
Abstract
Neddylation is a post-translational modification process, similar to ubiquitination, that controls several biological processes. Notably, it is often aberrantly activated in neoplasms and plays a critical role in the intricate dynamics of the tumor microenvironment (TME). This regulatory influence of neddylation permeates extensively and profoundly within the TME, affecting the behavior of tumor cells, immune cells, angiogenesis, and the extracellular matrix. Usually, neddylation promotes tumor progression towards increased malignancy. In this review, we highlight the latest understanding of the intricate molecular mechanisms that target neddylation to modulate the TME by affecting various signaling pathways. There is emerging evidence that the targeted disruption of the neddylation modification process, specifically the inhibition of cullin-RING ligases (CRLs) functionality, presents a promising avenue for targeted therapy. MLN4924, a small-molecule inhibitor of the neddylation pathway, precisely targets the neural precursor cell-expressed developmentally downregulated protein 8 activating enzyme (NAE). In recent years, significant advancements have been made in the field of neddylation modification therapy, particularly the integration of MLN4924 with chemotherapy or targeted therapy. This combined approach has demonstrated notable success in the treatment of a variety of hematological and solid tumors. Here, we investigated the inhibitory effects of MLN4924 on neddylation and summarized the current therapeutic outcomes of MLN4924 against various tumors. In conclusion, this review provides a comprehensive, up-to-date, and thorough overview of neddylation modifications, and offers insight into the critical importance of this cellular process in tumorigenesis.
Similar content being viewed by others

Introduction
Neddylation is a reversible post-translational modification akin to ubiquitination and is characterized by the reversible covalent conjugation of neural precursor cell-expressed developmentally downregulated protein 8 (NEDD8) with specific substrate proteins [1, 2]. NEDD8, a highly conserved protein in eukaryotes, exhibits predominant nuclear expression and comparatively weak cytoplasmic presence [3]. Initially cloned by Kumar et al. in 1992, NEDD8 shares 60% identity and 80% similarity with ubiquitin, making it the molecule most similar to ubiquitin among all ubiquitin-like proteins [4, 5]. The initial non-Cullin proteins implicated as substrates in NEDD8 research, were associated with Breast Cancer-Associated protein 3 (BCA3), a yeast-derived protein [6]. However, the most extensively studied substrates are cullin-RING ligases, which form the largest family of ubiquitin E3 ligases [7, 8]. CRLs are encoded by more than 600 human genes, of which 518 are protein kinase genes. The ubiquitin-proteasome system facilitates the ubiquitination and degradation of approximately 20% of proteins within cells. This mechanism significantly influences multiple cellular processes and is implicated in various human diseases [9, 10]. In 1998, researchers found that both cullin protein and NEDD8 were highly expressed in various cancer cell types, such as colon cancer and leukemia cells, thereby reinforcing the association of neddylation with cancer progression [11, 12]. In 2009, Soucy T. A. et al. recognized MLN4924 as a potent inhibitor of the NAE, disrupting cullin-RING ligase-mediated protein turnover, and inducing apoptosis in tumor cells [13]. MLN4924 inhibits neddylation by binding to the NAE enzyme, leading to its degradation. This inhibitory action prevents the activation of the NEDD8 protein, thereby obstructing the neddylation process in its entirety. This blockade results in the accumulation of unmodified cullin proteins, which subsequently inhibits the activity of the ubiquitin-proteasome system (UPS). This chain of events culminates in the accumulation of ubiquitinated proteins and triggers DNA damage responses in tumor cells, leading to cell cycle arrest, apoptosis, senescence, autophagy, and alterations in mitochondrial function [14,15,16,17].
The etiology of cancer is intrinsically tethered to the intricacies of its TME. This milieu, characterized by the amalgamation of cellular entities such as immune cells, fibroblasts, and endothelial cells embedded within a sophisticated extracellular matrix(ECM), has profound implications for neoplastic evolution [18]. Complex intercellular and matrix-associated interactions within the TME underlie tumor ontogenesis and contribute to the significant challenges of therapeutic refractoriness, such as drug resistance [19]. Historically, conventional therapeutic modalities have been myopic in addressing the protective sanctum that the TME proffers to malignant cells. This review aims to encapsulate and scrutinize the impact of neddylation mechanism on the TME and the anti-tumor effects of MLN4924 based on neddylation. Through this analysis, we sought to provide a comprehensive overview of this significant area of cancer biology.
Neddylation modification
NEDD8 is a key molecule in the neddylation process
NEDD8 shares structural and operational principles with ubiquitin, a regulatory protein involved in diverse cellular functions [20, 21]. Initially identified in mouse brain tissue as a developmentally downregulated gene contributing to its nomenclature, NEDD8 is not exclusive to neural precursor cells or the brain but is ubiquitously expressed across numerous tissues throughout the body, underscoring its essential biological role [22]. Despite its relatively small size (approximately 81 amino acids), human NEDD8 plays a pivotal role in cellular functions via neddylation, a mechanism similar to that of ubiquitination. Neddylation involves the covalent attachment of NEDD8 to target proteins, thereby modulating their function or stability [1, 5]. Although NEDD8 shares around 60% of sequence identity with ubiquitin and has a similar three-dimensional structure composed of a beta-grasp fold, the two are not interchangeable. They conjugate to different protein subsets and regulate distinct aspects of cellular biology [23, 24]. Thus, NEDD8, despite its small size, serves as a potent regulatory protein that functions akin to ubiquitin through neddylation. Further understanding its function would provide promising novel insights into cell biology and potential therapeutic strategies, given its involvement in various cellular processes.
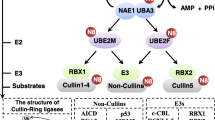
The cullin family
Proteins belonging to the cullin family serve as integral structural elements of CRLs. These modular E3 ubiquitin ligase complexes play an instrumental role in protein ubiquitination and degradation mediated by the 26 S proteasome [25, 26]. In mammals, this family encompasses proteins such as cullin-1, cullin-2, cullin-3, cullin-4 A, cullin-4B, cullin-5, cullin-7, and the p53-associated parkin-like cytoplasmic protein (PARC). Despite the structural similarity and shared conservation of the Cullin homology domains, these proteins participate in diverse complexes and target unique sets of substrates for degradation. Cullin1, for instance, forms the Skp1–Cul1–F-box (SCF) E3 ubiquitin ligase complex that targets proteins for degradation, including cell cycle regulators. It is also significantly involved in DNA damage response and repair processes [27]. Cullin2 and cullin5, on the other hand, form E3 ubiquitin ligase complexes with Elongin BC and SOCS box proteins, contributing to the hypoxia response and innate immunity [28, 29]. Similarly, cullin3 forms E3 ligase complexes with BTB domain proteins, regulating the oxidative stress response, neuronal morphology, and cardiovascular development [30]. Cullin-4 A and cullin4B are integral in forming E3 ligase complexes with DNA damage-binding protein 1 (DDB1) and DDB1- and CUL4-Associated Factor (DCAF) proteins. They play a role in DNA repair, replication, and cell cycle control, and cullin4A is involved in neddylation in the Hippo pathway [31, 32]. Cullin7, forming an E3 ligase complex with S-phase kinase-associated protein 1 (SKP1), F-box and WD repeat domain-containing 8 (FBXW8), and Regulator of cullins-1 (ROC1), is associated with growth regulation, embryonic development, and glucose metabolism [33]. Lastly, cullin-9, also known as PARC, is involved in the negative regulation of p53 and plays a crucial role in cellular responses to DNA damage and stress [34]. Thus, the diverse functions of Cullin proteins underscore their crucial roles in maintaining cellular homeostasis and response to various stimuli. These complexes are integral to regulating several cellular processes, encompassing cell cycle progression, DNA damage response, signal transduction, and development. This underscores the indispensable role of cullins in upholding cellular homeostasis [35, 36].
Cullin proteins, characterized by their elongated forms, serve as scaffolds that facilitate the assembly of other CRL components. The C-terminus of cullins binds to a RING-finger protein, typically either ring box protein 1 (RBX1)/ROC1 or ring box protein 2 (RBX2)/ROC2/ sensitive to apoptosis gene (SAG), facilitating the recruitment of an E2 ubiquitin-conjugating enzyme [7]. RBX1, also recognized as ROC1, is an essential component of CRL complexes, where it acts as a scaffold protein aiding the transfer of ubiquitin from E2 to the target protein [7]. Conversely, RBX2, also identified as ROC2 or SAG, mirrors the function of RBX1 but exhibits more restricted expression. Nonetheless, they play a unique role in safeguarding cells against apoptosis and fostering cell growth and survival, particularly in of cancer [37]. The N-terminus of cullins interacts with various adaptor proteins and substrate receptors, thereby dictating the substrate specificity of each CRL [7]. The regulatory mechanism of cullin proteins involves neddylation, a process in which the NEDD8, modifies cullins. The conjugation of NEDD8 inductes a conformational shift in the cullin protein, bringing the E2 enzyme close to the substrate thereby enhancing ubiquitin transfer efficiency [36, 38]. Given their pivotal roles in protein degradation, aberrations in cullin function or neddylation have been implicated in various pathological conditions, particularly cancer. This insight has led to the development of pharmaceutical inhibitors targeting the neddylation pathway as potential therapeutic interventions for cancer [13].
Neddylation modification process
Neddylation covalently binds the NEDD8 to specific lysine residues of target proteins post-translationally [39]. This process is similar to ubiquitination and involves the attachment of ubiquitin to target proteins to regulate their stability and function. The overall neddylation process is as follows (Fig. 1A):
-
Maturation: Before incorporation into the neddylation pathway, NEDD8 undergoes maturation. Initially synthesized as a precursor protein, NEDD8 contains additional amino acids following the C-terminal diglycine motif, which must be removed to expose this critical motif for subsequent conjugation steps. This maturation is achieved by specific proteases, notably NEDD8 Protease 1 (NEDP1/SENP8) and ubiquitin C-terminal hydrolase-L3 (UCH-L3), which cleave the precursor to yield the mature form of NEDD8. NEDP1, a cysteine protease, recognizes and binds to the precursor NEDD8 and cleaves the additional amino acids by catalyzing the hydrolysis of the peptide bond between the C-terminal glycine (Gly76) of the diglycine motif and the adjacent amino acid, thus exposing the C-terminal diglycine motif [40, 41]. UCH-L3, a member of the UCH family of deubiquitinating enzymes, is also involved in this process. Despite its primary role in the processing and recycling of ubiquitin or ubiquitin-like proteins, UCH-L3 also cleaves the precursor form of NEDD8 [42].
