
Abstract
Background
Immunotherapies, specifically those based on immune checkpoint inhibitors, have shown promising activity in multiple tumor types. Other than mifamurtide (MEPACT®) for osteosarcoma approved by European Medicines Agency, there are no approved immunotherapies for sarcomas.
Methods
We analyzed medical records of patients with advanced sarcoma who were referred to Phase 1 clinic at MD Anderson and received an immunotherapy (checkpoint inhibitors, vaccines, or cytokine based therapies). Clinical parameters including demographics, clinical history, toxicity, and response were abstracted.
Results
Among 50 patients enrolled in immunotherapy trials (Bone 10; Soft-tissue 40) we found 14 different subtypes of sarcomas. Royal Marsden Hospital (RMH) prognostic score was <2 (86%). Performance status (PS) was 0–1 in 48 patients (96%); median number of prior therapies was 3 (0–12). Immunotherapy consisted of checkpoint inhibitors (82%: PD1 = 7, PD-L1 = 11, CTLA4 = 22, other = 1) of which 42% were combinations, as well as vaccines (14%), and cytokines (4%). Median overall survival (OS) was 13.4 months (11.2 months: not reached). Median progression free survival (PFS) was 2.4 months (95% CI = 1.9–3.2 months). Best response was partial response (PR) in 2 patients with alveolar soft part sarcoma (ASPS) and stable disease (SD) in 11 patients (3 GIST, 3 liposarcomas (2 DDLS, 1 WDLS), 2 ASPS, 2 leiomyo, 1 osteo). PFS was 34% (23%, at 50%) at 3 months, 16% (8%, 30%) at 6 months, and 6% (2%, 20%) at 1 year. Pseudo-progression followed by stable disease was observed in 2 patients (4%). Grade 3/4 adverse events included rash (10%), fever (6%), fatigue (6%), and nausea/vomiting (6%).
Conclusion
Immunotherapies were well tolerated in advanced sarcoma patients enrolled in trials. All four ASPS patients had clinical benefit with checkpoint inhibitors and this was the only subtype experiencing partial response. Further evaluation of checkpoint inhibitors in ASPS is warranted.
Similar content being viewed by others

Background
Since the approval of anti-CTLA-4 antibody ipilimumab in 2011 and the anti-PD-1 drugs nivolumab and pembrolizumab in 2015, oncology has experienced a resurgence. Patients, clinicians, and drug companies have a new enthusiasm for immunotherapy not seen since the BATTLE trial elevated targeted therapy and small molecules [1]. In the last year melanoma, lung, head and neck, and bladder cancers have shown clinically significant improvement in response rate, progression free survival, and overall survival with the use of the immune checkpoint inhibitors.
Sarcomas are mesenchymal tumors of soft tissues and bone that are usually fatal when they progress beyond local control. Dating back to 1891, attempts have been made to treat sarcomas with immunotherapy [2,3,4,5]. These attempts were either with highly toxic “Coley’s toxins” or less potent vaccine therapies as well as unsuccessful trials with interferons. Interestingly, osteosarcoma was one of the first cancers to get regulatory approval with an immunotherapeutic agent. Mifamurtide (L-MTPPE) is an agent that increased circulating TNF-alpha and IL-6. It was approved in Europe for use in combination with adjuvant chemotherapy [6, 7]. Alveolar soft part sarcoma has similarly shown response to immunotherapy with interferon, but only at the case report level [8].
