
Summary
Purpose The therapeutic index of proteasome inhibitors may be improved through selective inhibition of a sub-component of the ubiquitin-proteasome system, such as the NEDD8-conjugation pathway. This multicenter, phase I, dose-escalation study assessed safety and the maximum tolerated dose (MTD), pharmacokinetics, pharmacodynamics, and antitumor activity of pevonedistat, an investigational NEDD8-activating enzyme (NAE) inhibitor, in patients with metastatic melanoma. Methods Patients received intravenous pevonedistat on Days 1, 4, 8, 11 (schedule A) or 1, 8, 15 (schedule B) of 21-day cycles. Results 26 patients received pevonedistat 50–278 mg/m2 on schedule A; 11 patients received pevonedistat 157 mg/m2 on schedule B. The schedule A MTD was 209 mg/m2: dose-limiting toxicities (DLTs) included grade 3 hypophosphatemia and grade 3 increased blood creatinine (associated with grade 3 hyperbilirubinemia). Two schedule A patients experienced acute organ failure toxicities, one of whom experienced grade 5 acute renal failure. Dose escalation did not occur in schedule B: DLTs included grade 3 myocarditis, grade 2 acute renal failure, and grade 2 hyperbilirubinemia in a single patient. Pevonedistat pharmacokinetics were approximately dose-proportional across the dose range studied, with a biphasic disposition profile characterized by a short elimination half-life (~10 h). Pharmacodynamic studies showed increases in NAE-regulated transcripts post-treatment; all post-dose biopsy samples were positive for pevonedistat-NEDD8 adduct. One schedule A patient achieved a partial response; 15 patients had stable disease (4 lasting ≥6.5 months). Conclusions Pevonedistat was generally well tolerated at the MTD. Anticipated pharmacodynamic effects of NAE inhibition were observed with single-agent pevonedistat in peripheral blood and tumor tissue.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ubiquitin–proteasome system (UPS) plays a critical role in regulating intracellular proteins in eukaryotic cells, including key substrate proteins that mediate cell growth and survival, cellular signaling, and transcription factor regulation [1]. Dysregulation of the UPS has been implicated in cancer development and progression [2]. The clinical success of proteasome inhibitors such as bortezomib and carfilzomib has validated the UPS as a rational target for cancer therapy [3]. However, these proteasome inhibitors result in broad inhibition of protein degradation, which accounts for their toxic effects. The therapeutic index of proteasome inhibitors may be improved through selective inhibition of a sub-component of the UPS, such as the neddylation pathway, as described below.
The UPS regulates degradation of intracellular proteins by tagging substrate proteins with a polyubiquitin chain, which marks them for subsequent degradation by the proteasome [4]. The polyubiquitination process is mediated by E3 ligases. Cullin-RING ligases (CRLs), a subgroup of the E3 ligases, are especially important in the degradation of several proteins relevant to oncology, including the cyclin-dependent kinase inhibitor p27, Cdt1 (chromatin licensing and DNA replication factor-1), and Nrf-2 [nuclear factor (erythroid derived 2)-related factor 2] [5]. CRLs are activated via the neddylation pathway [6], which involves conjugation of the ubiquitin-like protein NEDD8 (neural precursor cell expressed, developmentally down-regulated 8) to the CRLs. NEDD8 conjugation is essential for the E3 ligase activity of CRLs [5].
The NEDD8-activating enzyme (NAE) is a critical regulator of the neddylation pathway [6]. Inhibition of NAE can inhibit the activity of the CRLs and result in accumulation of CRL substrate proteins [5, 6]. Pevonedistat (TAK-924/MLN4924) is a first-in-class, investigational, small-molecule inhibitor of NAE [7]. It forms a covalently bound adduct with NEDD8 while bound to NAE [8]. NAE inhibition via pevonedistat prevents the proteasomal degradation of a fraction (~20 %) of the proteins regulated by the UPS, in contrast to the broad inhibition seen with proteasome inhibitors [5–7, 9, 10]. This fraction includes several proteins involved in tumorigenesis, such as the tumor suppressor ICER [11], and proteins involved in the modulation of transcription, cell cycle control, and apoptosis, such as ING3 [12].
