisphere already one month after TBI (Fig. 3d and e, open bars). Starting one month after trauma, hippocampal loss was significantly less pronounced in neuronal RIPK1 and global RIPK3 deficient mice (Fig. 3d, RIPK1: reduction to 74.9 ± 18.5%, wt: reduction to 40.3 ± 18.7% of contralateral hippocampus, p = 0.0004; Fig. 3e, RIPK3: reduction to 78.0 ± 30.7%, wt: reduction to 48.2% ± 19.6% of contralateral hippocampus, p = 0.01). This significant difference persisted until the end of the observation period three months after TBI and was corroborated by histology (Additional file 1: Figure S5e and f).
Astrogliosis and microglial activation are reduced in RIPK1 and RIPK3 deficient mice
The formation of a glial scar and the activation of microglia are other hallmarks of chronic brain damage after TBI [43, 44]. Indeed, we observed a marked increase in glial fibrillary acid protein (GFAP), an astrocyte marker, in the rim of the traumatic cavity and in perilesional tissue in wild type mice three months after TBI (Fig. 4a, left panel). Quantification of GFAP expression showed an almost four-fold increase in the rim of the lesion with a decreasing intensity towards perilesional areas (Fig. 4b and c, open bars). GFAP expression was far less pronounced in neuronal specific RIPK1 deficient mice (Fig. 4a, right panel). Quantification of astrocyte density by pixel-based analysis corroborated these findings and reveled that activation of astrocytes was significantly decreased by 25–35% in neuronal specific RIPK1 and in global RIPK3 deficient mice (Fig. 4b and c, closed bars).

RIPK1 and RIPK3 deficiency reduces reactive astrogliosis three months after TBI. a Exemplary GFAP stainings in a wild type (left) and a RIPK1 knockout (right) animal at different distances from the traumatic contusion. Both images show the right hemisphere, but the left image has been mirrored along the midline for better visualization. Scale bar = 20 µm. Significant astrogliosis is present in the WT mouse, while it is heavily reduced in the neuronal RIPK1 deficient mouse. b and c Quantification of astrocyte coverage at different distances from the lesion. Astrocyte coverage was decreased in neuronal RIPK1 (b) and global RIPK3 (c) knockout animals compared to controls. Data are presented as mean ± SD; n = 9–10 for RIPK1, n = 8–9 for RIPK3. Students t-test for parametric and Man-Whitney-Rank-Sum test for non-parametric data were used. *p < 0.05, **p < 0.005, ***p < 0.001
Staining for the microglia marker Ionized calcium-binding adaptor molecule 1 yielded similar but more lesion associated findings three months after TBI (Iba1; Fig. 5). In wild type mice Iba-1 staining was most pronounced within a distance of 100 µm from the rim of the traumatic cavity, while only subtle changes were observed in areas 300 µm away from the lesion site (Fig. 5, WT). The density of iba-1 staining was heavily reduced in neuronal specific RIPK1 deficient mice (Fig. 5, Ripk1 cKO). To quantify these changes, we assessed tissue coverage, area, circularity, and maximal span of microglia in the rim (100 µm) and in the vicinity (300 µm) of the traumatic lesion (Fig. 5b–i). In wild type mice all investigated parameters pointed towards a significant activation of microglia near the rim of the lesion, i.e. the coverage and circularity increased, while the area and the maximal span of microglia decreased (Fig. 5b–i, open bars). Microglia activation and the number of microglial branch points were significantly reduced and partly normalized in neuronal specific RIPK1 and in global RIPK3 deficient mice (Fig. 5b–m, closed symbols), suggesting that less neuronal cell death was associated with less microglial activation.