-
Activation: This process begins with the activation of NEDD8. The E1 activating enzyme for NEDD8 is a heterodimer of amyloid β precursor protein-binding protein 1, also known as NAE1 and ubiquitin-like modifier activating enzyme 3 (UBA3) [43, 44]. The NAE heterodimer binds to and activates NEDD8 in an ATP-dependent manner, consuming ATP to adenylate NEDD8’s C-terminal glycine and forming a thioester bond with UBA3’s catalytic cysteine [45].
-
Conjugation: Following activation, NEDD8 is converted to an E2 conjugating enzyme. There are two known E2 enzymes involved in neddylation: UBE2M, the ubiquitin-conjugating enzyme E2 M (also known as UBC12), and UBE2F, the ubiquitin-conjugating enzyme E2 F. The E2 enzyme carries activated NEDD8 to the E3 ligases [42]. The conjugation of NEDD8 to UBE2M or UBE2F involves a trans-thioesterification reaction that transfers NEDD8 from UBA3 to E2, creating a thioester linkage between the C-terminal glycine of NEDD8 and E2 [46, 47].
-
Ligation: The final step involves the transfer of NEDD8 from the E2 enzyme to the target protein, mediated by an E3 ligase. The best-characterized E3 ligase for neddylation is the CRL family, which consists of a cullin protein, a RING domain protein (RBX1 or RBX2), and a substrate recognition component. E3 ligase facilitates the formation of an isopeptide bond between the C-terminal glycine of NEDD8 and a lysine residue on the target protein. The precise lysine residue that is neddylated can varies depending on the specific substrate and context [9, 48].
-
Deneddylation: Deneddylation, the reverse of neddylation, involves the removal of NEDD8 from its conjugated proteins, which plays a crucial role in regulating protein function and cellular processes. Deneddylases are specific enzymes involved in this process. The NEDD8-specific protease NEDP1 (also known as DEN1 or SENP8), a primary enzyme involved in deneddylation, recognizes and binds to NEDD8-conjugated proteins and cleaves the isopeptide bond between NEDD8 and the substrate protein, thereby removing the NEDD8 moiety [41, 49]. Notably, deneddylation was not conducted solely by NEDP1. The COP9 signalosome (CSN), a multi-subunit protein complex, also exhibits deneddylase activity, primarily deneddylating the cullin subunits of CRL complexes, a key regulatory event in CRL activity [50]. This activity depends on the JAMM (JAB1/MPN/Mov34 metalloenzyme) motif located in the CSN5(COP9 Signalosome Subunit 5) subunit, which coordinates the necessary zinc ion for catalysis [50].
Neddylation can profoundly influence various aspects of a protein’s function, such as stability, localization, and activity [51]. The critical nature of neddylation makes it a tightly regulated process, ensuring a balanced and coordinated response to cellular demands [51]. However, the dysregulation of neddylation has serious implications, with increasing evidence pointing to its role in disease pathogenesis. Recently, we found that alterations in the neddylation process are present in the TME, where abnormal neddylation can drive uncontrolled cell growth and resistance to apoptosis [52]. This underlines the importance of further studies on the regulatory mechanisms of neddylation and its therapeutic potential in the TME (Fig. 1B).

Neddylation, a complex multi-step process involving the post-translational attachment of the NEDD8 protein to target proteins, carries out various cellular functions and protein degradation. This process involves the maturation, activation, conjugation, ligation, and deneddylation stages and is conducted by specialized enzymes such as NEDP1, UBA3, and UBE2M. On the other hand, the neddylation modification significantly influences on the tumor microenvironment. This modification can impact various factors, including VEGF, PDGFB, ANGPT2, the EMT process, CAFs, and the ECM. Together, these two figures highlight the critical role of neddylation in both general cell function and the specific context of tumor progression. NEDD8, neural precursor cell expressed developmentally downregulated protein 8; UCH-L3, ubiquitin C-terminal hydrolase-L3; NEDP1, NEDD8 Protease 1; NAE1, NEDD8-activating enzyme 1; UBA3, ubiquitin-like modifier activating enzyme 3; UBE2 M/F, ubiquitin conjugating enzyme E2 M/F, CSN, COP9 signalosome; VEGF, vascular endothelial growth factor; PDGFB, platelet-derived growth factor B; ANGPT2, angiopoietin 2; EMT, epithelial-to-mesenchymal transition; CAFs, cancer-associated fibroblasts; ECM, extracellular matrix. Created with BioRender.com
Neddylation regulates the TME through various signaling pathways: comprehensive frontier information update
The TME is a complex milieu that encapsulates tumor cells and is defined by intricate interactions between neoplastic cells and the various TME constituents [53]. Recent studies have highlighted neddylation’s pivotal role in shaping the TME, influencing malignant cells, immune cells, vascular networks, the epithelial-to-mesenchymal transition (EMT) and the ECM [54, 55]. Neddylation regulation within the TME has profound implications for tumor prognosis [56].
Regulation of tumor cells in TME by neddylation
Tumor cells reshape the TME to enhance cancer progression and resist therapies. They drive angiogenesis via angiogenic factors such as vascular endothelial growth factor (VEGF) [57], produce immunosuppressive factors, and amplify immunosuppressive cells such as myeloid-derived suppressor cells and regulatory T cells (Tregs) to evade the immune response [58]. Tumor cells also lay the groundwork for metastasis by secreting factors that condition distant tissues, establishing a favorable “pre-metastatic niche” for disseminated tumor cells to survive and proliferate [59]. Through metabolic reprogramming, these cells acidify the TME, promote tumor growth and therapy resistance, and impair immune function [60]. Research has shown that activating CRLs by neddylation of cullin proteins promotes cancer cell proliferation by regulating the cell cycle [5]. Furthermore, neddylation modulates apoptosis, affects the DNA damage response by influencing repair protein activity, and shapes the TME by directing cytokine and growth factor secretion [61, 62]. Given its extensive influence on tumor progression and treatment response, targeted neddylation has become a promising anti-cancer therapeutic strategy.
Regulatory mechanisms of neddylation in tumor cells: insights from the p53 and phosphoinositide 3-kinase (PI3K)/AKT/mechanistic Target of Rapamycin (mTOR) pathways
The p53 pathway is an important mechanism by which neddylation regulates cancer cells. In general, the p53 pathway is central to regulating cancer cells in response to DNA damage. Depending on the severity of the damage, p53 either halts the cell cycle for repair or induces apoptosis, preventing unchecked cell growth [63,64,65,66]. However, ribosomal protein L11 (RPL11), typically involved in protein synthesis, impacts the p53 pathway [67, 68]. When ribosome biogenesis is perturbed, RPL11 binds to Mouse double minute 2 homolog (MDM2), an E3 ubiquitin ligase that targets p53, thus preserving p53 by preventing its degradation [69, 70]. Notably, neddylation intervenes in this pathway by inhibiting the nucleolar release of RPL11, shielding it from degradation. This intervention obstructs the formation of the RPL11-MDM2 complex, which would otherwise inhibit p53 and indirectly cause p53 degradation [71, 72]. This mechanism can effectively shift the p53 pathway towards malignancy (Fig. 2).

The interplay between the RPL11-MDM2-p53 and PI3K/AKT/mTOR pathways can be regulated by neddylation. The binding of RPL11 to MDM2 inhibits MDM2’s E3 ligase activity, preventing p53 degradation, but neddylation can hinder this binding, indirectly causing p53 degradation and affecting the expression of various target genes. On the other hand, the PI3K/AKT/mTOR pathway activation initiates when growth factors or hormones bind to cell surface receptors like RTKs or GPCRs, leading to the recruitment and activation of PI3K, which then turns PIP2 into PIP3. PIP3 acts as a docking site for proteins such as AKT and PDK1, allowing PDK1 to activate AKT. Activated AKT inhibits the TSC, a negative regulator of mTORC1, thus enabling Rheb to activate mTORC1. This pathway can be negatively regulated by PTEN, which dephosphorylates PIP3 back to PIP2, removing AKT’s activation signal. Neddylation can enhance PTEN’s nuclear translocation, strengthening the pathway’s signal transduction, while deneddylation of PTEN, dependent on NEDP1, can inhibit the PI3K/AKT/mTOR pathway. MDM2, mouse double minute 2 homolog; RPL11, ribosomal Protein L11; Ub, ubiquitin; NEDD8, neural precursor cell expressed developmentally downregulated protein 8; RTKs, receptor tyrosine kinases; GPCRs, G protein-coupled receptors; PI3K, phosphoinositide 3-kinases; PIP2, Phosphatidylinositol 4,5-bisphosphate; PIP3, Phosphatidylinositol 3,4,5-trisphosphate; TSC, tuberous sclerosis complex; mTORC1, mTOR complex 1; AKT, AKT serine/threonine kinase; PDK1, 3-Phosphoinositide Dependent Protein Kinase-1; Rheb, Ras homolog enriched in brain. Created with BioRender.com
Neddylation has a similar regulatory effect on neoplastic cell behavior via the PI3K/AKT/mTOR signaling pathway. Initiated by the binding of RTKs or GPCRs [73], this pathway activates PI3K which converts PIP2 to PIP3 [74]. This conversion enables AKT kinase activation via PDK1 [75]. Activated AKT inhibits TSC, boosting mTORC1 activity, which is a pathological hallmark of various conditions, and allows Rheb to activate mTORC1 [76]. However, this pathway is modulated by PTEN, which reverts PIP3 to PIP2, thereby negating AKT’s activation signal [77]. Recent studies have shown that PTEN undergoes neddylation, potentially facilitating its translocation to the nucleus [78]. This modulation subsequently amplifies the signal transduction within the PI3K/AKT/mTOR pathway [78]. Notably the deneddylation of PTEN, mediated by NEDP1, can attenuate the signal propagation in the PI3K/AKT/mTOR pathway [78] (Fig. 2).
Regulation of infiltrated immune cells in the TME by neddylation
Neddylation within the TME critically modulates immune cell functions, impacting tumor-associated macrophages (TAMs), T-cells, B-cells, and dendritic cells, primarily through the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) and epidermal growth factor receptor (EGFR) pathways. This underscores its potential as a therapeutic target in cancer.