There are extensive preclinical data supporting the potential utility of pevonedistat in melanoma. Pevonedistat is cytotoxic in a range of hematologic and solid tumor cell lines [10, 13–18], and has shown antitumor activity in a number of in-vivo models [15, 16, 19, 20]. The mechanism of action primarily involves stabilization of Cdt1 and induction of the DNA damage response [9, 13, 21, 22], but likely also includes autophagy [14, 23] and inhibition of nuclear factor-κB through stabilization of I-κBα [16, 19]. Dysregulation of the UPS has been implicated in the development and progression of melanoma [11, 12]; thus, pevonedistat has been investigated in preclinical studies in melanoma cell lines and tumor xenograft models, and has demonstrated cytotoxicity and antitumor activity [24–28]. Pevonedistat-mediated cell death in melanoma cell lines appears to involve inhibition of cellular phase transition following the induction of DNA re-replication stress [24]. Pevonedistat was associated with tumor growth inhibition and regression in patient-derived melanoma tumor mouse xenograft explant models [27]. Preliminary clinical activity of pevonedistat in melanoma was also noted in the first-in-human phase I study; in nine melanoma patients, one achieved a partial response and another achieved stable disease lasting 6 months [29]. Investigation in melanoma patients also offered the feasibility of repeated skin tumor biopsies to facilitate pharmacodynamic (PD) investigations.
This phase I study (NCT01011530) was conducted to assess the safety, pharmacokinetics (PK), PD, and antitumor activity of pevonedistat in patients with metastatic melanoma. The study tested two administration schedules of pevonedistat. Paired pre-dose and post-dose tumor biopsies were acquired in patients treated at the maximum tolerated dose (MTD) to evaluate the PD effects of NAE inhibition in this clinical setting. The study results have helped inform the overall clinical development of pevonedistat.
Patients and methods
Patients
Eligible patients were ≥18 years old, and had a diagnosis of metastatic melanoma, an Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, adequate hematologic, hepatic, renal, and cardiovascular function, and radiographically or clinically evaluable tumor. Patients enrolled in the expansion cohort required measurable disease as defined by the Response Evaluation Criteria in Solid Tumors (RECIST) criteria (v1.1). Patients with brain metastases were eligible if they were asymptomatic and had stable neurologic status for ≥2 weeks after completion of local therapy (surgery or radiation).
Patients were excluded if they had received radiotherapy, systemic antineoplastic therapy, investigational agents, CYP3A inhibitors/inducers, major surgery, serious infection, or antibiotic therapy within 14 days of first dose of pevonedistat. Patients with known HIV or hepatitis B infection or known/suspected active hepatitis C infection were excluded, as were patients with a history of coagulopathy or bleeding disorder. The study was conducted in accordance with the ethical principles originating in or derived from the Declaration of Helsinki and its amendments and in accordance with 21 Code of Federal Regulations 50 / 56 / 312. Institutional review boards at each of the participating investigational centers approved the study. All patients provided written informed consent.
Study design
This was an open-label, multicenter, phase I dose-escalation study. The primary objectives were to determine the safety profile, establish the MTD, and inform the recommended phase II dose and dosing schedule for pevonedistat. Secondary objectives were to evaluate the PK and antitumor activity of pevonedistat, and to investigate its PD effects in blood and tumor samples.
Patients received pevonedistat as 1 h intravenous infusions either on Days 1, 4, 8, and 11 (schedule A) or on Days 1, 8, and 15 (schedule B, weekly dosing) of 21-day cycles. For schedule A, intermittent dosing on Days 1, 4, 8, and 11 was chosen to match the twice-weekly schedule of the proteasome inhibitor bortezomib [30]. The starting dose of 50 mg/m2 was selected based on the toxicities and the MTD observed in another phase I study that investigated pevonedistat administration on Days 1–5 of 21-day cycles in patients with advanced solid tumors. It was hypothesized that intermittent dosing – extending the dose over a 2-week period – would be better tolerated than the continuous dosing schedule, which was associated with severe hepatotoxicity [29]. Schedule B, which was added subsequently through a protocol amendment, was based on the rationale that weekly bortezomib has similar efficacy and is more convenient than the twice-weekly schedule [31, 32]. The starting dose for weekly dosing of pevonedistat on schedule B (157 mg/m2) was based on data indicating that this dose was well tolerated on a Day 1, 4, 8, and 11 schedule in a phase I study in multiple myeloma and lymphoma [33]. Patients could receive pevonedistat until disease progression or unacceptable drug-related toxicity, up to a maximum of 12 months.