RIPK1 and RIPK3 deficiency reduces microglia activation. a. Exemplary stainings for the microglia marker iba1 in WT (upper inserts) and neuronal RIPK1 deficient mice (lower inserts) 100 µm (left inserts) and 300 µm (right inserts) from the rim of the lesion. b-i. Coverage and fractal analysis of microglia. In areas closer to the lesion site (100 µm, left side of each panel), knockout animals of both lines showed a decrease in microglia coverage (b. RIP 1, d. RIPK3) compared to their wild type littermates. Fractal analysis revealed that microglia of knockout animals in proximity to the lesion have less processes (c. RIP 1, e. RIPK3) are less circular (f. RIP 1, h. RIPK3), and overall smaller (g. RIP 1, i. RIPK3). In the more distal region, cells resembled those in sham operated animals, with no differences between genotypes. j-m. Sholl analysis also shows increased ramification, i. e. more active cells, close to the lesion (j. RIP 1, l. RIPK3), but not further away from the lesion site (k. RIP 1, m. RIPK3). Data are presented as mean ± SD; n = 9–10 for RIPK1, n = 8–9 for RIPK3. Student t-test for normalized and Man-Whitney-Rank-Sum-test for non-normalized data was used. *p < 0.05, **p < 0.005, ***p < 0.001. n.s. indicates no significant statistical difference between groups
Neuronal RIPK1 and RIPK3 deficiency improves cognitive outcome three months after TBI
To investigate whether the reduction of lesion size, hippocampal damage, scar formation, and microglial activation had an effect of functional outcome, we investigated motor function by beam walk, depression-like behavior by the Tail Suspension test, and long-term memory using the Barnes Maze test. TBI significantly deteriorated motor function and induced depression-like behavior compared to pretrauma performance as previously described [23], genetic deletion of RIPK1 or RIPK3, however, had no effect on these parameters (Fig. 6a–d). Long-term memory was normal in non-traumatized Ripk1 or Ripk3 knock-out mice; it was, however, significantly disturbed in traumatized animals, i.e. TBI increased the time needed to find the home cage, the latency to goal, by more than ten times and memory loss progressed over time (Fig. 6e and f, triangles and open circles). In neuronal specific RIPK1 and in global RIPK3 deficient mice, however, long-term memory function was almost completely preserved and resembled that of not traumatized animals (Fig. 6e and f, closed circles). Hence, protection of hippocampal neurons by genetic deletion of RIP 1 or RIPK3 resulted in preserved long-term memory function.

RIPK1 and RIPK3 deficiency improves neurocognitive performance three months after TBI. a and b Motor impairment after TBI. Beam Walk Test revealed long term impaired motor function of the left hind limb in TBI animals (missteps compared to respective baseline, # for WT, * for KO), but there were no differences between a RIPK1 or b RIPK3 knockout animals and their respective controls. c and d Depression-like behavior after TBI. Mice of both strains showed an increase of total immobility time throughout the time course of three months, however no significant differences could be detected between RIPK1 (c) and RIPK3 (d) knockout animals compared to wild type. e and f Learning and memory dysfunction after TBI. CCI induces severe long-term memory deficits in WT mice (open circles) while RIPK1 or RIPK3 knockout animals (grey circles) show similar long-term memory function as uninjured littermate controls. Data are presented as mean ± SD; n = 9–10 for RIPK1, n = 8–9 for RIPK3. Two-way RM ANOVA with Tukey’s multiple comparisons test was used. *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.0001
Chronic lesion progression after TBI is associated with iron deposits and reduced in RIP-deficient mice
Since our data suggest that necroptotic signaling is important for chronic brain damage after TBI, we were interested to identify the mechanisms triggering this process. Traumatic contusions are associated with hemorrhage and subsequent deposition of iron in perilesional brain parenchyma. Since free iron is well-known to trigger ferroptotic [45] and possibly necroptotic cell death signaling [46], we hypothesized that chronic posttraumatic brain damage may be associated with iron deposition. Since iron can alter MRI signals, we looked for signal alterations using MRI scans. Indeed, we found hyperintense signals at the border of the lesion one month after TBI by T1-weighted MRI (Fig. 7a, upper panel) and could demonstrate that these signals showed a close spatial correlation with iron deposits as identified by Prussian blue staining (Fig. 7a, lower panel and Fig. 7b). Comparison of the volume of iron deposits between wild type and RIP deficient mice revealed equal amounts of iron in all animals, suggesting that the amount of hemorrhage was equal in all experimental groups (Fig. 7c and d). In a next step, we investigated the spatial and temporal relationship between chronic lesion expansion and iron deposits. For this purpose, we recorded iron deposits one month and lesion area three months after TBI, a time point when the lesion already expanded. In wild type mice the rim of the lesion area, the site of lesion progression, colocalized with iron deposits, while in neuronal specific RIPK1 deficient mice colocalization was minimal (Fig. 7e). The quantification of lesion area and iron deposition in wild type, RIPK1 and RIPK3 deficient mice, demonstrated that tissue loss colocalizing with histopathological detection of iron was significantly reduced in RIP deficient animals, suggesting that free iron may be involved in the pathophysiology of chronic neuronal necroptosis following TBI (Fig. 7f and g).