Regulation of the TAMs by neddylation
TAMs are the predominant leukocytes within tumors and are derived from circulating monocytes drawn to the tumor by chemotactic signals. Once in the TME, these cells differentiate and often adopt M2-like phenotypes. This phenotype is modulated by specific local environmental factors and plays a pivotal role in facilitating tumor progression. Studies have shown that neddylation mediates the production of pro-inflammatory cytokines by macrophages. Multiple studies have pointed out that inhibiting the neddylation process can inhibit lipopolysaccharide (LPS)-induced inflammatory cytokine production, such as interleukin (IL)-6, tumor necrosis factor-alpha (TNF-α), and IL-1β [79, 80]. Additionally, inactivating neddylation curtails inflammation by disrupting lipid metabolism via cullin-6-mediated inhibitor of κB (IκB) degradation, blocking NF-κB activation, which not only modulates macrophage function but also influences cell cycle, apoptosis, and macrophage survival [81, 82]. In conclusion, targeting the neddylation pathway in macrophages, owing to its significant role in tumor progression, offers a promising cancer therapeutic strategy.
Regulation of T-cells by neddylation
T-cells are actively triggered to kill cancer cells when their receptors identify unique malignancy-specific antigens. However, the TME can hinder T-cell function using inhibitory molecules such as programmed death-ligand 1 (PD-L1) or Tregs [83, 84]. This underscores the critical role of T-cell modulation in the trajectory of tumor progression. The neddylation pathway governs T-cell function via several mechanisms, with research indicating that its inhibition hampers T-cell receptor/CD28-driven proliferation and cytokine production both in vitro and in vivo, concurrent with diminished extracellular signal-regulated kinase (ERK) activation, underscoring the regulatory involvement of ERK [85]. Pharmacological and genetic manipulations of the neddylation pathway have been shown to modulate T-cell mediated immune responses [86]. Additionally, emerging evidence suggests that modulation of the neddylation pathway, such as MLN4924 treatment, influences T-cell growth, cytokine production, and differentiation, emphasizing its significant role in T-cell function [87, 88]. In summary, the neddylation pathway plays a crucial role in T-cell functionality and modulation, affecting their response to tumor antigens and overall tumor progression. This serves as a direction for future research exploring the regulatory implications of neddylation on T-cell activity.
Regulation of B-cells by neddylation
As an anti-neoplastic countermeasure, B-cells operationalize a cascade of antigen-specific antibody synthesis and meticulous antigenic presentation to T-lymphocytes. However, -regulatory B-cells contravene this immune propitiousness by promoting the action of immunosuppressive cytokine IL-10, thereby influencing tumor progression and shaping therapeutic outcomes [89]. Studies have suggested that using MLN4924 to inhibit CRLs results in accumulating CRL substrates like IκB and in a CD40L-expressing stromal co-culture system. Both continuous and pulse treatments with MLN4924 suppress NF-κB activity in CLL B-cells ex vivo, and alkylating agents bendamustine and chlorambucil amplify MLN4924-induced DNA damage and apoptosis thereby improving therapeutic efficacy [90,91,92]. In conclusion, the aforementioned research underscores the promising potential of MLN4924 for augmenting the therapeutic effectiveness of B-cell targeted interventions.
Regulation of dendritic cells (DCs) by neddylation
DCs serve as sentinels for presenting tumor-derived antigens to T-cells [93, 94]. However, the TME can impair their maturation and function by producing immunosuppressive molecules such as IL-10 and VEGF [95]. Dysregulated neddylation leads to aberrant DC responses and is implicated in the pathogenesis of multiple malignancies [86]. Neddylation targeting impedes DC maturation by reducing cytokine production and down-regulating costimulatory molecules while promoting caspase-dependent apoptosis, a process linked to mTOR inactivation due to cullin-1 deneddylation-induced deptor accumulation [96]. By inhibiting neddylation, there is a marked reduction in proinflammatory cytokine release from DCs, outperforming the effects of bortezomib or dexamethasone, and a diminished capacity of DCs to activate murine and human allogeneic T cells, positioning neddylation blockade as a promising approach for modulating immune-mediated diseases [97]. In conclusion, targeting neddylation in dendritic cells offers a potential therapeutic strategy for modulating immune responses in various malignancies.
Regulatory mechanisms of neddylation in infiltrated immune cells: insights from the NF-κB and EGFR pathways
The NF-κB pathway plays a pivotal role in immune function. When danger signals are detected, innate immune cells activate NF-κB, promoting anti-tumor activity by releasing inflammatory cytokines. However, continuous activation can enhance tumor growth and survival [98]. In its inactive state, NF-κB is confined to the cytoplasm by the inhibitor protein IκB [99]. The recognition of pathogen-associated molecules like LPS, TNF, or IL-1 by toll-like receptors activates the IκB kinase (IKK) complex [100,101,102]. Once activated, IKK facilitates the degradation of IκB, freeing NF-κB to enter the nucleus [103] and stimulating the transcription of genes essential for immune responses and cell survival [104, 105]. The SCF complex, an integral E3 ubiquitin ligase in the NF-κB pathway, comprises four key components: Skp1, an adaptor molecule linking Cul1 and the F-box protein; Cul1, a scaffold protein that connects to Skp1 and the F-box protein at its N-terminal end and to ring-box 1 (Rbx1), also known as ROC1, at its C-terminal end; F-box proteins, responsible for guiding the SCF complex to its specific targets [106]; and Rbx1, which eases the ubiquitination of the target protein by attracting an E2 ubiquitin-conjugating enzyme to the complex. Neddylation, a process that involves the covalent attachment of NEDD8 to Cul1, amplifies the activity of the SCF complex [107, 108]. This enhancement allows the SCF complex to ubiquitinate its target proteins more effectively, such as the IκB protein [7]. The activated IKK complex can phosphorylate IκB to pIκB, enabling its recognition by the ubiquitin-binding enzyme SCF and degradation through ubiquitination and proteases. The neddylation process may boost SCF activity by activating Cul1 [108], thereby indirectly modulating the expression of the NF-κB pathway. Amplified neddylation boosts the polyubiquitination and proteasomal degradation of the IκB protein, directing the TME’s immune response toward cancer promotion (Fig. 3).

Neddylation plays a crucial role in the regulation of the NF-κB pathway and EGFR pathway, affecting several immune cells. In the NF-κB pathway, IκB inhibition and subsequent proteasomal degradation occur via IKK complex activation. The SCF complex, whose function is enhanced by neddylation, is instrumental in IκB ubiquitination. In the EGFR pathway, neddylation helps regulate the function of Tregs, dendritic cells, M2 macrophages, and CD8 + T cells. NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; IκB, inhibitor of κB; IKK, IκB kinase; TLRs, Toll-like receptors; c-Rel, proto-oncogene c-Rel; SCF, Skp1-Cul1-F-box protein; Skp1, S-phase kinase-associated protein 1; Rbx1, ring-box 1; ROC1, regulator of Cullins 1; NEDD8, neural precursor cell expressed developmentally downregulated protein 8; UBE2M, ubiquitin-conjugating enzyme E2 M; c-CBL, casitas B-lineage lymphoma; Tregs, regulatory T cells. Created with BioRender.com
Similarly, EGFR significantly modulates immune cell behavior. Upon ligand binding, EGFR activation stimulates Treg activation, creating an immunosuppressive microenvironment that promotes tumor growth [109, 110]. It also promptes autophosphorylation, recruiting molecules like Janus kinase 2, which regulates the transcription of cytokine genes, including IL-6, IL-8, IL-10, and VEGF [73, 111,112,113]. These cytokines hinder dendritic cell maturation and functionality, thereby reducing their tumoricidal capacity [114]. Additionally, the EGFR/PI3K/AKT/mTOR pathway directs macrophage polarization towards the M2 phenotype, which secretes growth factors such as EGF, platelet-derived growth factor (PDGF), and transforming growth factor (TGF)-β, favoring tumor cell proliferation and survival [115, 116]. This pathway further diminishes the cytotoxicity of CD8 + T-cells, weakens the immune response against cancer cells, and promotes tumor progression [117,118,119,120]. The EGFR pathway is amplified by Casitas B-lineage lymphoma (c-Cbl), a ubiquitin ligase that modifies EGFR using the ubiquitin-like molecule NEDD8 [121]. This action triggers EGFR neddylation, leading to the endocytic internalization of EGFR and further augmentation of pathway expression [121]. In summary, neddylation of the EGFR indirectly promotes the occurrence and development of tumors by regulating the expression of various immune cells in the TME (Fig. 3).
Regulation of angiogenesis in TME by neddylation
Angiogenesis, a vital process in the TME, ensures nutrient and oxygen supply to tumors but often results in abnormal vasculature and hypoxia [57]. Such changes foster tumor malignancy and metastasis by augmenting vascular permeability [122, 123]. Although new vessels attract immune cells, their abnormalities can deter immune infiltration and assist tumor evasion [124]. Studies have shown that neddylation modulates the VEGF pathway by regulating the stability and degradation of VEGF receptors, thus affecting endothelial cell behavior crucial for new blood vessel formation [62]. Furthermore, neddylation stabilizes HIF-1α, a central player in the cellular response to hypoxia, ensuring the sustained activation of angiogenic genes such as VEGF [125]. Additionally, by targeting the enzymes responsible for ECM remodeling, neddylation influences ECM restructuring, a fundamental step in angiogenesis [126].
Regulatory mechanisms of neddylation in angiogenesis: insights from the hypoxia-inducible factor (HIF) pathways
The HIF pathway is pivotal for tumor angiogenesis. Primarily composed of HIF-1α and HIF-1β, HIF-1’s activity is oxygen-sensitive [127]. Under normal oxygen levels, HIF-1α is degraded due to hydroxylation by prolyl hydroxylase and is also restricted by factors inhibiting HIF-1 (FIH-1) [128]. In hypoxic TMEs, this degradation is halted, allowing HIF-1α to accumulate and pair with HIF-1β. This combined entity activates genes like VEGF, platelet-derived growth factor B (PDGFB), and angiopoietin 2, thereby promoting vessel formation, stability, and sprouting [129, 130]. Additionally, HIF-1 regulates genes that are essential for glucose metabolism and cell survival under hypoxic conditions [131]. Interestingly, von Hippel-lindau is a neddylation target. When neddylation occurs, it inhibits the subsequent degradation of HIF-1α, fostering its stabilization [132]. Such conditions may augment tumor malignancy. Therefore, maintaining the homeostasis of neddylation processes can enhance angiogenesis within the TME by regulating HIF-1α. This, in turn, profoundly impacts tumor development (Fig. 4).