Dose escalation proceeded via a Bayesian continual reassessment method using 2-patient cohorts and 1.33-fold dose increments over the previous dose level. Dose escalation decisions were based on occurrence of DLTs in Cycle 1 and the predicted MTD from the continual reassessment model (Supplementary Fig. S1). The MTD was defined as the dose level at which 6 patients had been treated and at which the algorithm did not recommend dose de-escalation. Additional patients were to be enrolled at the MTD to further evaluate safety.
DLTs were defined as: grade 4 neutropenia for >7 consecutive days or grade 3 neutropenia with fever (oral temperature ≥38.5 °C) and/or infection; grade 4 thrombocytopenia for >7 consecutive days, platelets <10,000/mm3 at any time, or grade 3 thrombocytopenia with bleeding; grade ≥3 nausea or diarrhea despite optimal anti-emetic prophylaxis or supportive therapy, or any other grade ≥3 nonhematologic toxicity (except arthralgia/myalgia, brief [<1 week] fatigue, or fever occurring in the absence of grade ≥3 neutropenia); a decrease in LVEF to <40 % or an absolute reduction of ≥10 % to <50 %; an increase in pulmonary artery systolic pressure, as determined by echocardiogram, to >50 mmHg or three times that at baseline; treatment delay of >1 week due to lack of recovery from drug-related toxicities, or other drug-related toxicity requiring doses to be missed or therapy to be discontinued.
Assessments
Adverse events (AEs) were graded using the National Cancer Institute’s Common Terminology Criteria for AEs version 3.0. Response was assessed using the modified RECIST guideline (v1.1) [34]. Computed tomography or magnetic resonance imaging scans were performed at screening, at the end of Cycle 2, and then every other cycle.
Blood samples (3 mL) for PK analysis were collected during Cycle 1. On schedule A, serial samples were collected within 1 h before dosing on Day 1, immediately after completion of infusion (immediately before switching off infusion pump), and at 1, 4, and 7 h after completion of infusion. Samples were also collected on Days 2 and 3; on Days 4 and 8 (immediately before dosing and immediately after completion of infusion); on Day 11 immediately before dosing, immediately after completion of infusion, and at 4 and 7 h after completion of infusion; and on Day 15. On schedule B, blood samples were collected within 1 h before dosing on Day 1, immediately after completion of infusion (immediately before switching off infusion pump), and at 1, 2, 4, and 7 h after completion of infusion. Samples were also collected on Days 2, 3, and 4; and on Days 8 and 15 (immediately before dosing and immediately after completion of infusion). Samples were analyzed at Tandem Labs (West Trenton, NJ) for pevonedistat plasma concentrations using Good Laboratory Practice-validated liquid chromatography/tandem mass spectrometry methods. The dynamic ranges were 1–500 ng/mL for the low-range assay and 75–7500 ng/mL for the high-range assay.
Blood samples for PD analysis were collected during Cycle 1. On schedule A samples were collected: at screening; on Days 1 and 11 within 1 h before dosing and at 4 and 7 h after completion of infusion; and on Days 4 and 15. On schedule B samples were collected: at screening; on Day 1 within 1 h before dosing and 1, 2, 4, and 8 h after completion of infusion; on Day 8 within 1 h before dosing; and on Day 15 within 1 h before dosing and 1, 2, and 4 h after completion of infusion. Gene expression of NAE-regulated transcriptional targets (ATF3, GCLM, GSR, MAG1, NQ01, SLC7A11, SRXN1, and TXNRD1) in whole blood was analyzed by reverse-transcription polymerase chain reaction (RT-PCR). Raw data were transformed prior to calculation of percent change and assumptions were made for missing data, as previously described [33]. Summary statistics were generated for the percent change from baseline at each time point for each gene.
For patients in the MTD expansion cohort, paired tumor biopsies were obtained at screening and at 3–6 h post-dosing on Day 4 (schedule A) or Day 8 (schedule B). Immunohistochemistry was used to detect pevonedistat–NEDD8 adduct (to demonstrate penetration of the drug into tumor tissue and the formation of the expected entity upon NAE inhibition), and to determine levels of Cdt1 and Nrf-2 expression (to demonstrate anticipated PD effects arising from NAE inhibition). Immunohistochemical analyses were performed at Millennium Pharmaceuticals, Inc. as previously described [33].