Lesion progression occurs in areas with iron deposits. a T1-weighted MRI (upper panel) and Prussian blue staining (lower panel) three months after injury. There is a close spatial correlation between the T1-hyperintense signal and iron staining (arrowheads). b There is high spatial correlation between the area of T1 hyperintensities and iron deposits as assessed by Prussian blue staining. Pearson product-moment correlation analysis. c and d Iron deposits in pericontusional brain tissue assessed by longitudinal MRI. Extent of hemorrhage is comparable in RIPK1 (c) and RIPK3 (d) knockout animals and controls, indicating no differences in hemorrhage size after TBI between groups. T1 hyperintensities decreased over time in all groups, suggesting a very slow resorption of iron over time. e. Co-localization of iron deposits (red) observed at 1 month after TBI (upper panels) and lesion size assessed at the end of the observation period (3 months, middle row, green) suggests a progressive expansion of the lesion towards the regions with iron deposits. f and g Quantification of overlap between iron deposits and lesion. The higher the overlap of iron deposits and lesion size at three months, the higher the rate of tissue loss/ cell death in iron containing tissue. Co-localization is significantly less pronounced in RIPK1 (f) or RIPK3 (g) deficient mice, suggesting a reduced lesion growth in RIP knockouts due to toxic iron residues. Data are presented as mean ± SD; n = 9–10 for RIPK1, n = 8–9 for RIPK3. Two-way RM ANOVA with Tukey’s multiple comparisons test was used. **p < 0.005, ***p < 0.001
Discussion
It is increasingly recognized that next to its acute sequelae, traumatic brain injury is a chronic disease [14]. Chronic post-trauma brain damage is associated with inflammation, persists for years after the initial insult, and may spread to areas initially not affected by the initial impact [47]. Affected patients often suffer from neuro-cognitive and mood disorders, personality changes, neurocognitive dysfunction, or even dementia [48,49,50,51,52,53,54]. So far, no therapeutic concepts targeting the long-term sequelae of TBI exist as the pathophysiology of chronic traumatic brain injury is still poorly understood.
Here, we propose, to our knowledge for the first time, that programmed cell death signaling mediates chronic neuronal injury after TBI. More specifically, we identified the necroptosis signaling molecules RIPK1 and RIPK3 to be major players in this process. Neurons affected by chronic traumatic damage showed necroptotic signaling as evidenced by enhanced levels of pMLKL. Moreover, Ripk3 global knockout animals as well as neuronal RIPK1 deficient mice were significantly protected from chronic brain injury and showed improved neurocognitive function up to three months after TBI. Of note, no protection was observed within the first days after TBI, suggesting that necroptosis is not involved in acute injury, but specifically mediates chronic traumatic brain damage. Hence, our data further suggest that mechanistically acute and chronic neuronal cell death seem to be mediated by different processes.
Longitudinal MRI and histological assessment revealed that lesion progression was associated with parenchymal iron deposition and that neuronal or global deletion of RIPK1 or RIPK3 prevented lesion progression. These findings suggest that chronic traumatic brain damage may be triggered by iron and mediated by necroptotic signaling. The association of lesion progression with iron deposits is intriguing and may indicate that iron plays an important role in this process, however, further experiments addressing this issue in more detail will need to further evaluate whether there is a causal or just a correlative relationship between iron deposition and neuronal necroptosis.
Hemorrhage and the subsequent degradation of red blood cells releases large amounts of hemoglobin, heme, and free iron, i.e. molecules with high cytotoxic activity, into brain tissue [55]. From numerous studies investigating intraparenchymal hemorrhage, a subtype of hemorrhagic stroke, it is well known that specifically free iron generates reactive oxygen species thereby damaging cell membranes and causing tissue damage and neurological dysfunction [36, 56,57,58,59]. Cerebral macro- and microhemorrhages are common after TBI [60,61,62]. Specifically, microbleeds have been shown to exert toxic effects on endothelial cells, astrocytes, neurons, oligodendrocytes, and microglia and may thus lead to blood–brain barrier damage, neuronal cell death, demyelination, and chronic inflammation [63]. The importance of blood degradation products for the pathophysiology of TBI is further demonstrated by the fact that the presence and extent of hemorrhage show a close correlation with injury severity and long-term clinical outcome in TBI patients [63, 64]. In line with these clinical studies, we previously demonstrated that the TBI model used in current study shows acute macro-hemorrhage, which, however, resolves within the first week after injury [23]. In the current study we now show by MRI and Prussian blue staining that iron persists in pericontusional tissue for up to three months after the initial impact. Iron deposits identified in still viable pericontusional brain tissue one month after TBI co-localized with damaged tissue three months after injury, suggesting that chronic lesion progression preferably occurred in areas with previous hemorrhage and subsequent iron deposition. Since this process was significantly attenuated in neuronal Ripk1 and global Ripk3 knockout animals, our findings suggest that pericontusional iron may be involved in chronic posttraumatic lesion expansion and that this process is mediated by necroptotic signaling in neurons.