With the advent of modern immune checkpoint inhibitors several trials are ongoing to test the safety and efficacy of immunotherapy in sarcomas. Pre-clinical data suggests that tumor infiltrating lymphocytes (TILs) are an important positive prognostic indicator in multiple soft tissue sarcoma subtypes [9] including angiosarcoma [10] and gastrointestinal stromal tumor (GIST) [11]. Especially with GIST, there is preclinical data to suggest that checkpoint blockade enhances activity of imatinib [12]. PD-L1 expression, which is an important biomarker of response to anti-PD-1 therapy for certain malignancies [13], has been investigated in sarcomas. One study found PD-L1 to be relatively uncommon except in GIST, spindle cell, and radiation associated sarcomas [14]. Others have shown strong expression of PD-L1 in dedifferentiated chondrosarcoma [15], epithelioid, synovial, Ewing, rhabdomyosarcoma [16], and others [17] including bone and leiomyosarcomas [18]. However, as with other sarcoma studies the numbers were small for each subtype. Unfortunately, an early phase II trial of nivolumab in metastatic uterine leiomyosarcomas failed to show a response [19]. However, this may be an issue of patient selection as genomic sequencing has shown lynch syndrome associated gene mutations in a subset of these patients [20]. Other phase II studies have shown partial responses to anti-PD-1 directed therapy in some bone and soft tissue sarcomas [21, 22]. The SARC-028 study in particular showed signals of pembrolizumab activity in undifferentiated pleomorphic sarcoma and dedifferentiated liposarcoma. These two sarcoma cohorts are now undergoing expansion to further evaluate activity [22]. The results have been inconsistent across studies and while the overall trend is that immunotherapy doesn’t have overwhelming activity in sarcomas as observed with melanoma or non-small cell lung cancer, it is clear that patients with certain sarcoma subtypes may respond and investigators continue to evaluate the use of immunotherapy in specific sarcomas such as alveolar soft part sarcoma [23]. We undertook a retrospective review of our own sarcoma patients treated on phase I trials with various immunotherapy agents. The hope is that with more published data on responses, we can begin to intelligently design prospective immunotherapy trials targeted at a specific sarcoma histology or molecular subtype.
Methods
We reviewed the charts of 50 sequential patients with metastatic or unresectable advanced sarcoma. Pathology had previously been reviewed and verified by an MD Anderson pathologist with expertise in bone and soft tissue sarcomas. All patients had been referred to Investigational Therapeutics Department at MD Anderson Cancer Center (MDACC) and participated in an immunotherapy clinical trial. Trial choice and enrollment was dependent on availability at clinic visit. Immunotherapy was defined as any drug that primes the immune system against the tumor. Our study allowed checkpoint inhibitors, vaccines, and cytokine therapy. Combinations with other drugs were also included. Patient charts were reviewed for age at enrollment on study, race, sex, tumor histology, LDH, albumin, metastatic sites, performance status, prior therapies, toxicity, number of treatment cycles and response based on imaging. Other data collected included the date patient started an investigational therapy, their best response, and duration of that response. Response Evaluation Criteria in Solid Tumors (RECIST V1.1) as well as irRC (immune related response criteria) were used to assess response. Last known follow-up or date of death were also recorded. Progression free survival was measured from date of first dose to date of documented progression on imaging or onset of symptoms. Subsequent scans after progression were reviewed to ensure pseudo-progression was not missed. Toxicities were graded based on NCI CTCAE v4.0.
MD Anderson institutional review board (IRB) independently reviewed and approved each clinical trial presented in our study. Patients were provided with written informed consent prior to treatment with an investigational therapy. MD Anderson IRB also approved this retrospective review.
Results
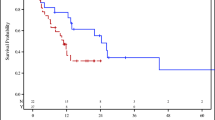
Between September 2012 and May 2016 fifty patients with multiple types of sarcoma were enrolled on immunotherapy trials. The clinical characteristics of patients are shown in Table 1. Male to female ratio was 42 to 58%. Median age was 53.5 years (range 18–84). Most patients had favorable RMH score (less than two 43/50, 86%); the remaining 14% of patients had an RMH score of two. Overall survival based on RMH score trended toward favoring lower scores, (Hazard Ratio = 2.0 (0.9, 4.6) for 1, 2 vs. 0, but was statistically inconclusive). Median OS was 24 months for RMH score 0 and was 12 months for patients with score 1–2 (P = 0.08). Performance status was also favorable with 48 patients (96%) with an ECOG 0–1. Patients received a median of 3 prior therapies (0–12) and those with two or fewer therapies had improved overall survival compared to those with greater than two prior therapies (P = 0.001). (Fig. 1).