Statistics
All results were summarized descriptively, and no formal hypothesis testing was conducted. The safety population included all patients who received ≥1 dose of pevonedistat. The DLT-evaluable population comprised patients who received all scheduled doses during Cycle 1 or experienced a DLT during Cycle 1. The PK-evaluable population included all patients who received all protocol-defined doses in Cycle 1, had sufficient plasma concentration–time data to reliably estimate PK parameters, and had not received any excluded concomitant medications per protocol. Individual pevonedistat plasma concentration–time data were analyzed by noncompartmental methods using WinNonlin software (Version 6.2, Pharsight Corporation, Cary, NC). Plasma concentrations below the lower limit of quantification were set to zero for analysis. The response-evaluable population included all patients who received ≥1 dose of pevonedistat, had measurable disease at baseline per RECIST [34], and had ≥1 post-baseline disease assessment.
Results
Patients
A total of 37 patients were enrolled and received at least one dose of pevonedistat at one of three sites in the United States from December 2009 to May 2012. Twenty six patients received pevonedistat on schedule A and 11 patients received pevonedistat on schedule B. Patients’ baseline demographics and disease characteristics are summarized in Table 1. Thirty one (84 %) patients had skin melanoma as the primary site. Of the 15 patients whose tumor could be assessed for BRAF mutation status, 10 (67 %) patients had wild-type BRAF, and 5 (33 %) patients had a BRAF V600E mutation.
All patients had discontinued pevonedistat at the time of data cut-off. On schedule A, 21 (81 %) patients came off study upon experiencing progressive disease (n = 20) or symptomatic deterioration (n = 1); the remaining 5 (19 %) patients discontinued treatment due to AEs (n = 2), withdrawal (n = 2), and lengthy treatment hold unrelated to study drug (n = 1). On schedule B, 8 (73 %) patients came off study upon experiencing progressive disease, and 3 (27 %) patients discontinued due to AEs (n = 2) or withdrawal (n = 1).
Dose escalation, DLTs, and MTD determination
On schedule A, 26 patients received pevonedistat at one of 7 different dose levels, including 2 patients each at 50, 67, and 89 mg/m2, 5 at 118 mg/m2, 2 at 157 mg/m2, 11 at 209 mg/m2, and 2 at 278 mg/m2. A total of 19 patients were DLT-evaluable. No DLTs were reported at the first three dose levels. At the 118 mg/m2 dose level, one patient experienced asymptomatic drug-related grade 3 hypophosphatemia on Day 4 of Cycle 1, which resulted in a dose reduction to 89 mg/m2; the hypophosphatemia did not recur at the lower dose. A total of 5 patients were enrolled at 118 mg/m2 and no other DLTs were recorded. One patient at the 118 mg/m2 dose level was hospitalized on Day 3 of Cycle 1 due to grade 3 acute respiratory failure, which resolved on Day 5; this patient resumed pevonedistat at a reduced dose of 89 mg/m2 and tolerated it well. Although the investigator did not initially report this as a DLT, attributing it to underlying lung metastases, it was eventually concluded that this event had met the protocol-specified definition of a DLT. Subsequently, no DLTs were reported at the 157 or 209 mg/m2 dose levels. At the 278 mg/m2 dose level, one patient experienced drug-related grade 3 increased blood creatinine and grade 3 increased blood bilirubin on Day 3 of Cycle 1, which resulted in hospitalization and treatment discontinuation. These events subsequently resolved without long-term sequelae. No further patients were enrolled at 278 mg/m2 due to the severity of the DLT at this dose level. Four additional patients were enrolled at the 209 mg/m2 dose level. In the absence of any recorded DLTs among the initial 6 patients enrolled at this dose level, the MTD of pevonedistat on schedule A was determined to be 209 mg/m2, and another 5 patients were enrolled to further evaluate safety. Among these 5 dose-expansion patients, two experienced AEs that met the definition for DLTs; both patients had dose reductions to 157 mg/m2. One patient had drug-related grade 1 increased aspartate aminotransferase, and one patient had drug-related grade 1 increased B-type natriuretic peptide. Additionally, one dose-expansion patient with a history of renal insufficiency was hospitalized on Day 2 of Cycle 1 due to grade 4 acute renal failure and grade 4 acute hepatic failure, and subsequently died on day 9 due to acute renal failure.
On schedule B, all 11 patients received pevonedistat at the 157 mg/m2 dose level. Dose escalation did not occur due to the Sponsor’s decision to stop enrollment following a program-wide review. Ten patients were DLT-evaluable. One patient experienced DLTs of grade 3 myocarditis on Day 1, and grade 2 acute renal failure and grade 2 hyperbilirubinemia on Day 2 of Cycle 1, which resulted in hospitalization and treatment discontinuation. The exact cause of these DLTs was unclear, but was thought to be secondary to direct effects of pevonedistat on the kidneys, biliary tree, and myocardium.