So far, neuroinflammation was believed to be the main cause of chronic brain damage after TBI [44]. However, the mechanisms by which microglial activation promotes neuronal injury and death remain elusive. Further, microglial activation after TBI may exert beneficial as well as detrimental effects, i.e. tissue regeneration versus accelerated damage, respectively. Therefore, disentangling these opposite functions of microglia may have important therapeutic consequences. Our current data suggest that the final steps causing neuronal cell death during chronic post-trauma brain damage depend on RIPK1 and RIPK3 activity. Based on these findings, we suggest a hypothetical scenario in which chronic post-trauma brain damage is initiated by the ongoing production of reactive oxygen species (ROS) by inflammatory cells. Physiological concentrations of ROS are usually well tolerated by cells since they are detoxified to water and oxygen by the glutathione system and catalases [65]. However, in the presence of iron, hydrogen peroxide is converted to highly reactive hydroxyl radicals by the Fenton reaction and may initiate a form of programmed cell death called ferroptosis [66, 67]. The link between ferroptosis and RIPK1/3-mediated necroptosis in neurons is not fully established, but may be mediated by cylindromatosis (Cyld), a deubiquitinase able to activate RIPK1 and downstream necroptosome formation under conditions of oxidative stress as we recently demonstrated [21]. We showed that CYLD-dependent RIPK1/RIPK3 necrosome-formation occurred in neuronal cells exposed to ferroptosis activators and knockdown of the necroptosis-mediators CYLD, RIPK1 or RIPK3 attenuated cell death. Interestingly, the role for CYLD in ferroptosis also translated into neuroprotective effects in vivo, since CYLD knockout mice showed reduced secondary brain damage after TBI compared to controls [21]. These findings are corroborated by results in models of hemorrhagic stroke suggesting that necroptosis and ferroptosis are indeed interconnected under conditions of blood-induced tissue damage [68].
Despite its obvious strengths, the current study also has some notable limitations. We studied only young male animals and are therefore not able to make any statements on the role of necroptosis in the aged or female brain. Further, due to technical limitations, such as the lack of specific antibodies for the study of necroptosis in brain tissue, we were only able to demonstrate the involvement of a single signaling molecule downstream of RIPK activation, namely pMLKL. Thus, future studies using novel experimental tools will need to define necroptotic signaling after TBI in more detail. Another shortcoming of the current study is that we demonstrate only a spatial correlation between iron deposition and necroptosis. Thus, further studies are needed to further clarify whether there is a causal relationship between iron deposition and neuronal necroptosis. Finally, we want to point out that memory tests in mice are sometimes hard to interpret since results may be influenced by differences in motor function or the level of disinhibition which are well known to occur after TBI. We controlled for differences in motor function and the intensity of exploratory behavior between groups and are confident that the presented data indeed reflect memory function, however, the results need nevertheless to be interpreted with caution.
In conclusion, the current study provides evidence that RIPK1 and RIPK3 are critically involved in chronic post-trauma brain damage. Further, our findings suggest that free iron may be involved in this process. Our results therefore help to better understand the mechanisms of chronic post-trauma brain damage and suggest that RIPK1- and RIPK3-mediated necroptosis may represent a novel therapeutic target for the treatment of patients suffering from the long-term sequels of TBI.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Change history
04 November 2021
This article has been updated to add the missing funding information.