Overall survival by RMH score Hazard Ratio = 2.0 (0.9, 4.6) for 1, 2 vs. 0. “Inconclusive” results. Overall survival by for 0–2 prior therapies median was 24 months and for those with >2 prior therapies was 8 months
Sarcoma subtypes
We found 14 different subtypes of sarcomas with 10 bone sarcomas and 40 soft tissue sarcomas (Table 2).
Therapy
Checkpoint inhibitor based trials made up of single agents in addition to combinations comprised the majority of trials (41/50, 82% (PD1 = 7, PD-L1 = 11, CTLA4 = 22, other = 1). Other therapies included vaccines (7/50, 14%) and cytokines (2/50, 4%). Ten sarcoma patients were enrolled in a clinical trial with ipilimumab and imatinib. (Table 1) Radiation therapy was used in combination with ipilimumab for five patients.
Adverse events
Adverse events as graded by Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0 included rash (5/50, 10%), fever (3/50, 6%), fatigue (3/50, 6%), and nausea/vomiting (2/50, 4%). Other toxicities included grade 3 hypothyroidism, grade 3 transaminitis, grade 4 pancreatitis, pituitary hypophysitis, pneumonitis and mucositis.
Responses
The median overall survival was 13.4 months, 95% CI = (11.2, not reached). Twenty-six patients died after a median 15 months’ follow-up. Overall survival was 76% (65%, 90%) at 6 months, 59% (45%, 76%) at 1 year, and 27% (13%, 54%) at 2 years. The median progression free survival was 2.4 months, 95% CI = (1.9, 3.2). Forty-six patients progressed; progression free survival was 34% (23%, at 50%) at 3 months, 16% (8%, 30%) at 6 months, and 6% (2%, 20%) at 1 year. (Fig. 2).

Median OS = 13.4 months, 95% CI = (11.2, not reached); 26 patients died after median 15 months follow-up; OS was 76% (65%, 90%) at 6 months, 59% (45%, 76%) at 1 year, and 27% (13%, 54%) at 2 years (Left). Median PFS = 2.4 months, 95% CI = (1.9, 3.2); 46 patients progressed; PFS was 34% (23%, at 50%) at 3 months, 16% (8%, 30%) at 6 months, and 6% (2%, 20%) at 1 year. (Right)
Best response (Table 3) was partial response in 2 patients (4%); both patients had alveolar soft part sarcoma. These two patients both received anti-PD-L1 based therapy (Table 4). Stable disease was observed in 11 patients (22%) with a median of 7.4 months (1.3 to 9.1). GIST (3/ 9 GIST, 33%), well-differentiated liposarcoma (1/2 WDLS, 50%), de-differentiated liposarcoma (2/5 DDLS, 40%), leiomyosarcoma (2/12 LMS, 17%), and osteosarcoma (n = 1/5 osteo, 20%) represented patients with stable disease. PFS was 34% (23%, at 50%) at 3 months, 16% (8%, 30%) at 6 months, and 6% (2%, 20%) at 1 year. One of the ASPS patients with stable disease received the same combination as the two partial responders; the other received single agent anti-PD-1. The patient with osteosarcoma received anti-CTLA-4 and radiation; the patients with DDLS received anti-PD-1 or anti-PD-L1; the patients with LMS received anti-CTLA-4 or anti-PD-1. Some patients were treated past progression (16/50, 32%). Pseudo-progression followed by stable disease per ir-RECIST was observed in 2 patients (4%, overall 12.5% of patients treated beyond progression). Both were well-differentiated liposarcomas. One patient with GIST had hyper-progression after treatment with a checkpoint inhibitor. He had continually growing disease prior to therapy, but his rate of tumor growth accelerated upon initiation of immunotherapy (Fig. 3).