Pevonedistat exposure and safety profile
All 37 patients received at least one dose of pevonedistat and were included in the safety population. The median number of treatment cycles of pevonedistat administered was 2 (range 1–16) for the entire study population, with a median of 4 (range 1–16) on schedule A and a median of 2 (range 1–11) on schedule B. The intensity of pevonedistat dosing, defined as the dose received as a proportion of the dose expected, was ≥80 % in 19 (73 %) patients on schedule A and 8 (73 %) patients on schedule B.
The safety profile of pevonedistat is summarized in Table 2. All patients experienced at least one AE. The most common AEs regardless of causality are shown in Supplementary Table S2 and included fatigue (n = 25; 68 %), diarrhea (n = 18; 49 %), anemia (n = 15; 41 %), myalgia (n = 15; 41 %), nausea (n = 13; 35 %), constipation (n = 12; 32 %), vomiting (n = 12; 32 %), arthralgia (n = 11; 30 %), decreased appetite (n = 11; 30 %), dizziness (n = 10; 27 %), and peripheral neuropathy (n = 10; 27 %). Peripheral neuropathy was mild (grade 1) in all 10 patients; due to a lack of comprehensive data on prior therapies, we could not address the possibility that prior treatment with neurotoxic agents (such as taxanes, platinum-based therapies, or vinca alkaloids) predisposed these patients to develop neuropathy with pevonedistat. Common drug-related AEs are listed in Table 2. Overall, 18 (49 %) patients experienced grade ≥3 AEs; the only grade ≥3 AEs, regardless of causality, that were reported in more than one patient were anemia (n = 5; 14 %), benign/malignant neoplasms, and small intestinal obstruction (each n = 2; 5 %) (Supplementary Table S2). Grade ≥3 AEs assessed as drug-related by the investigators are shown in Table 2.
Overall, 14 (38 %) patients experienced at least one serious AE (SAE), with 6 (16 %) experiencing at least one drug-related SAE. Four (11 %) patients had AEs that resulted in discontinuation: a patient receiving pevonedistat 209 mg/m2 on schedule A had drug-related grade 4 acute renal failure; a patient receiving pevonedistat 278 mg/m2 on schedule A discontinued due to the DLT of drug-related grade 3 increased blood creatinine and drug-related grade 3 increased blood bilirubin; a patient receiving pevonedistat 157 mg/m2 on schedule B discontinued due to grade 3 small intestinal obstruction (associated with multifocal abdominal subcutaneous metastatic deposits), which was considered unrelated to treatment; a second patient on schedule B discontinued due to the DLTs of grade 3 myocarditis, grade 2 acute renal failure, and grade 2 hyperbilirubinemia.
Three patients died on study, within 30 days of their last dose of pevonedistat. One patient treated at 118 mg/m2 on schedule A received four doses of pevonedistat, discontinued due to symptomatic deterioration not related to treatment, and died 26 days after the Cycle 1, Day 11 dose. A patient treated at 209 mg/m2 on schedule A received one dose of pevonedistat and died due to drug-related acute renal failure on Day 9 of Cycle 1. One patient on schedule B died 30 days after the Cycle 2, Day 15 dose due to progressive disease.
Pharmacokinetics
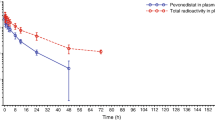
A total of 34 patients were evaluable for PK, including 24 patients on schedule A (2 patients each treated at pevonedistat 50 and 67 mg/m2, 1 at 89 mg/m2, 5 at 118 mg/m2, 1 at 157 mg/m2, 11 at 209 mg/m2, and 2 at 278 mg/m2) and 10 on schedule B. Mean pevonedistat plasma concentration–time profiles on Cycle 1, Day 1 for all patients are shown in Fig. 1. On schedule A, the majority of individual PK profiles were truncated at the 7-h post-infusion time point due to missing subsequent samples. Therefore, pevonedistat systemic exposure (as assessed by area under the plasma concentration–time curve [AUC]) could not be accurately estimated, except at the MTD. Based on limited data availability in both schedules, mean plasma exposure of pevonedistat (maximum plasma concentration or AUC when available) increased approximately proportionally with dose from 50 to 278 mg/m2 after Day 1 intravenous infusion. Individual PK profiles across schedules A and B showed a biphasic disposition phase following completion of the intravenous infusion. Plasma concentrations were generally quantifiable between 24 and 72 h after dosing across the dose range studied. Pevonedistat PK parameters at the schedule A MTD of 209 mg/m2 and at 157 mg/m2 on schedule B are summarized in Table 3.