The HIF signaling pathway plays a vital role in tumor angiogenesis by adjusting HIF-1α levels based on oxygen availability, leading to angiogenesis-related gene activation under hypoxic conditions, while neddylation, by inhibiting HIF-1α degradation, can promote tumor growth and angiogenesis. HIF, hypoxia-inducible factor; VHL, von Hippel-Lindau; HREs, hypoxia response elements; VEGF, vascular endothelial growth factor; PDGFB, platelet-derived growth factor B; ANGPT2, angiopoietin 2; NEDD8, neural precursor cell expressed developmentally downregulated protein 8. Created with BioRender.com
Regulation of ECM and cancer-associated fibroblasts (CAFs) in TME by neddylation
The ECM and CAFs are essential for modulating tumor growth, metastasis, and therapeutic resistance. The ECM is a complex structural lattice composed of diverse biomolecules that serve not only as a physical scaffold but also as a mediator of tissue compartmentalization and intercellular signaling [133]. Alterations in the neddylation pathway within tumor cells can significantly reshape the communication dynamics between the tumor and its stroma, particularly influencing cytokine expression and release patterns and growth factors vital for stromal interactions [126]. Recent research highlights an upsurge in neddylation expression in tumor cell, intensifying tumor-stroma crosstalk and potentially hastening cancer progression [134, 135]. Conversely, reduced neddylation impedes pathways linked to fibroblast activation, specifically HMGA1 and HMGA2, angiogenesis markers like annexinA2 and agrin, and pivotal oncogenic routes such as NIK/NF-κB, TNF, Wnt, TGFβ, and mitogen-activated protein kinase [135]. Furthermore, there is an impact on ECM architecture, as neddylation can determine the activity of enzymes such as matrix metalloproteinases (MMPs) [126]. Altered neddylation of specific targets can lead to changes in the turnover of ECM components, affecting tissue stiffness, porosity, and the overall architecture [136, 137]. This modified ECM can directly promote malignancy by inducing mechanotransduction pathways [138], ECM degradation products can have bioactive properties that promote tumor growth and migration [139], and the ECM can control the availability of growth factors to tumor cells [140]. The above studies indicate that within the TME, the ECM is pivotal, and alterations in the neddylation pathway significantly influence tumor-stroma communication, ECM structural dynamics, and the mechanisms of tumor progression.
Similarly, in the TME, CAFs evolve from the transformation of typical fibroblasts and are instrumental in promoting tumor growth and invasion. As prevalent components of the tumor stroma, CAFs can stem from various sources [141] and secrete factors that stimulate cancer cell proliferation, enhance angiogenesis, modulate the immune response, and remodel the ECM [139]. CAFs also induce tumor-promoting inflammation, contribute to cancer cell metabolic reprogramming [142], and aid in therapeutic resistance, either by obstructing drug delivery or by producing factors that counteract drug-induced apoptosis [143]. Neddylation has emerged as a key regulator of CAFs in the TME [144]. This process potentially modulates CAF activation, altering their secretion profiles which influence cancer cell behaviors such as growth and invasiveness [145]. Additionally, it may influence CAF metabolic reprogramming, facilitating the metabolic needs of cancer cells and playing a role in the therapeutic resistance conferred by CAFs [135]. Furthermore, neddylation can shape CAF interactions with other TME cells, affecting overall cancer progression [144]. In summary, the neddylation pathway has emerged as a central modulator of CAF function, directing its interactions with neoplastic cells and subsequently affecting tumor behavior, offering a novel avenue for therapeutic interventions in oncology.
Regulatory mechanisms of neddylation in ECM and CAFs: insights from the TGF-β pathways
TGF-β significantly modulates the ECM within the TME. It stimulates the production of key ECM proteins, leading to denser ECM deposition and characteristic stromal rigidity in many solid tumors [146, 147]. Additionally, TGF-β regulates ECM-remodeling enzymes, influencing ECM integrity and promoting tumor cell invasion [148]. Through its promotion of EMT, TGF-β enhances epithelial cell motility and invasiveness, facilitating tumor progression [149]. In addition to its direct impact on the ECM, TGF-β indirectly influences ECM remodeling by activating CAFs. TGF-β, therefore, induces the differentiation of fibroblasts into myofibroblasts, a CAF subtype that produces substantial amounts of ECM components and ECM-remodeling enzymes [150]. A recent study demonstrated that the phosphorylation of TGFβ receptor 2 (TGFβRII) instigates the RING E3 ligase c-CBL activation, subsequently stabilizing and prolonging its signaling [151]. This mechanism targets TGFβRII for clathrin-mediated endocytosis under endogenous conditions, modulated by neddylation [151]. Thus, neddylation indirectly regulates the expression of the TGF-β pathway. Enhanced neddylation leads to amplified TGF-β pathway expression, increasing the invasiveness and migratory capabilities of the tumor, and consequently, fostering a higher degree of malignancy (Fig. 5).

The TGF-β pathway, modulated by NEDD8, contributes to tumor progression by increasing ECM protein production, regulating ECM remodeling enzymes, promoting epithelial-to-mesenchymal transition, and activating CAFs. Furthermore, neddylation, facilitated by NEDD8, indirectly regulates the TGF-β pathway by stabilizing its signaling through c-CBL, potentially enhancing tumor invasiveness and malignancy. Simultaneously, the Hippo pathway, through the ubiquitination of MST1 and LATS1/2 by CUL7 and CUL4 respectively, plays a crucial role in tumorigenesis. MST1 inhibits the kinase cascade, including LATS1 and LATS2 activation, leading to the phosphorylation of the transcriptional co-activators YAP and TEAD, key downstream effectors of the Hippo pathway, thereby modulating tumor cell growth. Thus, both the TGF-β and Hippo pathways together form a complex network influencing tumor development. N8, neural precursor cell expressed developmentally downregulated protein 8; TGF-β, transforming growth factor-β; CAFs, cancer-associated fibroblasts; c-CBL, casitas b-lineage lymphoma; UBE2M, ubiquitin-conjugating enzyme E2 M; ECM, extracellular matrix; MST1, mammalian STE20-like protein kinase 1; LATS1 and LATS2, Large tumor suppressor kinase 1 and 2; YAP, Yes-associated protein; TEAD, TEA domain. Created with BioRender.com.
Regulation of EMT in TME by neddylation
EMT significantly affects the TME, thereby advancing cancer progression, metastasis, and treatment resistance. Initiated by TME factors such as cytokines and hypoxia, EMT shifts cancer cells from epithelial to mesenchymal states, enhancing their invasiveness and resistance to apoptosis [152, 153]. This process aids in metastasis by promoting ECM degradation [154] and contributes to therapeutic resistance in various cancers, including lung cancer and melanoma [155,156,157], while also inducing cancer stem cell-like properties that intensify treatment challenges and recurrence [158]. Recent findings indicate that neddylation, pivotal for cancer cell migration via the PI3K-Akt pathway, when inhibited, upregulates HIF-1α, modulating EMT-activator ZEB1 in various cancer cell lines, underscoring its significant role in cancer progression and metastasis [159]. In breast cancer, neddylation modulates basal MKK7 activity, which affects the EMT phenotype [160]. Simultaneously, neddylation inhibitors (MLN4924) combined with celecoxib showed promising results in treating urothelial carcinoma, with celecoxib further enhancing the EMT-inhibitory effects of MLN4924 [161]. Thus, understanding the multifaceted role of neddylation in the EMT and its interactions with various drugs may pave the way for improved therapeutic strategies.
Regulatory mechanisms of neddylation in EMT: insights from the Hippo-YAP pathways
The Hippo- yes-associated protein (YAP) signaling pathway is a key regulator of EMT, orchestrating tissue homeostasis under normal conditions and driving tumor formation and progression when dysregulated. Typically, an active Hippo pathway phosphorylates YAP, sequesters and degrades them in the cytoplasm to suppress gene transcription, thereby promoting cell proliferation and inducing apoptosis [162]. Conversely, deregulation of the Hippo pathway triggers YAP dephosphorylation, causing nuclear translocation [163]. YAP forms complexes with TEA domains and other transcription factors, promoting the transcription of pro-proliferative and anti-apoptotic genes, thereby fostering tumor initiation, progression, and drug resistance [163]. This regulatory landscape is also shaped by neddylation, where the NEDD8 substrates CUL7 and CUL4, both ubiquitin ligases, promote the ubiquitination of mammalian STE20-like protein kinase 1 and potentially of large tumor suppressor kinase 1 and 2 (LATS1/2) [164, 165]. These events activate YAP signaling, suggesting that neddylation can amplify the transcription of genes that promote proliferation and inhibit apoptosis [166]. In addition to its role in tumorigenesis, YAP orchestrates cancer metastasis, guiding key processes such as tumor cell invasion and migration by remodeling the actin cytoskeleton and promoting EMT. YAP activates the transcription of genes governing cell motility, invasion, and EMT, such as connective tissue growth factors, fibroblast growth factors, and MMPs [167]. Their upregulation in circulating tumor cells, which are critical players in the metastatic process, underscores their crucial role in cancer spread [168] (Fig. 5).
Targeting the neddylation pathway: emerging strategies in cancer therapeutics
Investigating deneddylating enzymes as potential therapeutics in oncology
Deneddylation, a counter-process to neddylation, is driven by the pivotal enzymes, CSN and SENP8 [169, 170]. Therapeutically, deneddylating enzymes are in the spotlight; inhibitors may have anti-cancer properties, while activators might be beneficial where enhanced deneddylation is required [171]. The CSN is a multifaceted multi-protein complex essential for cellular homeostasis [172]. Comprising eight distinct subunits, with CSN5 and CSN6 exhibiting isopeptidase activity that is pivotal for deneddylation, CSN plays a critical role in regulating the neddylation status of cullin proteins in CRLs [171, 173]. By modulating this status, CSN directly affects the ubiquitin-proteasome system, governing protein degradation, stability, and several of cellular processes such as cell cycle progression and DNA damage response [174]. Given the significance of CSNs in cellular processes, their targeting, especially in pathologies such as cancer, presents an intriguing therapeutic opportunity [171]. Emerging data suggest that CSN inhibitors, such as CSN5i-3 and the natural compound curcumin, operate predominantly by obstructing the CSN5’s deneddylase activity, leading to the hyper-neddylation of cullin proteins and consequent disruption of CRL function [171, 175]. Although these inhibitors have exhibited potential anti-cancer properties, challenges such as off-target effects, cellular redundancy, and potential toxicity underscore the need for meticulous research and optimization [49]. SENP8, also known as DEN1 or NEDP1, is a vital cysteine protease responsible for deneddylation of proteins [176]. By regulating the activity of CRLs, SENP8 ensures proper cellular homeostasis; however, its misregulation can lead to tumorigenesis by stabilizing pro-oncogenic proteins [176]. Consequently, SENP8 has emerged as a potential therapeutic target. Nonetheless, achieving specificity remains paramount, given the extensive array of neddylated proteins. In conclusion, although the roles of CSN and SENP8 in cellular dynamics are undeniable, translating them into mature, therapeutic solutions for tumors requires further refinement to mitigate potential risks.