GIST patient with hyper-progression on immunotherapy (based on ir RECIST)
Discussion
Metastatic, relapsed and refractory sarcomas continue to have a grave prognosis. There is considerable enthusiasm for developmental therapeutics in sarcomas with recent approvals of pazopanib, eribulin, trabectedin and olaratumab with doxorubicin. There are multiple trials ongoing with the combinations of these agents [24,25,26]. The exceptional success of immunotherapy in other cancer types spurred us to examine our own records for potential responses to immunotherapy in sarcoma patients. As is frequently the case with sarcomas, our dataset is small and has many different sarcoma subtypes. Admittedly, this mix of low-grade and high-grade sarcomas makes comparison difficult. Complicating matters are the multiple different immunotherapies. Certain observationsemerge even in this heterogeneous group of sarcomas and therapies.
The most remarkable response was that of alveolar soft part sarcomas (ASPS) to immunotherapy. Even with a limited sample of four patients, half had a strong partial response bordering on complete response. The other two patients had stable disease. This is far outside the normal behavior for a biologically indolent but relentless tumor [27] and raises the question of mechanism. Is this a question of PD-L1 blockade and cytotoxic T-cell activation? We know that most tumors with FDA approved anti-PD-1 immunotherapy have response rates in the 10–20% range. This would imply that either our four patients are unusual responders such as those seen in prior interferon trials, or that other mechanisms exist. Tanaka et al. [28] created a mouse model of alveolar soft part sarcoma based on the characteristic ASPSCR1-TFE3 fusion protein. The model demonstrated a highly vascular tumor with genes expressed in transendothelial migration. This vascularity is key to the early metastatic potential of this tumor. Additionally, ASPS lines these new blood vessels with hemangiopericytes that prevent leakage of nutrients and oxygen out of the blood vessels. We know that chemokines and their ligands are often involved in vascular recognition and targeting of microvascular endothelial cells [29]. Perhaps chemokines play an important role in the action of immunotherapy in ASPS; our group is undertaking further studies to elucidate this mechanism. Alternatively, the TFE3 fusion may be immunogenic itself or act via TGF-β or CD40 ligand to stimulate T-cells and antigen presenting cells [30]. Others have reported that mismatch repair pathway aberrations may be responsible for ASPS response to immunotherapy [31].
Another interesting observationwas seen in the patients with stable disease. It is entirely possible that some of the patients simply had indolent disease, such as the GIST and well-differentiated liposarcoma. However, osteosarcoma, dedifferentiated liposarcoma, and leiomyosarcoma are generally not considered indolent diseases and their stabilization in response to immunotherapy may serve as an indication of activity. While next generation sequencing (NGS) data was not available for the liposarcoma or leiomyosarcoma patients, clinical grade NGS was performed on the osteosarcoma patient. This testing did not reveal a particularly high mutational load which is thought to increase response to immunotherapy. The response of the patients in our study along with recently reported abstracts of positive anti-PD-1 activity in diverse sarcomas suggests that earlier immunotherapy trials in sarcomas were not entirely correct in their negative experience. For example, a recently completed phase II trial of pembrolizumab showed activity in undifferentiated pleomorphic sarcoma and dedifferentiated liposarcoma [32]. Another trial with advanced soft tissue sarcomas treated with pembrolizumab and metronomic cyclophosphamide yielded only one responder out of 50 treated patients [33]. While immunotherapy in sarcomas has shown small promise, we can say that it is unlikely to be the success that it has been in melanoma and non-small cell lung cancer.
GIST is a tumor with great preclinical data for immunotherapy that did not materialize into results for patients. Patients were enrolled on a trial of imatinib and ipilimumab based on convincing pre-clinical rationale that showed imatinib reduced levels of indoleamine 2,3-dioxygenase (Ido). Ido is an immunosuppressive enzyme and inhibition of Ido led to regulatory T-cell destabilization, deactivation, and apoptosis. Treatment naïve mice with KIT mutant GIST treated with imatinib showed decreased regulatory T cell activity [34]. In the clinical trial testing this hypothesis, the combination of imatinib and ipilimumab did not translate into improved response rates for patients or even a signal of synergistic activity [35].