References
Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM et al (2018) Estimating the global incidence of traumatic brain injury. J Neurosurg. https://doi.org/10.3171/2017.10.JNS17352
Injury GBDTB, Spinal Cord Injury C (2019) Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet Neurology 18: 56-87https://doi.org/10.1016/S1474-4422(18)30415-0
Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, Bragge P, Brazinova A, Buki A, Chesnut RM et al (2017) Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol 16:987–1048. https://doi.org/10.1016/S1474-4422(17)30371-X
Rabinowitz AR, Levin HS (2014) Cognitive sequelae of traumatic brain injury. Psychiatr Clin North Am 37:1–11. https://doi.org/10.1016/j.psc.2013.11.004
Selassie AW, Zaloshnja E, Langlois JA, Miller T, Jones P, Steiner C (2008) Incidence of long-term disability following traumatic brain injury hospitalization, United States, 2003. J Head Trauma Rehabil 23:123–131. https://doi.org/10.1097/01.HTR.0000314531.30401.39
Silver JM, McAllister TW, Arciniegas DB (2009) Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry 166:653–661. https://doi.org/10.1176/appi.ajp.2009.08111676
Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S (2018) Traumatic brain injury and Alzheimer’s disease: the cerebrovascular link. EBioMedicine 28:21–30. https://doi.org/10.1016/j.ebiom.2018.01.021
MacKenzie JD, Siddiqi F, Babb JS, Bagley LJ, Mannon LJ, Sinson GP, Grossman RI (2002) Brain atrophy in mild or moderate traumatic brain injury: a longitudinal quantitative analysis. AJNR Am J Neuror 23:1509–1515
Reider-Groswasser I, Cohen M, Costeff H, Groswasser Z (1993) Late CT findings in brain trauma: relationship to cognitive and behavioral sequelae and to vocational outcome. AJR Am J Roentgenol 160:147–152. https://doi.org/10.2214/ajr.160.1.8416613
Cardoso ER, Galbraith S (1985) Posttraumatic hydrocephalus–a retrospective review. Surg Neurol 23:261–264. https://doi.org/10.0090/3019(85)90092-8[pii]
Guyot LL, Michael DB (2000) Post-traumatic hydrocephalus. Neurol Res 22:25–28
Honeybul S, Ho KM (2012) Incidence and risk factors for post-traumatic hydrocephalus following decompressive craniectomy for intractable intracranial hypertension and evacuation of mass lesions. J Neurotrauma 29:1872–1878. https://doi.org/10.1089/neu.2012.2356
Mazzini L, Campini R, Angelino E, Rognone F, Pastore I, Oliveri G (2003) Posttraumatic hydrocephalus: a clinical, neuroradiologic, and neuropsychologic assessment of long-term outcome. Arch Phys Med Rehabil 84:1637–1641
Faden AI, Loane DJ (2015) Chronic neurodegeneration after traumatic brain injury: alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 12:143–150. https://doi.org/10.1007/s13311-014-0319-5
Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136:28–42. https://doi.org/10.1093/brain/aws322
Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014) Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73:14–29
Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G et al (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70:374–383. https://doi.org/10.1002/ana.22455
Ritzel RM, Doran SJ, Barrett JP, Henry RJ, Ma EL, Faden AI, Loane DJ (2018) Chronic alterations in systemic immune function after traumatic brain injury. J Neurotrauma 35:1419–1436. https://doi.org/10.1089/neu.2017.5399
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323. https://doi.org/10.1016/j.cell.2008.10.044
Moquin DM, McQuade T, Chan FK (2013) CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 8:e76841. https://doi.org/10.1371/journal.pone.0076841
Ganjam GK, Terpolilli NA, Diemert S, Eisenbach I, Hoffmann L, Reuther C, Herden C, Roth J, Plesnila N, Culmsee C (2018) Cylindromatosis mediates neuronal cell death in vitro and in vivo. Cell Death Differ 25:1394–1407. https://doi.org/10.1038/s41418-017-0046-7
Krieg SM, Voigt F, Knuefermann P, Kirschning CJ, Plesnila N, Ringel F (2017) Decreased secondary lesion growth and attenuated immune response after traumatic brain injury in Tlr2/4(-/-) mice. Front Neurol 8:455. https://doi.org/10.3389/fneur.2017.00455
Mao X, Terpolilli NA, Wehn A, Chen S, Hellal F, Liu B, Seker B, Plesnila N (2019) Progressive histopathological damage occurring up to one year after experimental traumatic brain injury is associated with cognitive decline and depression-like behavior. J Neurotrauma: Doi https://doi.org/10.1089/neu.2019.6510
Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. https://doi.org/10.1371/journal.pbio.1000412
Guillen J (2012) FELASA guidelines and recommendations. J Am Assoc Lab Anim Sci 51:311–321
Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, Divert T, Goncalves A, Sze M, Gilbert B, Kourula S et al (2014) RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513:95–99. https://doi.org/10.1038/nature13706