Mean pevonedistat plasma concentration–time profiles on Cycle 1, Day 1 following 1-h intravenous infusion of pevonedistat in patients receiving different dose levels on schedule A (Days 1, 4, 8, and 11) and patients receiving the one dose level on schedule B (Days 1, 8, and 15)
Pharmacodynamics
A panel of NAE-regulated transcripts was measured (by RT-PCR) in whole blood samples collected on study. All eight NAE-regulated gene transcripts were significantly increased post-treatment among patients receiving pevonedistat at the MTD on schedule A and patients receiving pevonedistat on schedule B (Supplementary Fig. S2, Supplementary Table S1). Induction of all NAE-regulated transcriptional targets was observed at pevonedistat doses of 50 mg/m2 and higher (data not shown). On schedule A, changes were characterized by rapid increases in gene transcript levels that remained increased compared with baseline during the first 7 h following pevonedistat dosing on Days 1 and 11, when assessments were conducted. Changes were heterogeneous between patients at a given dose level and between gene transcripts at a given dose level, as shown for the pevonedistat MTD of 209 mg/m2 in Supplementary Figure S2. SLC7A11 and NQ01 were consistently the most robustly increased gene transcripts. Findings were similar on schedule B (Supplementary Fig. S2).
Tumor biopsies were collected at screening and at 3–6 h after the Cycle 1, Day 4 dose of pevonedistat from 3 patients in the Schedule A MTD expansion cohort. All of the Day 4 post-dose biopsy samples were positive for the pevonedistat–NEDD8 adduct, demonstrating penetration of pevonedistat into the tumors. Additionally, in one patient with adequate biopsy tissue, it was feasible to analyze the immunohistochemical expression of Cdt1 and Nrf-2, both of which are substrates of CRLs; expression of both proteins was increased post-treatment (Fig. 2).

Representative immunohistochemistry images of tumor formalin-fixed paraffin-embedded serial sections stained for pevonedistat–NEDD8 adduct, Cdt1, and Nrf-2, at screening/baseline and at 3–6 h after the Day 4, Cycle 1 dose, indicating increases in post-dose staining for all three markers (patient received pevonedistat 209 mg/m2 on schedule A)
Clinical activity
Thirty-one of 37 patients were evaluable for response, including 23 in schedule A and 8 in schedule B. Figure 3 indicates the duration of pevonedistat exposure among all 37 patients across both schedules, and highlights the patients who achieved best responses of partial response or stable disease. These included one (3 %) partial response, achieved by a patient on schedule A who received pevonedistat at the MTD (209 mg/m2). Another 15 (48 %) patients on schedule A and B had a best response of stable disease; the remaining 15 (48 %) patients had progressive disease.

Duration of pevonedistat exposure among all 37 patients on schedules A and B, including the 15 patients achieving stable disease and the 1 patient achieving a partial response
The patient who achieved a partial response was a 61-year-old white female with stage IV malignant melanoma (tumor sites in the brain, lung, and skin). She had received prior antineoplastic therapy (tremelimumab), radiation therapy, and surgery, and received 6 cycles of pevonedistat 209 mg/m2. The partial response was reported at Cycle 4 (Supplementary Fig. S3 includes PET scans showing tumor reduction) and was maintained until an assessment of progressive disease after Cycle 6, which resulted in discontinuation of study treatment. The duration of response was 1.55 months, and the overall duration of stable disease or better was 4.4 months. Among the 15 patients who achieved stable disease, 4 had stable disease for 6.5 months or longer (Fig. 3); none of these 4 patients had a BRAF V600E mutation.