The tolerability of immunotherapy in sarcomas appears to be similar to other patients. Rash, fever, and fatigue were the most common adverse events. As with other experiences with immunotherapeutic agents, some unusual toxicities were observed necessitating discontinuation of drug and administration of steroids [36]. The Royal Marsden Hospital prognostic scoring system continues to be a valid predictor of survival in a phase 1 trial (Fig. 1) [37]. Our trial had substantially longer overall survival than has been reported in other phase 1 trials of sarcoma patients. Previous trial experiences by our own group as well as groups from the Royal Marsden Hospital and the European phase 1 database have reported consistent OS in the 7.6–9.8 month range with a PFS between 2.1 and 3.5 months [38, 39]. Our extended overall survival with immunotherapy of 13.4 months can be explained either by patient selection for more indolent tumors, since PFS was similar at 2.4 months. Alternatively, there may be some downstream effect of immunotherapy that goes beyond response rates to contribute to an improved overall survival.
It is notable to point out that we experienced one patient with hyper-progression upon initiation of checkpoint blockade (Fig. 3). Others have reported a similar disturbing phenomenon in as many as 9% of patients. This hyper-progression is unrelated to tumor type or burden of disease, but did portend a poor prognosis especially for elderly patients [40]. One group reported that in their experience mutations in MDM2/MDM4 and EGFR predisposed to a hyper-progressor phenotype [41]. Our patient proceeded to another clinical trial where he had continued progression. Ultimately he succumbed to his disease. This reminds us that while adverse events are manageable with immunotherapy there is much we still do not know about the mechanism of these drugs and their ultimate potential.
Conclusion
In our small and limited retrospective study of various immunotherapies in diverse sarcomas we found encouraging early signals of activity. The strongest clinical responses came from combinations of checkpoint inhibitors. Alveolar soft part sarcomas benefitted particularly well from immune checkpoint inhibitors. Further study and evaluation needs to be done in the heterogeneous and rare group of diseases. Perhaps the biopsy specimens collected from SARC 028 will shed light on mechanisms of response. In addition to understanding the sarcoma immune microenvironment, we need to pursue the early leads in activity found in this study as well as SARC 028 prospective study for patients with high grade undifferentiated pleomorphic sarcoma, dedifferentiated liposarcoma, and alveolar soft part sarcoma.
Abbreviations
- ASPS:
-
Alveolar soft part sarcoma
- CR:
-
Complete response
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- DDLS:
-
Dedifferentiated liposarcoma
- GIST:
-
Qastrointestinal stromal tumor
- IRB:
-
IInstitutional review board
- irRC:
-
Immune-related response criteria
- LMS:
-
Leiomyosarcoma
- OS:
-
Overall survival
- PD1:
-
Programmed cell death protein
- PD-L1:
-
Programmed death ligand −1
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- PS:
-
Performance Status
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- RMH:
-
Royal Marsden Hospital
- SD:
-
Stable disease
- TIL:
-
Tumor infiltrating lymphocytes
- WDLS:
-
Well-differentiated liposarcoma
References
Kim ES, Herbst RS, Wistuba II, et al. The BATTLE Trial: Personalizing Therapy for Lung Cancer. Cancer Discov. 2011;1(1):44–53.
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8.
Ghisoli M, Barve M, Mennel R, et al. Three-year follow up of GMCSF/bi-shRNAfurin DNA-transfected Autologous tumor immunotherapy (vigil) in metastatic advanced Ewing's sarcoma. Mol Ther. 2016;24(8):1478–83.
McCaughan GJB, Fulham MJ, Mahar A, et al. Programmed cell death-1 blockade in recurrent disseminated Ewing sarcoma. J Hematol Oncol. 2016;9:48.
Miwa S, Nishida H, Tanzawa Y, et al. Phase 1/2 study of immunotherapy with dendritic cells pulsed with autologous tumor lysate in patients with refractory bone and soft tissue sarcoma. Cancer. 2017:n/a-n/a.