Newton K, Sun X, Dixit VM (2004) Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24:1464–1469. https://doi.org/10.1128/mcb.24.4.1464-1469.2004
Terpolilli NA, Zweckberger K, Trabold R, Schilling L, Schinzel R, Tegtmeier F, Plesnila N (2009) The novel nitric oxide synthase inhibitor 4-amino-tetrahydro-L-biopterine prevents brain edema formation and intracranial hypertension following traumatic brain injury in mice. J Neurotrauma 26:1963–1975. https://doi.org/10.1089/neu.2008-0853
Zweckberger K, Eros C, Zimmermann R, Kim SW, Engel D, Plesnila N (2006) Effect of early and delayed decompressive craniectomy on secondary brain damage after controlled cortical impact in mice. J Neurotrauma 23:1083–1093. https://doi.org/10.1089/neu.2006.23.1083
Zweckberger K, Stoffel M, Baethmann A, Plesnila N (2003) Effect of decompression craniotomy on increase of contusion volume and functional outcome after controlled cortical impact in mice. J Neurotrauma 20:1307–1314. https://doi.org/10.1089/089771503322686102
Barnes CA (1979) Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93:74–104. https://doi.org/10.1037/h0077579
Gawel K, Gibula E, Marszalek-Grabska M, Filarowska J, Kotlinska JH (2019) Assessment of spatial learning and memory in the Barnes maze task in rodents-methodological consideration. Naunyn Schmiedebergs Arch Pharmacol 392:1–18. https://doi.org/10.1007/s00210-018-1589-y
Cryan JF, Mombereau C, Vassout A (2005) The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29:571–625. https://doi.org/10.1016/j.neubiorev.2005.03.009
Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U, Plesnila N (2015) Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol 78:887–900. https://doi.org/10.1002/ana.24512
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Hua Y, Xi G, Keep RF, Hoff JT (2000) Complement activation in the brain after experimental intracerebral hemorrhage. J Neurosurg 92:1016–1022. https://doi.org/10.3171/jns.2000.92.6.1016
Young K, Morrison H (2018) Quantifying microglia morphology from photomicrographs of immunohistochemistry prepared tissue using imageJ. J Vis Exp: Doi https://doi.org/10.3791/57648
Ferreira TA, Blackman AV, Oyrer J, Jayabal S, Chung AJ, Watt AJ, Sjostrom PJ, van Meyel DJ (2014) Neuronal morphometry directly from bitmap images. Nat Methods 11:982–984. https://doi.org/10.1038/nmeth.3125
Karperien A (1999–2013) FracLac for ImageJ.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227. https://doi.org/10.1016/j.cell.2011.11.031
Ariza M, Serra-Grabulosa JM, Junque C, Ramirez B, Mataro M, Poca A, Bargallo N, Sahuquillo J (2006) Hippocampal head atrophy after traumatic brain injury. Neuropsychologia 44:1956–1961. https://doi.org/10.1016/j.neuropsychologia.2005.11.007
Jorge RE, Acion L, Starkstein SE, Magnotta V (2007) Hippocampal volume and mood disorders after traumatic brain injury. Biol Psychiatry 62:332–338. https://doi.org/10.1016/j.biopsych.2006.07.024
Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J (2017) Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron 95:1246–1265. https://doi.org/10.1016/j.neuron.2017.07.010
Morganti-Kossmann MC, Semple BD, Hellewell SC, Bye N, Ziebell JM (2019) The complexity of neuroinflammation consequent to traumatic brain injury: from research evidence to potential treatments. Acta Neuropathol 137:731–755. https://doi.org/10.1007/s00401-018-1944-6
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517:311–320. https://doi.org/10.1038/nature14191
Plesnila N (2016) The immune system in traumatic brain injury. Curr Opin Pharmacol 26:110–117. https://doi.org/10.1016/j.coph.2015.10.008
Al-Khindi T, Macdonald RL, Schweizer TA (2010) Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 41:e519–e536. https://doi.org/10.1161/STROKEAHA.110.581975
Alghnam S, AlSayyari A, Albabtain I, Aldebasi B, Alkelya M (2017) Long-term disabilities after traumatic head injury (THI): a retrospective analysis from a large level-I trauma center in Saudi Arabia. Inj Epidemiol 4:29. https://doi.org/10.1186/s40621-017-0126-7[pii]
Andelic N, Howe EI, Hellstrom T, Sanchez MF, Lu J, Lovstad M, Roe C (2018) Disability and quality of life 20 years after traumatic brain injury. Brain Behav. https://doi.org/10.1002/brb3.1018
Draper K, Ponsford J (2008) Cognitive functioning ten years following traumatic brain injury and rehabilitation. Neuropsychology 22:618–625. https://doi.org/10.1037/0894-4105.22.5.618
Hackett ML, Anderson CS (2000) Health outcomes 1 year after subarachnoid hemorrhage: an international population-based study. The australian cooperative research on subarachnoid hemorrhage study group. Neurology 55:658–662