Therapeutic effect of MLN4924: a current update
MLN4924, also known as pevonedistat, is a small-molecule inhibitor targeting NAE, a critical enzyme in the neddylation pathway [1]. Structurally, MLN4924, also known by its IUPAC name: (1 S,2 S,4R)-4-(4-((S)-2,3-dihydro-1 H-inden-1-ylamino)-7 H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate, has a complex polycyclic arrangement. This includes a 7 H-pyrrolo[2,3-d]pyrimidinyl group linked to a cyclopentylmethyl sulfamate unit and an indenylaminyl substituent, which adds to its structural integrity and bioactivity [13].
Focusing on its mechanism of action, MLN4924 uniquely inhibits NAE, a key facilitator of the neddylation pathway that activates CRLs. MLN4924’s inhibition of NAE hinders the activation of CRLs, leading to the accumulation of CRL substrates and disruption of regular cellular processes [13]. This compound inhibits NAE by selectively forming a covalent NEDD8-MLN4924 adduct in situ that binds to NAE’s active site, thereby maintaining its activity [177]. This selectivity and the often dysregulated neddylation pathway in several cancers highlight MLN4924’s therapeutic potential [178].
This compound has shown promise in preclinical studies of numerous cancer types, including lymphoma, leukemia, and solid tumors, owing to its capacity to induce cell cycle arrest, apoptosis, senescence, and autophagy [179]. Its therapeutic efficacy and safety are currently being evaluated in clinical trials. Despite the potential of MLN4924 (pevonedistat) as a novel therapeutic for various cancer types in both preclinical and clinical studies, it is not without limitations. One such problem is drug resistance, which may arise from mutations in NAE1. These mutations alter the binding site of the drug, thereby dampening its inhibitory effects [180]. In addition, the increased expression of NEDD8-conjugated proteins has been observed in some MLN4924-resistant cancer cells [181]. Adverse side effects such as fatigue, nausea, vomiting, diarrhea, and anemia have also been reported in phase I trials [182]. Furthermore, while MLN4924 selectively inhibits NAE, it may not completely block all NEDD8 conjugation pathways, leaving other neddylation targets such as p53 and MDM2 unaffected [183]. Lastly, although MLN4924 has demonstrated efficacy in preclinical models, it may not always suffice as a standalone therapy. Certain cancer types may need to be treated in conjunction with other treatments to enhance their therapeutic efficacy [184].
Therapeutic effect of pevonedistat on patients with acute Myeloid Leukemia (AML) and Myelodysplastic Syndrome (MDS)
AML and MDS are both myeloid malignancies; however, they differ in their clinical presentations and challenges. MDS typically progresses slowly and does not require immediate therapeutic interventions [185]. By contrast, AML is characterized by a diverse clinical and molecular landscape, underscored by the uncontrolled proliferation of abnormally differentiated myeloid progenitor cells [186, 187]. Despite these differences, both MDS and AML are notorious for frequent relapses after chemotherapy and resistance to conventional treatments owing to the abnormal activation of various signaling pathways [188, 189]. These complications highlight the urgent need for innovative therapeutic strategies.
For a long time, the combined use of azacitidine and other chemotherapeutic drugs has had a certain effect, but owing to its non-negligible cytotoxicity, more complete drug combination therapy is warranted [190, 191]. Simultaneously, pevonedistat has emerged as a promising drug for treating acute myeloid leukaemia and myelodysplastic syndromes, demonstrating feasible administration and modest clinical efficacy in a phase 1 clinical trial (NCT00911066), despite hepatotoxicity and multi-organ failure identified as dose-limiting constraints [182]. Recent clinical trials (NCT03862157) have provided promising evidence for the role of pevonedistat in AML and MDS. When tested in combination with azacitidine and venetoclax in a phase 1/2 trial, the study targeted older adults recently diagnosed with secondary AML, MDS, or chronic myelomonocytic leukemia (CMML) and assessed their response rates to the specified drug combination [192]. Notably, an impressive complete remission or incomplete hematological recovery (CR/CRi) rate of 66% was observed in the AML cohort, whereas the MDS/CMML cohort demonstrated a robust overall response rate of 75%, signaling the potency of this drug combination [192]. However, the prevalence of common adverse events, such as infection and febrile neutropenia, must be considered [192]. Moreover, the research findings suggest potential molecular alterations that may contribute to the development of therapeutic resistance. As such, the ensuing implications from these results suggest that this innovative triplet combination could offer a beneficial treatment pathway for patients presenting with high-risk AML, MDS, or CMML.
In conclusion, an increasing number of clinical studies have begun to focus on combining pevonedistat and chemotherapeutic drugs such as azacitidine, offering a promising new direction for treating MDS and AML (Table 1).
Therapeutic effect of pevonedistat on patients with malignant Lymphoma
Despite advancements in treatment, the prognosis of patients with relapsed or refractory lymphoma remains poor. Although targeted therapies have cured some patients, managing refractory and relapsed conditions remains challenging [193]. Notably, although targeted therapeutic strategies have led to curative outcomes in a subset of patients with lymphoma, managing refractory and relapsed disease persistently presents a substantial challenge [194]. Therefore, pevonedistat has emerged as a promising therapeutic agent. Pevonedistat induces intrinsic apoptosis or senescence in diverse lymphoma cells in a cell line-dependent manner [195] and triggers G2 cell-cycle arrest in lymphoma cells, leading to apoptosis or senescence, while concurrently upregulating pro-apoptotic factors and downregulating anti-apoptotic factors [196]. It is also known for its inhibitory effect on NFκB activity, thereby re-sensitizing diffuse large B-cell lymphoma and primary chronic lymphocytic leukemia cells to extrinsic apoptosis [197]. Further evidence of the therapeutic potential of pevonedistat has been observed in preclinical lymphoma models. Particularly in activated B-cell diffuse large B-cell lymphoma cell lines, pevonedistat enhances the activity of various chemotherapeutic agents and inhibitors; when used in combination with ibrutinib or cytarabine, it improves survival rates in severe combined immunodeficiency mouse xenograft models [198].
Pevonedistat has shown promise not only in preclinical experiments but also in clinical trials. A phase I study investigating its effects in patients with relapsed or refractory myeloma and lymphoma demonstrated a well-tolerated profile with minimal myelosuppression and no treatment-related deaths [184]. These outcomes suggest that pevonedistat could potentially be effective in managing refractory lymphoma, as evidenced by some patients achieving disease stability or partial response. In conclusion, this drug has shown encouraging results in clinical trials, both as a standalone treatment and in conjunction with other chemotherapies or targeted therapy regimens (Table 1).
Therapeutic effect of pevonedistat on patients with solid tumors
Primarily effective against various solid tumors such as those of the colon and lung as exhibited in preclinical studies, pevonedistat’s potent anti-tumor activity extends across multiple tumor types [199,200,201,202,203], and it has been proven efficacious in tumor xenograft mouse models, where it curtails tumor growth through the inhibition of NEDD8 conjugation and increasing NAE inhibition following both single and repeated doses [204]. As with hematological tumors, pevonedistat continues to be used in conjunction with chemotherapeutic drugs to treat solid tumors with significant success (Table 1). An earlier study revealed that pevonedistat, under phase I/II clinical trials as a potential glioblastoma treatment, was found that when combined with anti-PD-L1 therapy, the therapeutic efficacy significantly improves in vivo by effectively restoring T cell sensitivity [205]. These findings provide investigators with increased confidence in potentially combining pevonedistat with targeted therapies.
The latest Clinical Trial (NCT03486314) demonstrated that MLN4924 not only inhibited cell viability and induced apoptosis in human umbilical vascular endothelial cells but also disrupted cell cycle checkpoint regulators suppressed angiogenic activity and cell migration, decreased UBC12 levels (indicating VEGF-activated neddylation pathway involvement), and inhibiteds tumor growth in mouse models of four different types of cancer [206]. Given the significant anti-tumor effects observed in various solid tumors with the neddylation inhibitor, pevonedistat can be used in combination treatments for advanced solid tumors, potentially paving the way for new therapeutic strategies.
Conclusions
In oncological research, there has been a marked transition from an exclusive concentration on malignant cells to a comprehensive exploration of the TME, encompassing the interplay of malignant cells, immune cells, stromal cells, the ECM, and the molecular constituents that interface with the tumor [207,208,209]. The TME plays an instrumental role in tumorigenesis, metastasis, and the response to therapy, making it a rich source of potential therapeutic targets. However, future studies must address several challeng to harness this potential fully. Both intra- and inter-tumoral, heterogeneity pose significant therapeutic resistance challenges [210]. Intra-tumoral variations encompass genetic, epigenetic, and phenotypic disparities within a tumor, which are influenced by factors such as distinct cancer cell evolutionary paths and microenvironmental variations [211, 212]. Inter-tumor differences arise from variances in genetics, environment, and immune responses among tumors, even within the same patient [213], and can foster the development of resistant clones, complicating treatment outcomes [214]. To address the challenges posed by tumor heterogeneity, it is essential to understand the underlying signaling mechanisms within the TME and identify potential therapeutic targets. Emphasizing the need for in-depth mechanistic studies, our research reveals the extensive involvement of neddylation in the TME’s regulatory processes, encompassing pathways like p53, PI3K/AKT/mTOR, NF-κB, EGFR, HIF, TGF-β, and Hippo-YAP. The centrality of neddylation in tumor progression underscores its potential as a novel therapeutic intervention.