Discussion
This phase I study investigated the safety of two different administration schedules of pevonedistat, a first-in-class NAE inhibitor, in patients with metastatic melanoma. The study also provided useful insights into the efficacy, PK, and PD profile of pevonedistat in these patients. NAE inhibition with pevonedistat resulted in the anticipated PD effects in the tumor microenvironment in patients with metastatic melanoma. These findings, along with data from other phase I studies of pevonedistat in other solid tumors and hematologic malignancies [29, 33, 35], have helped inform the clinical development of this agent, which is ongoing in combination with azacitidine in patients with acute myeloid leukemia (AML) (NCT01814826) [36] and in combination with docetaxel, gemcitabine, and carboplatin–paclitaxel in patients with solid tumors (NCT01862328). A randomized phase II study of pevonedistat plus azacitidine versus single-agent azacitidine in patients with higher risk myelodysplastic syndromes (MDS), chronic myelomonocytic leukemia, and low blast AML is currently recruiting patients (NCT02610777).
The pevonedistat MTD achieved in the present study on schedule A (Days 1, 4, 8, and 11 of 21-day cycles), of 209 mg/m2, appears similar to the MTD of 196 mg/m2 established using the same dosing schedule in a study in patients with multiple myeloma or lymphoma [33]. This contrasts with the lower MTDs seen with slightly more dose-intensive schedules in other trials. The MTDs were 110 mg/m2 using a Day 1, 2, 8, and 9 schedule in patients with multiple myeloma or lymphoma [33] and 50–67 mg/m2 using Days 1–5 and Days 1, 3, and 5 schedules in a study in patients with solid tumors [29]. Of particular note, the MTD of single-agent pevonedistat was also lower when administered on Days 1, 3, and 5 of 21-day cycles in patients with relapsed/refractory AML or MDS (50/59 mg/m2; compared with 83 mg/m2 using a Days 1, 4, 8, and 11 schedule in the same study) [35, 37]. The MTD was even lower (20 mg/m2) when pevonedistat was investigated on the same schedule in combination with azacitidine in treatment-naïve elderly patients with AML [36].
The reasons for this substantial range in the established MTDs are not clear, although the lower MTDs were reported in studies employing more frequent dosing, predominantly on Days 1, 3, and 5. It is plausible that dosing every other day may not have allowed sufficient organ recovery time between doses. This is supported by the safety profile on schedule B (weekly dosing) in the present study; a dose of 157 mg/m2 resulted in only 1 DLT in 11 patients. However, the MTDs may also have been influenced by individual patient- and disease-specific characteristics and their association with the key pevonedistat-related toxicities. Thus, comparisons between different studies in different patient populations should be made with caution. Of note, ongoing studies of pevonedistat in combination are employing a Days 1, 3, and 5 dosing schedule [36].
The DLTs and other key toxicities observed on schedule A in the present study, which included hypophosphatemia, acute renal and hepatic toxicity, and acute respiratory failure, appear similar to those reported in other phase I studies. These toxicities were usually observed soon after the first dose and did not occur in patients who tolerated the first few doses well. In the phase I study of pevonedistat in multiple myeloma and lymphoma, DLTs included febrile neutropenia, AST elevation, muscle cramps, and thrombocytopenia [33]. The phase I study of pevonedistat in solid tumors reported DLTs of ALT elevation, AST elevation, and hyperbilirubinemia [29]. In the AML/MDS phase I study, multi-organ failure, reversible ALT elevation, AST/ALT elevation, cardiac failure, and acute renal failure were among the reported DLTs [35, 37].
Most patients tolerated pevonedistat well. The precise reasons for the occurrence of acute severe toxicities at higher pevonedistat doses in a few patients is unclear at this time. Preclinical studies of mechanism-based toxicity with pevonedistat showed increased levels of markers of tissue injury, such as ALT/AST, bilirubin, and creatinine, increased circulating cytokines, and organ damage in rodents receiving pevonedistat combined with tumor necrosis factor-alpha [38]. This suggests a mechanism potentially giving rise to the acute organ failure toxicities reported in the present study and other phase I studies. Based on a program-wide safety review, doses of pevonedistat at the higher end of the range studied (>100 mg/m2) are not being considered for further investigation.
The limited PK data from this study indicate that plasma exposure of pevonedistat following completion of the intravenous infusion was approximately dose-proportional across the 50–278 mg/m2 dose range. Pevonedistat showed a biphasic disposition profile characterized by a short elimination half-life of approximately 10 h in plasma. These findings are consistent with the PK profile of pevonedistat obtained from other phase I studies in different tumor types using similar or different dosing schedules [29, 33, 35, 37].