Hart T, Whyte J, Polansky M, Millis S, Hammond FM, Sherer M, Bushnik T, Hanks R, Kreutzer J (2003) Concordance of patient and family report of neurobehavioral symptoms at 1 year after traumatic brain injury. Arch Phys Med Rehabil 84:204–213. https://doi.org/10.1053/apmr.2003.50019
Himanen L, Portin R, Isoniemi H, Helenius H, Kurki T, Tenovuo O (2005) Cognitive functions in relation to MRI findings 30 years after traumatic brain injury. Brain Inj 19:93–100
Regan RF, Panter SS (1996) Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma 13:223–231. https://doi.org/10.1089/neu.1996.13.223
Bhasin RR, Xi G, Hua Y, Keep RF, Hoff JT (2002) Experimental intracerebral hemorrhage: effect of lysed erythrocytes on brain edema and blood-brain barrier permeability. Acta Neurochir Suppl 81:249–251. https://doi.org/10.1007/978-3-7091-6738-0_65
Derry PJ, Vo ATT, Gnanansekaran A, Mitra J, Liopo AV, Hegde ML, Tsai AL, Tour JM, Kent TA (2020) The chemical basis of intracerebral hemorrhage and cell toxicity with contributions from eryptosis and ferroptosis. Front Cell Neurosci 14:603043. https://doi.org/10.3389/fncel.2020.603043
Letarte PB, Lieberman K, Nagatani K, Haworth RA, Odell GB, Duff TA (1993) Hemin: levels in experimental subarachnoid hematoma and effects on dissociated vascular smooth-muscle cells. J Neurosurg 79:252–255. https://doi.org/10.3171/jns.1993.79.2.0252
Wan J, Ren H, Wang J (2019) Iron toxicity, lipid peroxidation and ferroptosis after intracerebral haemorrhage. Stroke Vasc Neurol 4:93–95. https://doi.org/10.1136/svn-2018-000205
Hasiloglu ZI, Albayram S, Selcuk H, Ceyhan E, Delil S, Arkan B, Baskoy L (2011) Cerebral microhemorrhages detected by susceptibility-weighted imaging in amateur boxers. AJNR Am J Neuroradiol 32:99–102. https://doi.org/10.3174/ajnr.A2250
Park JH, Park SW, Kang SH, Nam TK, Min BK, Hwang SN (2009) Detection of traumatic cerebral microbleeds by susceptibility-weighted image of MRI. J Korean Neurosurg Soc 46:365–369. https://doi.org/10.3340/jkns.2009.46.4.365
van der Horn HJ, de Haan S, Spikman JM, de Groot JC, van der Naalt J (2018) Clinical relevance of microhemorrhagic lesions in subacute mild traumatic brain injury. Brain Imaging Behav 12:912–916. https://doi.org/10.1007/s11682-017-9743-6
Glushakova OY, Johnson D, Hayes RL (2014) Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma 31:1180–1193. https://doi.org/10.1089/neu.2013.3080
Lawrence TP, Pretorius PM, Ezra M, Cadoux-Hudson T, Voets NL (2017) Early detection of cerebral microbleeds following traumatic brain injury using MRI in the hyper-acute phase. Neurosci Lett 655:143–150. https://doi.org/10.1016/j.neulet.2017.06.046
Conrad M, Pratt DA (2019) The chemical basis of ferroptosis. Nat Chem Biol 15:1137–1147. https://doi.org/10.1038/s41589-019-0408-1
Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C et al (2017) Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI insight 2:e90777. https://doi.org/10.1172/jci.insight.90777
Ratan RR (2020) The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol 27:479–498. https://doi.org/10.1016/j.chembiol.2020.03.007
Zille M, Karuppagounder SS, Chen Y, Gough PJ, Bertin J, Finger J, Milner TA, Jonas EA, Ratan RR (2017) Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48:1033–1043. https://doi.org/10.1161/strokeaha.116.015609
Acknowledgements
We want to thank Dr. Ulrike Schillinger for her support during the analysis of the MRI data. Research in the Vandenabeele group is supported by Flemish grants (EOS MODEL-IDI, FWO Grant 30826052), FWO research grants (G.0E04.16N, G.0C76.18N, G.0B71.18N, G.0B96.20N), Methusalem (BOF16/MET_V/007), iBOF20/IBF/039 ATLANTIS, Foundation against Cancer (FAF-F/2016/865, F/2020/1505), CRIG and GIGG consortia, and VIB. The Plesnila group was funded by Munich University´s Förderprogramm für Forschung und Lehre (FöFoLe), by the BMBF-funded research consortium TRAINS (Project ID: 01EW1709) and by the Munich Cluster of Systems Neurology (SyNergy; Project ID EXC 2145 / ID 390857198).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conception and study design: ACW, NP, NAT. Surgery, genotyping, neurological testing, histology: ACW, IK. MR imaging: ACW, MD. Data analysis & interpretation: ACW, IK, MD, NP, NAT. Statistical analysis: ACW, NAT. Manuscript preparation: ACW, NP, NAT. Critical revision of the manuscript: all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All procedures were reviewed and approved by the respective institutional and governmental authorities and performed according to all regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Conflict of interest
There is no conflict of interest for any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional File 1 Supplementary Fig. S1.