Metastasis, a primary cause of cancer-related fatalities, depends on the formation of pre-metastatic niches. These niches, which are altered regions within potential metastatic sites, are primed by the primary tumor to facilitate metastatic cell growth. Tumors achieve this through factor secretion, cell recruitment [59], induction of inflammation, and modification of vascular and extracellular structures [215, 216]. Although progress has been made in understanding these niches, the exact mechanisms and pathways involved, especially those related to the EMT within the TME, remain elusive [168]. Through an in-depth analysis, we determined that neddylation critically influences the formation of pre-metastatic niches. It directly governs cancer cell migration, invasion, and interactions within the TME, particularly by modulating cytokine secretion and growth factors [144]. Neddylation also plays a crucial role in signaling pathways related to metastasis and has noteworthy implications for immune evasion [5]. Its significant impact on ECM remodeling, essential for fostering a favorable environment for metastatic cells, further underscores the potential of neddylation as a therapeutic target [135].
Recently, Mln4924 emerged as a major breakthrough. By directly inhibiting neddylation modifications within cell pathways, MLN4924 disrupts many signaling pathways that could potentially interfere with the TME and promote tumor progression [217]. The efficacy of MLN4924, particularly when used in conjunction with chemotherapy, has been demonstrated in numerous clinical trials. These insights may aid in the development of more effective cancer treatment strategies. In summary, our findings highlight the profound impact of neddylation on TME, offering promising avenues for enhanced cancer treatment strategies.
Availability of data and materials
The datasets supporting the conclusions are included within this article.
References
Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272(45):28557–62.
Xirodimas DP. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem Soc Trans. 2008;36(Pt 5):802–6.
Mergner J, Schwechheimer C. The NEDD8 modification pathway in plants. Front Plant Sci. 2014;5:103.
Kumar S, Yoshida Y, Noda M. Cloning of a cDNA which encodes a novel ubiquitin-like protein. Biochem Biophys Res Commun. 1993;195(1):393–9.
Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44.
Zheng YC, Guo YJ, Wang B, Wang C, Mamun MAA, Gao Y, et al. Targeting neddylation E2s: a novel therapeutic strategy in cancer. J Hematol Oncol. 2021;14(1):57.
Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20.
Zhao Y, Sun Y. Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19(18):3215–25.
Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434.
Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6(5):369–81.
Du M, Sansores-Garcia L, Zu Z, Wu KK. Cloning and expression analysis of a novel salicylate suppressible gene, Hs-CUL-3, a member of cullin/Cdc53 family. J Biol Chem. 1998;273(38):24289–92.
Hori T, Osaka F, Chiba T, Miyamoto C, Okabayashi K, Shimbara N, et al. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999;18(48):6829–34.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–6.
Zhao Y, Xiong X, Jia L, Sun Y. Targeting cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3(9):e386.
Zhou Q, Li H, Li Y, Tan M, Fan S, Cao C, et al. Inhibiting neddylation modification alters mitochondrial morphology and reprograms energy metabolism in cancer cells. JCI Insight. 2019;4(4): e121582.
Jia L, Li H, Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13(6):561–9.
Lin S, Shang Z, Li S, Gao P, Zhang Y, Hou S, et al. Neddylation inhibitor MLN4924 induces G(2) cell cycle arrest, DNA damage and sensitizes esophageal squamous cell carcinoma cells to cisplatin. Oncol Lett. 2018;15(2):2583–9.
Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019;20(4): 840.
Quail DF, Joyce JA. Microenvironmental regulation of Tumor progression and Metastasis. Nat Med. 2013;19(11):1423–37.
Nath D, Shadan S. The ubiquitin system. Nature. 2009;458(7237):421.
Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26(4):399–422.
Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, sine quibus non. Cancer Cell. 2011;19(2):168–76.
Whitby FG, Xia G, Pickart CM, Hill CP. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J Biol Chem. 1998;273(52):34983–91.
Hjerpe R, Thomas Y, Kurz T. NEDD8 overexpression results in neddylation of ubiquitin substrates by the ubiquitin pathway. J Mol Biol. 2012;421(1):27–9.
Lu G, Wang L, Zhou J, Liu W, Shen HM. A destiny for degradation: interplay between cullin-RING E3 ligases and autophagy. Trends Cell Biol. 2021;31(6):432–44.
Nguyen HC, Wang W, Xiong Y. Cullin. -RING E3 ubiquitin ligases: bridges to destruction. Subcell Biochem. 2017;83:323–47.
Wang Z, Zhang H, Liu J, Cheruiyot A, Lee JH, Ordog T, et al. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016;30(8):946–59.
Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18(24):3055–65.
Liu J, Zhang C, Wang XL, Ly P, Belyi V, Xu-Monette ZY, et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014;21(11):1792–804.
Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. Embo J. 2013;32(17):2307–20.
Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8(11):1277–83.
Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22(18):2507–19.
Kim SS, Shago M, Kaustov L, Boutros PC, Clendening JW, Sheng Y, et al. CUL7 is a novel antiapoptotic oncogene. Cancer Res. 2007;67(20):9616–22.
Li Z, Pei XH, Yan J, Yan F, Cappell KM, Whitehurst AW, et al. CUL9 mediates the functions of the 3 M complex and ubiquitylates survivin to maintain genome integrity. Mol Cell. 2014;54(5):805–19.
Bosu DR, Kipreos ET. Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 2008;3: 7.
Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134(6):995–1006.
Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11(3):347–56.
Lydeard JR, Schulman BA, Harper JW. Building and remodelling cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14(12):1050–61.
Gai W, Peng Z, Liu CH, Zhang L, Jiang H. Advances in cancer treatment by targeting the neddylation pathway. Front Cell Dev Biol. 2021;9: 653882.
Gan-Erdene T, Nagamalleswari K, Yin L, Wu K, Pan ZQ, Wilkinson KD. Identification and characterization of DEN1, a deneddylase of the ULP family. J Biol Chem. 2003;278(31):28892–900.
Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278(31):28882–91.
Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251(3):688–92.
Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999;274(17):12036–42.
Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10(5):319–31.
Walden H, Podgorski MS, Huang DT, Miller DW, Howard RJ, Minor DL Jr, et al. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol Cell. 2003;12(6):1427–37.
Huang DT, Ayrault O, Hunt HW, Taherbhoy AM, Duda DM, Scott DC, et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol Cell. 2009;33(4):483–95.
Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8’s E1. Mol Cell. 2005;17(3):341–50.
Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014;21(17):2383–400.
Mergner J, Kuster B, Schwechheimer C. DENEDDYLASE1 protein counters automodification of neddylating enzymes to maintain NEDD8 protein homeostasis in Arabidopsis. J Biol Chem. 2017;292(9):3854–65.
Verma R, Aravind L, Oania R, McDonald WH, Yates JR 3, Koonin EV, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298(5593):611–5.
Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9(10):969–76.
Ying J, Zhang M, Qiu X, Lu Y. Targeting the neddylation pathway in cells as a potential therapeutic approach for diseases. Cancer Chemother Pharmacol. 2018;81(5):797–808.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Zhou L, Zhang W, Sun Y, Jia L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018;44:92–102.
He ZX, Yang WG, Zengyangzong D, Gao G, Zhang Q, Liu HM, et al. Targeting cullin neddylation for cancer and fibrotic diseases. Theranostics. 2023;13(14):5017–56.
Tanaka T, Nakatani T, Kamitani T. Inhibition of NEDD8-conjugation pathway by novel molecules: potential approaches to anticancer therapy. Mol Oncol. 2012;6(3):267–75.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307.
Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25.
Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer. 2017;17(5):302–17.
Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer. 2017;17(10):577–93.
Pellegrino NE, Guven A, Gray K, Shah P, Kasture G, Nastke MD, et al. The next frontier: translational development of ubiquitination, SUMOylation, and NEDDylation in cancer. Int J Mol Sci. 2022;23(7): 3480.
Duncan K, Schäfer G, Vava A, Parker MI, Zerbini LF. Targeting neddylation in cancer therapy. Future Oncol. 2012;8(11):1461–70.
Hafner A, Bulyk ML, Jambhekar A, Lahav G. The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol. 2019;20(4):199–210.
Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–70.
Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062–78.
Labuschagne CF, Zani F, Vousden KH. Control of metabolism by p53 - cancer and beyond. Biochim Biophys Acta Rev Cancer. 2018;1870(1):32–42.
Zhou X, Hao Q, Zhang Q, Liao JM, Ke JW, Liao P, et al. Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ. 2015;22(5):755–66.
Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, et al. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23(23):8902–12.
Bursać S, Brdovčak MC, Pfannkuchen M, Orsolić I, Golomb L, Zhu Y, et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc Natl Acad Sci U S A. 2012;109(50):20467–72.
Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16(5):369–77.
Mahata B, Sundqvist A, Xirodimas DP. Recruitment of RPL11 at promoter sites of p53-regulated genes upon nucleolar stress through NEDD8 and in an Mdm2-dependent manner. Oncogene. 2012;31(25):3060–71.
Sundqvist A, Liu G, Mirsaliotis A, Xirodimas DP. Regulation of nucleolar signalling to p53 through NEDDylation of L11. EMBO Rep. 2009;10(10):1132–9.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–34.
Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405.
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 Tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–62.
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–96.
Xie P, Peng Z, Chen Y, Li H, Du M, Tan Y, et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021;31(3):291–311.
Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Mulligan G, Pickard MD, et al. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Invest New Drugs. 2016;34(4):439–49.
Jiang Y, Li L, Li Y, Liu G, Hoffman RM, Jia L. Neddylation regulates macrophages and implications for cancer therapy. Front Cell Dev Biol. 2021;9: 681186.
Chang FM, Reyna SM, Granados JC, Wei SJ, Innis-Whitehouse W, Maffi SK, et al. Inhibition of neddylation represses lipopolysaccharide-induced proinflammatory cytokine production in macrophage cells. J Biol Chem. 2012;287(42):35756–67.
Li L, Liu B, Dong T, Lee HW, Yu J, Zheng Y, et al. Neddylation pathway regulates the proliferation and survival of macrophages. Biochem Biophys Res Commun. 2013;432(3):494–8.
Pauken KE, Wherry EJ. Overcoming T cell exhaustion in Infection and cancer. Trends Immunol. 2015;36(4):265–76.
De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45(5):1135–47.
Jin HS, Liao L, Park Y, Liu YC. Neddylation pathway regulates T-cell function by targeting an adaptor protein shc and a protein kinase erk signaling. Proc Natl Acad Sci U S A. 2013;110(2):624–9.