Frampton JE. Mifamurtide: A Review of its Use in the Treatment of Osteosarcoma. Pediatr-Drugs. 2010;12(3):141–53.
Kleinerman ES, Jia SF, Griffin J, Seibel NL, Benjamin RS, Jaffe N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol. 1992;10(8):1310–6.
Roozendaal KJ, de Valk B, ten Velden JJA, van der Woude HJ, Kroon BBR. Alveolar soft-part sarcoma responding to interferon alpha-2b. Br J Cancer. 2003;89(2):243–5.
Sorbye SW, Kilvaer T, Valkov A, et al. Prognostic impact of lymphocytes in soft tissue sarcomas. PLoS One. 2011;6(1):e14611.
Fujii H, Arakawa A, Utsumi D, et al. CD8+tumor-infiltrating lymphocytes at primary sites as a possible prognostic factor of cutaneous angiosarcoma. Int J Cancer. 2014;134(10):2393–402.
Rusakiewicz S, Semeraro M, Sarabi M, et al. Immune infiltrates are prognostic factors in localized gastrointestinal Stromal tumors. Cancer Res. 2013;73(12):3499–510.
Seifert AM, Zeng S, Zhang JQ, et al. PD-1/PD-L1 blockade enhances T-cell activity and antitumor efficacy of Imatinib in gastrointestinal Stromal tumors. Clin Cancer Res. 2017;23(2):454–65.
Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33.
D’Angelo SP, Shoushtari AN, Agaram NP, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2015;46(3):357–65.
Kostine M, Cleven AH, de Miranda NF, Italiano A, Cleton-Jansen AM, Bovee JV. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod Pathol. 2016;29(9):1028–37.
Kim C, Kim EK, Jung H, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer. 2016;16:434.
Kim JR, Moon YJ, Kwon KS, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS One. 2013;8(12):e82870.
Raj S, Bui R, Gonzales D, Letson SJ. Impact of PDL1 expression on clinical outcomes in subtypes of sarcoma. 2014/09/27/, 2014.
George S, Barysauskas CM, Solomon S, et al. Phase 2 study of nivolumab in metastatic leiomyosarcoma of the uterus. Journal of Clinical Oncology. 2016;34(suppl; abstr 11007).
Groisberg R, Hong DS, Holla V, et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget. 2017;5:39254–67.
Paoluzzi L, Cacavio A, Ghesani M, et al. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin Sarcoma Res. 2016;6
Tawbi HA-H, Burgess MA, Crowley J, et al. Safety and efficacy of PD-1 blockade using pembrolizumab in patients with advanced soft tissue (STS) and bone sarcomas (BS): Results of SARC028—A multicenter phase II study. Journal of Clinical Oncology. 2016;34(suppl; abstr 11006).
Wilky B. Axitinib and Pembrolizumab in Subjects With Advanced Alveolar Soft Part Sarcoma and Other Soft Tissue Sarcomas. 2015 - Full Text View - ClinicalTrials.gov.
Subbiah V, Meyer C, Zinner R, et al. Phase Ib/II Study of the Safety and Efficacy of Combination Therapy with Multikinase VEGF Inhibitor Pazopanib and MEK Inhibitor Trametinib In Advanced Soft Tissue Sarcoma. Clin Cancer Res. 2017;17:0272.
Subbiah V, Holmes O, Gowen K, et al. Activity of c-met/ALK inhibitor Crizotinib and multi-Kinase VEGF inhibitor Pazopanib in metastatic gastrointestinal Neuroectodermal tumor harboring EWSR1-CREB1 fusion. Oncology. 2016;91(6):348–53.
Subbiah V, Kurzrock R. Phase 1 clinical trials for sarcomas: the cutting edge. Curr Opin Oncol. 2011;23(4):352–60.
Folpe AL, Deyrup AT. Alveolar soft-part sarcoma: a review and update. J Clin Pathol. 2006;59(11):1127–32.
Tanaka M, Homme M, Yamazaki Y, Shimizu R, Takazawa Y, Nakamura T. Modeling alveolar soft part sarcoma unveils novel mechanisms of metastasis. Cancer Res. 2017;77(4):897–907.
Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol. 2015;27(1):11–20.
Conley AP, Trinh VA, Zobniw CM, et al. Positive Tumor Response to Combined Checkpoint Inhibitors in a Patient With Refractory Alveolar Soft Part Sarcoma: A Case Report. JGO. 2017:JGO.2017.009993.
Samer Salah JHL, Scott Davidson, Nathaniel Anderson, Sergio L Periera, Beatrice Lau, Adam Shlien, Brendan Dickson, Peter Ferguson, Jay Wunder, Albiruni Ryan Abdul Razak. Immunoprofiling in alveolar soft part sarcoma. J Clin Oncol 35, 2017 (suppl; abstr 11059). 2017.
Burgess MA. Multicenter phase II study of pembrolizumab (P) in advanced soft tissue (STS) and bone sarcomas (BS): Final results of SARC028 and biomarker analyses. Paper presented at: ASCO 2017; 2017/06/02/, 2017.
Toulmonde M, Penel N, Adam J, et al. Use of pd-1 targeting, macrophage infiltration, and ido pathway activation in sarcomas: a phase 2 clinical trial. JAMA Oncol. 2017;
Balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17(9):1094–100.
Reilley MJ, Bailey A, Subbiah V, et al. Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J Immunother Cancer. 2017;5:35.
Duvic M, Pinter-Brown LC, Foss FM, et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood. 2015;125(12):1883–9.
Garrido-Laguna I, Janku F, Vaklavas C, et al. Validation of the Royal Marsden Hospital prognostic score in patients treated in the phase I clinical trials program at the MD Anderson Cancer Center. Cancer. 2012;118(5):1422–8.
Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
Livingston JA, Hess KR, Naing A, et al. Validation of prognostic scoring and assessment of clinical benefit for patients with bone sarcomas enrolled in phase I clinical trials. Oncotarget. 2016;7(39):64421–30.
Champiat S, Dercle L, Ammari S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 2017;23(8):1920–8.
Kato S, Goodman AM, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyper-progressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23(15):4242–50.
Acknowledgements
We thank the patients and their families for enrolling on clinical trials.
Funding
We thank the patients and their families for enrolling on clinical trials. The University of Texas MD Anderson Cancer Center is supported by the National Institutes of Health Cancer Center Support Grant CA016672. VS acknowledges the Shannon Wilkes Sarcoma Research funds. This work was supported in part by Cancer Prevention Research Institute of Texas Grant RP110584 and National Center for Advancing Translational Sciences Grant UL1 TR000371 (Center for Clinical and Translational Sciences).The funding sources had no input into the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Availability of data and materials
All of the relevant data is presented within this manuscript.
Author information
Authors and Affiliations
Contributions
RG and VS contributed to the conception, design, and writing. AB contributed to figure production. KH contributed to interpretation of data. DH, FJ, SPP, AN, SF, RB, SP, NS, AC, FMB, VS were involved in the care of patients. All authors were involved in revising the manuscript and have given final approval of the version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This was a retrospective exploratory evaluation of outcomes and, as such, ethical approval was not required to conduct this analysis. Clinical outcomes in the primary analysis of the individual trials have been or will be reported elsewhere. As previously described, all patients provided their written informed consent to participate in the clinical studies and study procedures were conducted in accordance with the ethical principles founded in the Declaration of Helsinki, as well as the demands of the national drug and data protection laws and any other applicable regulatory requirements. Approval was obtained from the appropriate regulatory authorities of each participating country before sites were initiated. The study protocols were approved by an institutional review board (IRB)/independent ethics committee (IEC) at each participating institution, and all patients provided written informed consent before initiating any study procedures.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Groisberg, R., Hong, D.S., Behrang, A. et al. Characteristics and outcomes of patients with advanced sarcoma enrolled in early phase immunotherapy trials. j. immunotherapy cancer 5, 100 (2017). https://doi.org/10.1186/s40425-017-0301-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40425-017-0301-y