Genotyping of RIPK1 and RIPK3 deficient mice and proof of neuronal specific RIPK1 knock-out in RIPK1flox/floxCamk2CreERT2 mice. a. Neuron specific RIPK1 deficient mice used for experiments were heterozygous for Camk2CreERT2 and homozygous for the floxed RIPK1 allele. Littermate controls were also homozygous for the floxed RIPK1 allele, but did not express the Cre recombinase. b. Global RIPK3 deficient mice were homozygous for disrupted allele, while control mice expressed only the wild type gene. c. and d. To demonstrate specific neuron specific RIPK1 deficiency in induced RIPK1flox/floxCamk2CreERT2 mice, we performed immunohistochemistry for RIPK1 and NeuN, a neuronal marker. In the cortex of control mice RIPK1 was almost exclusively expressed in neurons (upper panels), while in induced RIPK1flox/floxCamk2CreERT2 mice RIPK1 staining was significantly reduced (lower panels) to 20% of baseline (d).
Additional File 1 Supplementary Fig. 2 Body weight and physical condition after experimental TBI. a. and b. Weight after TBI. Animals recovered from weight loss directly after trauma within one week after injury; in the following observation period weight constantly increased. No differences were detected between CCI and sham-operated animals in the RIPK1 (a) and RIPK3 (b) groups. c. and d. General health score to assess recovery. All animals’ general condition transiently worsened in the perioperative phase with a peak at day 1 after TBI, but returned to baseline within one week. There was no difference between groups, c. RIPK1, d. RIPK3. Data are presented as mean ± SD; n = 5 for sham, n = 8–10 for TBI.
Additional File 1 Supplementary Fig. 3. Individual lesion volume progression for a. RIP1 and b. RIP3 deficient mice.
Additional File 1 Supplementary Fig. 4. RIPK3 deficiency does not affect acute brain injury after TBI. No differences in lesion volume as assessed by histology was detected between RIPK3 knockout mice and C57BL/6 wild type controls at 24 h after TBI. Data are presented as mean ± SD; n = 10.
Additional File 1 Supplementary Fig. 5. Lesion volumes by MRI and histology. a. T2-weighted MRI and Nissl stained coronal section three months after injury. Lesion volume was quantified in T2-weighted MRI images as well as Nissl stained sections obtained in the same animals three months post injury. b. Correlation analysis of lesion volumes assessed by histomorphometry or T2-weighted MRI revealed a strong positive correlation between both methods. Differences in measured lesion size are due to shrinking of tissue in the fixation process, with a shrinking factor of 0.77 ± 0.23. c.—f. Quantification of lesion volume (c. RIPK1, d. RIPK3) as well as hippocampal damage (e. RIPK1, f. RIPK3) using both methods. Data are presented as mean ± SD; n = 8–10 for TBI. Pearson correlation, Students t-test for parametric and Man-Whitney-Rank-Sum test for non-parametric data were used. *p < 0.05, ***p < 0,001.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wehn, A.C., Khalin, I., Duering, M. et al. RIPK1 or RIPK3 deletion prevents progressive neuronal cell death and improves memory function after traumatic brain injury. acta neuropathol commun 9, 138 (2021). https://doi.org/10.1186/s40478-021-01236-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-021-01236-0