Lu Y, Yang X. The pivotal roles of neddylation pathway in immunoregulation. Immun Inflamm Dis. 2020;8(4):782–92.
Biedermann T, Röcken M, Carballido JM. TH1 and TH2 lymphocyte development and regulation of TH cell-mediated immune responses of the skin. J Investig Dermatol Symp Proc. 2004;9(1):5–14.
Cheng Q, Liu J, Pei Y, Zhang Y, Zhou D, Pan W, et al. Neddylation contributes to CD4 + T cell-mediated protective immunity against blood-stage Plasmodium Infection. PLoS Pathog. 2018;14(11):e1007440.
Rosser EC, Mauri C. Regulatory. B cells: origin, phenotype, and function. Immunity. 2015;42(4):607–12.
Godbersen JC, Paiva C, Danilova OV, Berger A, Brown JR, Danilov AV. Targeting neddylation effectively antagonizes nuclear factor-κB in chronic lymphocytic Leukemia B-cells. Leuk Lymphoma. 2015;56(5):1566–9.
Paiva C, Godbersen JC, Berger A, Brown JR, Danilov AV. Targeting neddylation induces DNA damage and checkpoint activation and sensitizes chronic lymphocytic Leukemia B cells to alkylating agents. Cell Death Dis. 2015;6(7):e1807.
Best S, Lam V, Liu T, Bruss N, Kittai A, Danilova OV, et al. Immunomodulatory effects of pevonedistat, a NEDD8-activating enzyme inhibitor, in chronic lymphocytic leukemia-derived T cells. Leukemia. 2021;35(1):156–68.
Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019;19(2):89–103.
Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, et al. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. 2013;38(2):336–48.
Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res. 2017;5(1):3–8.
Cheng M, Hu S, Wang Z, Pei Y, Fan R, Liu X, et al. Inhibition of neddylation regulates dendritic cell functions via deptor accumulation driven mTOR inactivation. Oncotarget. 2016;7(24):35643–54.
Mathewson N, Toubai T, Kapeles S, Sun Y, Oravecz-Wilson K, Tamaki H, et al. Neddylation plays an important role in the regulation of murine and human dendritic cell function. Blood. 2013;122(12):2062–73.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
Hayden MS, Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–34.
Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16(1):3–9.
Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733.
Hinz M, Scheidereit C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014;15(1):46–61.
Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034.
Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5(5):392–401.
Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–66.
Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14(6):369–81.
Saha A, Deshaies RJ. Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol Cell. 2008;32(1):21–31.
Read MA, Brownell JE, Gladysheva TB, Hottelet M, Parent LA, Coggins MB, et al. Nedd8 modification of cul-1 activates SCF(beta(TrCP))-dependent ubiquitination of IkappaBalpha. Mol Cell Biol. 2000;20(7):2326–33.
Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Macciò A. EGFR-mutated non-small cell Lung cancer and resistance to immunotherapy: role of the Tumor microenvironment. Int J Mol Sci. 2022;23(12):6489.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9.
Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117(Pt 8):1281–3.
Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: receptors, functions, and roles in Diseases. J Allergy Clin Immunol. 2016;138(4):984–1010.
Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437(2):169–83.
Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108(5):1435–40.
Lian G, Chen S, Ouyang M, Li F, Chen L, Yang J. Colon Cancer cell secretes EGF to promote M2 polarization of TAM through EGFR/PI3K/AKT/mTOR pathway. Technol Cancer Res Treat. 2019;18:1533033819849068.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8 + T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–88.
Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70.
Oved S, Mosesson Y, Zwang Y, Santonico E, Shtiegman K, Marmor MD, et al. Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem. 2006;281(31):21640–51.
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64.
Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873–87.
Motz GT, Coukos G. The parallel lives of angiogenesis and immunosuppression: cancer and other tales. Nat Rev Immunol. 2011;11(10):702–11.
Curtis VF, Ehrentraut SF, Colgan SP. Actions of adenosine on cullin neddylation: implications for inflammatory responses. Comput Struct Biotechnol J. 2015;13:273–6.
Kreienbühl J, Changkhong S, Orlowski V, Kirschner MB, Opitz I, Meerang M. Cullin 4B ubiquitin ligase is important for cell survival and regulates TGF-β1 expression in pleural Mesothelioma. Int J Mol Sci. 2023;24(17): 13410.
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–4.
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–71.
Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56.
Hellström M, Kalén M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–55.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85.
Russell RC, Ohh M. NEDD8 acts as a ‘molecular switch’ defining the functional selectivity of VHL. EMBO Rep. 2008;9(5):486–91.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–200.
Vava A, Paccez JD, Wang Y, Gu X, Bhasin MK, Myers M, et al. DCUN1D1 is an essential regulator of Prostate cancer proliferation and tumour growth that acts through neddylation of cullin 1, 3, 4A and 5 and deregulation of Wnt/Catenin pathway. Cells. 2023;12(15): 1973.
Olaizola P, Lee-Law PY, Fernandez-Barrena MG, Alvarez L, Cadamuro M, Azkargorta M, et al. Targeting NAE1-mediated protein hyper-NEDDylation halts cholangiocarcinogenesis and impacts on tumor-stroma crosstalk in experimental models. J Hepatol. 2022;77(1):177–90.
Hryniewicz-Jankowska A, Wierzbicki J, Tabola R, Stach K, Sossey-Alaoui K, Augoff K. The effect of neddylation inhibition on inflammation-induced MMP9 gene expression in esophageal squamous cell carcinoma. Int J Mol Sci. 2021;22(4): 1716.
Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406.
Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9(2):108–22.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98.
Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–9.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–47.
Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–4.
Zhou L, Jiang Y, Luo Q, Li L, Jia L. Neddylation: a novel modulator of the Tumor microenvironment. Mol Cancer. 2019;18(1):77.
Wang Y, Weng X, Wang L, Hao M, Li Y, Hou L, et al. HIC1 deletion promotes Breast cancer progression by activating Tumor cell/fibroblast crosstalk. J Clin Invest. 2018;128(12):5235–50.
Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and Metastasis. Semin Cancer Biol. 2014;25:15–22.
Lodyga M, Hinz B. TGF-β1 - A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev Biol. 2020;101:123–39.
Jobling MF, Mott JD, Finnegan MT, Jurukovski V, Erickson AC, Walian PJ, et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat Res. 2006;166(6):839–48.
Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–72.
Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122(1):103–11.
Zuo W, Huang F, Chiang YJ, Li M, Du J, Ding Y, et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-β type II receptor. Mol Cell. 2013;49(3):499–510.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67.
Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–60.
Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in Melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23(6):811–25.
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the War on cancer. Oncogene. 2010;29(34):4741–51.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15.
Park JB, Seo J, Park JW, Chun YS. Neddylation blockade induces HIF-1α driven cancer cell migration via upregulation of ZEB1. Sci Rep. 2020;10(1):18210.
Zhu T, Wang J, Pei Y, Wang Q, Wu Y, Qiu G, et al. Neddylation controls basal MKK7 kinase activity in breast cancer cells. Oncogene. 2016;35(20):2624–33.
Xiong S, Huang W, Liu X, Chen Q, Ding Y, Huang H, et al. Celecoxib synergistically enhances MLN4924-induced cytotoxicity and EMT inhibition via AKT and ERK pathways in human urothelial carcinoma. Cell Transpl. 2022;31:9636897221077920.
Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29(6):783–803.
Cunningham R, Hansen CG. The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin Sci (Lond). 2022;136(3):197–222.
Cooper J, Xu Q, Zhou L, Pavlovic M, Ojeda V, Moulick K, et al. Combined inhibition of NEDD8-Activating enzyme and mTOR suppresses NF2 Loss-driven tumorigenesis. Mol Cancer Ther. 2017;16(8):1693–704.
Zou J, Ma W, Li J, Littlejohn R, Zhou H, Kim IM, et al. Neddylation mediates ventricular chamber maturation through repression of Hippo signaling. Proc Natl Acad Sci U S A. 2018;115(17):E4101-4110.
Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163(4):811–28.
Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20(6):638–46.
Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of Metastasis. Cell. 2017;168(4):670–91.
Lin H, Zhang X, Liu L, Fu Q, Zang C, Ding Y, et al. Basis for metabolite-dependent cullin-RING ligase deneddylation by the COP9 signalosome. Proc Natl Acad Sci U S A. 2020;117(8):4117–24.
Li Y, Zhou H, Liu P, Lv D, Shi Y, Tang B, et al. SHP2 deneddylation mediates Tumor immunosuppression in colon cancer via the CD47/SIRPα axis. J Clin Invest. 2023;133(4): e162870.
Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun. 2016;7;13166.
Du W, Zhang R, Muhammad B, Pei D. Targeting the COP9 signalosome for cancer therapy. Cancer Biol Med. 2022;19(5):573–90.
Birol M, Enchev RI, Padilla A, Stengel F, Aebersold R, Betzi S, et al. Structural and biochemical characterization of the Cop9 signalosome CSN5/CSN6 heterodimer. PLoS ONE. 2014;9(8):e105688.
Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14(11):1209–25.
Dubiel D, Wang J, Hartig R, Chaithongyot S, Dubiel W, Naumann M. Latent CSN-CRL complexes are crucial for curcumin-induced apoptosis and recruited during adipogenesis to lipid droplets via small GTPase RAB18. iScience. 2023;26(4):106468.
Coleman KE, Békés M, Chapman JR, Crist SB, Jones MJ, Ueberheide BM, et al. SENP8 limits aberrant neddylation of NEDD8 pathway components to promote cullin-RING ubiquitin ligase function. Elife. 2017;6:e24325.
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102–11.
Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell Lymphoma models: rationale for treatment of NF-{kappa}B-dependent Lymphoma. Blood. 2010;116(9):1515–23.
Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115(18):3796–800.
Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, et al. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102(36):12873–8.
Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, et al. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-κB activation and induces apoptosis in chronic lymphocytic Leukemia B cells. Clin Cancer Res. 2014;20(6):1576–89.
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid Leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169(4):534–43.
Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118(1):83–97.
Shah JJ, Jakubowiak AJ, O’Connor OA, Orlowski RZ, Harvey RD, Smith MR, et al. Phase I study of the Novel Investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22(1):34–43.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120(12):2454–65.
Halik A, Arends CM, Bullinger L, Damm F, Frick M. Refining AML treatment: the role of genetics in response and resistance evaluation to new agents. Cancers (Basel). 2022;14(7):1689.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute Myeloid Leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–76.
Derissen EJ, Beijnen JH, Schellens JH. Concise drug review: azacitidine and decitabine. Oncologist. 2013;18(5):619–24.
Koenig KL, Sahasrabudhe KD, Sigmund AM, Bhatnagar B. AML with myelodysplasia-related changes: development, challenges, and treatment advances. Genes (Basel). 2020;11(8):845.
Ravandi F, Roboz GJ, Wei AH, Döhner H, Pocock C, Selleslag D, et al. Management of adverse events in patients with acute Myeloid Leukemia in remission receiving oral azacitidine: experience from the phase 3 randomized QUAZAR AML-001 trial. J Hematol Oncol. 2021;14(1):133.
Cogle CR, Scott BL, Boyd T, Garcia-Manero G. Oral azacitidine (CC-486) for the treatment of myelodysplastic syndromes and acute Myeloid Leukemia. Oncologist. 2015;20(12):1404–12.
Short NJ, Muftuoglu M, Ong F, Nasr L, Macaron W, Montalban-Bravo G, et al. A phase 1/2 study of azacitidine, venetoclax and pevonedistat in newly diagnosed secondary AML and in MDS or CMML after failure of hypomethylating agents. J Hematol Oncol. 2023;16(1):73.
Booth S, Collins G. Epigenetic targeting in Lymphoma. Br J Haematol. 2021;192(1):50–61.
Wang L, Qin W, Huo YJ, Li X, Shi Q, Rasko JEJ, et al. Advances in targeted therapy for malignant Lymphoma. Signal Transduct Target Ther. 2020;5(1):15.
Shankland KR, Armitage JO, Hancock BW. Non-hodgkin Lymphoma. Lancet. 2012;380(9844):848–57.
Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS, et al. Targeting protein neddylation with an NEDD8-activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human Lymphoma cells. Cancer Biol Ther. 2015;16(3):420–9.
Paiva C, Godbersen JC, Rowland T, Danilova OV, Danes C, Berger A, et al. Pevonedistat, a Nedd8-activating enzyme inhibitor, sensitizes neoplastic B-cells to death receptor-mediated apoptosis. Oncotarget. 2017;8(13):21128–39.
Torka P, Mavis C, Kothari S, Belliotti S, Gu J, Sundaram S, et al. Pevonedistat, a NEDD8-activating enzyme inhibitor, induces apoptosis and augments efficacy of chemotherapy and small molecule inhibitors in pre-clinical models of diffuse large B-cell Lymphoma. EJHaem. 2020;1(1):122–32.
Zhang W, Liang Y, Li L, Wang X, Yan Z, Dong C, et al. The Nedd8-activating enzyme inhibitor MLN4924 (TAK-924/Pevonedistat) induces apoptosis via c-Myc-noxa axis in head and neck squamous cell carcinoma. Cell Prolif. 2019;52(2):e12536.
Jones TM, Carew JS, Bauman JE, Nawrocki ST. Targeting NEDDylation as a novel approach to improve the treatment of Head and Neck cancer. Cancers (Basel). 2021;13(13):3250.
Wan J, Zhu J, Li G, Zhang Z. Radiosensitization of human Colorectal cancer cells by MLN4924: an inhibitor of NEDD8-activating enzyme. Technol Cancer Res Treat. 2016;15(4):527–34.
Ferris J, Espona-Fiedler M, Hamilton C, Holohan C, Crawford N, McIntyre AJ, et al. Pevonedistat (MLN4924): mechanism of cell death induction and therapeutic potential in Colorectal cancer. Cell Death Discov. 2020;6:61.
Zheng W, Luo Z, Zhang J, Min P, Li W, Xu D, et al. Neural precursor cell expressed, developmentally downregulated 8–activating enzyme inhibitor MLN4924 sensitizes Colorectal cancer cells to oxaliplatin by inducing DNA damage, G2 cell cycle arrest and apoptosis. Mol Med Rep. 2017;15(5):2795–801.
Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15(12):3912–6.
Zhou S, Zhao X, Yang Z, Yang R, Chen C, Zhao K, et al. Neddylation inhibition upregulates PD-L1 expression and enhances the efficacy of immune checkpoint blockade in glioblastoma. Int J Cancer. 2019;145(3):763–74.
Shi CS, Kuo KL, Lin WC, Chen MS, Liu SH, Liao SM, et al. Neddylation inhibitor, MLN4924 suppresses angiogenesis in huvecs and solid cancers: in vitro and in vivo study. Am J Cancer Res. 2020;10(3):953–64.
Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–99.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the Tumor microenvironment. Nat Immunol. 2013;14(10):1014–22.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the Tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613–28.
Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–34.
Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169–75.
Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(8):846–53.
Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94.
Sceneay J, Smyth MJ, Möller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32(3–4):449–64.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7.
Zhou Q, Sun Y. MLN4924: additional activities beyond neddylation inhibition. Mol Cell Oncol. 2019;6(5):e1618174.
Funding
This project is supported by the Scientific Research Fund of the Liaoning Provincial Education Department (No. LZ2020071), the Doctoral Start-up Foundation of Liaoning Province (No. 2021-BS-209), the Dalian Youth Science and Technology Star (No. 2021RQ010).
Author information
Authors and Affiliations
Contributions
G.W. provided the theme and conceived the manuscript’s structure. D.L. participated in the research and wrote the draft. X.C. was responsible for the visualization of the article content and the manuscript’s revision. All authors contributed to the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agree with the content of the manuscript and consent to publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- AEs
-
Adverse events.
- ALL
-
Acute lymphoblastic leukemia.
- AML
-
Acute myeloid leukemia.
- ANGPT2
-
Angiopoietin 2.
- BCA3
-
Breast cancer-associated protein 3.
- CAFs
-
Cancer-associated fibroblasts.
- c-CBL
-
Casitas b-lineage lymphoma.
- CLL
-
Chronic lymphocytic leukemia.
- CMML
-
Chronic myelomonocytic leukemia.
- CR
-
Complete remission.
- CRi
-
Complete remission with incomplete blood count recovery.
- CRLs
-
Cullin-RING ligases.
- CUL
-
Cullin.
- CSN5
-
COP9 signalosome subunit 5.
- DCs
-
Dendritic cells.
- DCAF
-
DDB1- and CUL4-Associated Factor.
- DDB1
-
DNA damage-binding protein 1.
- DLTs
-
Incidence of dose-limiting toxicities.
- ECM
-
Extracellular matrix.
- EFS
-
Event-free survival.
- EMT
-
Epithelial-to-mesenchymal transition.
- FIH
-
Factor inhibiting HIF.
- GPCRs
-
G protein-coupled receptors.
- HIF
-
Hypoxia-inducible factor.
- HR MDS
-
Higher-risk myelodysplastic syndromes.
- HREs
-
Hypoxia response elements.
- IKK
-
IκB kinase.
- IL
-
Interleukin.
- JAB1
-
Jun activation domain-binding brotein 1.
- JAMM
-
JAB1/MPN/Mov34 metalloenzyme.
- LATS1/2
-
Large tumor suppressor kinase 1 and 2.
- LPS
-
Lipopolysaccharides.
- MDM2
-
Mouse double minute 2 homolog.
- MDS
-
Myelodysplastic syndromes.
- MMPs
-
Matrix metalloproteinases.
- Mov34
-
Modifier of variegation 3–4.
- MPN
-
Mpr1/Pad1 N-terminal domain.
- Mpr1
-
Multidrug resistance-associated protein 1.
- MRD
-
Measurable residual disease.
- MST1
-
Mammalian STE20-like protein kinase 1.
- MTD
-
Maximum tolerated dose.
- mTORC1
-
mTOR complex 1.
- NAE
-
NEDD8 activating enzyme.
- NCI
-
National cancer institute.
- NEDD8
-
Neural precursor cell expressed developmentally downregulated protein 8.
- NEDP1
-
NEDD8 protease 1.
- NF-κB
-
Nuclear factor kappa-light-chain-enhancer of activated B cells.
- NHL
-
Non-Hodgkin lymphoma.
- NSCLC
-
Non-small cell lung cancer.
- OS
-
Overall survival.
- Pad1
-
Phenylacrylic acid decarboxylase 1.
- PARC
-
p53-associated parkin-like cytoplasmic protein.
- PDGFB
-
Platelet-derived growth factor B.
- PDK1
-
3-Phosphoinositide Dependent Protein Kinase-1.
- PD-L1
-
Programmed death-ligand 1.
- PHD
-
Prolyl hydroxylase domain.
- PI3K
-
Phosphoinositide 3-kinases.
- PIP2
-
Phosphatidylinositol 4,5-bisphosphate.
- PIP3
-
Phosphatidylinositol 3,4,5-trisphosphate.
- RBX
-
RING box protein.
- Rheb
-
Ras homolog enriched in brain.
- ROC
-
Regulator of Cullins.
- RP2D
-
Recommended phase 2 dose.
- RPL11
-
Ribosomal protein L11.
- RTKs
-
Receptor tyrosine kinases.
- SAEs
-
Serious adverse events.
- SAG
-
Sensitive to apoptosis gene.
- SCF
-
Skp1-Cul1-F-box protein.
- SENP8
-
Sentrin/SUMO-specific protease 8.
- Skp1
-
S-phase kinase-associated protein 1.
- TAMs
-
Tumor-associated macrophages.
- TEAD
-
TEA domain.
- TEAEs
-
Treatment-emergent adverse events.
- TGFβ
-
Transforming growth factor-β.
- TME
-
Tumor microenvironment.
- TNF
-
Tumor necrosis factor.
- Tregs
-
Regulatory T cells.
- TSC
-
Tuberous sclerosis complex.
- UBA3
-
Ubiquitin-like modifier activating enzyme 3.
- UBC12
-
Ubiquitin conjugating enzyme E2 M.
- UBE2F
-
Ubiquitin-conjugating enzyme E2 F.
- UBE2M
-
Ubiquitin-conjugating enzyme E2 M.
- UBL
-
Ubiquitin-like.
- UCH-L3
-
Ubiquitin C-terminal hydrolase-L3.
- UPS
-
Ubiquitin-proteasome system.
- VEGF
-
Vascular endothelial growth factor.
- VHL
-
Von hippel-lindau.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, D., Che, X. & Wu, G. Deciphering the role of neddylation in tumor microenvironment modulation: common outcome of multiple signaling pathways. Biomark Res 12, 5 (2024). https://doi.org/10.1186/s40364-023-00545-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00545-x