PD studies demonstrated the anticipated effects of NAE inhibition at all doses tested in this study. For example, the RT-PCR data showing increases in NAE-regulated gene transcripts and the tumor biopsy stains for the CRL substrates Cdt1 and Nrf-2 are supportive of the mechanism of action of NAE inhibition and subsequent CRL inactivation. Increases in these CRL substrates have also been seen in preclinical studies of pevonedistat [7, 13, 16, 19, 21, 22], and similar PD effects have been reported from other clinical studies [29, 33, 35–37]. Measurement of NAE-regulated gene transcript levels provides a simple, indirect way of observing the anticipated build-up of CRL target proteins as a result of NAE inhibition. Our findings further validate the use of NAE-regulated transcriptional targets as PD markers of NAE inhibition in the clinic. The PD studies also confirmed that pevonedistat was reaching its intended target; immunohistochemical stains of tumor biopsies indicated that the drug penetrated into the tumor tissue and formed the anticipated pevonedistat–NEDD8 adduct in the presence of activated NAE [8].
The relevance of the efficacy results of this study to the current therapeutic landscape of metastatic melanoma is unclear. At the time of study initiation, there was a substantial unmet need for novel treatment approaches in this setting, and the rationale for the investigation of pevonedistat in metastatic melanoma was supported by preclinical studies showing the relevance of the mechanism of action, as evidenced by cytotoxicity and antitumor activity [24–28]. In the present study, a partial response was reported in one patient and stable disease was reported in 15 patients (lasting for 6.5 months or more in 4 patients). Pevonedistat monotherapy currently has limited utility in the context of the broader melanoma treatment landscape, which has evolved rapidly due to the recent emergence of a number of new therapies, offering improved outcomes [39]. These new drugs, including ipilimumab (an immune checkpoint inhibitor targeting CTLA-4) [40], pembrolizumab and nivolumab (monoclonal antibodies that inhibit the PD-1 receptor) [41, 42], vemurafenib [43] and dabrafenib [44] (BRAF inhibitors), and trametinib (MEK inhibitor) [45], have resulted in substantial efficacy and improvements in outcomes. Pevonedistat, with its good safety profile in most patients and clinically meaningful antitumor activity in some patients, may be useful as a combination partner for use with other melanoma therapies. Interestingly, all of the patients with a partial response or durable stable disease had wild-type BRAF melanoma. This subgroup of melanoma has a clear unmet need for effective molecularly targeted therapies and could benefit from further investigation of pevonedistat in combination with other therapies.
In conclusion, this study has provided further evidence, in addition to phase I studies in other solid tumors and hematologic malignancies, that NAE inhibition with pevonedistat results in the anticipated PD effects and that pevonedistat reaches the tumor target in metastatic melanoma patients. The data from this study have contributed to the characterization of the safety profile of this first-in-class agent, studies of which are currently ongoing in AML in combination with azacitidine [36] and in solid tumors in multiple combinations.
References
Ciechanover A (2012) Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Neurodegener Dis 10(1–4):7–22
Shen M, Schmitt S, Buac D, Dou QP (2013) Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets 17(9):1091–1108
Esseltine DL, Mulligan G (2012) An historic perspective of proteasome inhibition. Semin Hematol 49(3):196–206
Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE (2011) Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov 10(1):29–46
Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE (2010) The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 1(7):708–716
Petroski MD, Deshaies RJ (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6(1):9–20
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458(7239):732–736
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, Dick LR (2010) Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 37(1):102–111
Li L, Liu B, Dong T, Lee HW, Yu J, Zheng Y, Gao H, Zhang Y, Chu Y, Liu G, Niu W, Zheng S, Jeong LS, Jia L (2013) Neddylation pathway regulates the proliferation and survival of macrophages. Biochem Biophys Res Commun 432(3):494–498
Li L, Wang M, Yu G, Chen P, Li H, Wei D, Zhu J, Xie L, Jia H, Shi J, Li C, Yao W, Wang Y, Gao Q, Jeong LS, Lee HW, Yu J, Hu F, Mei J, Wang P, Chu Y, Qi H, Yang M, Dong Z, Sun Y, Hoffman RM, Jia L (2014) Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst 106(6):dju083
Healey M, Crow MS, Molina CA (2013) Ras-induced melanoma transformation is associated with the proteasomal degradation of the transcriptional repressor ICER. Mol Carcinog 52(9):692–704
Chen G, Wang Y, Garate M, Zhou J, Li G (2010) The tumor suppressor ING3 is degraded by SCF(Skp2)-mediated ubiquitin-proteasome system. Oncogene 29(10):1498–1508
Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A (2010) NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res 70(24):10310–10320
Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, Pan Y, Ding C, Qian J, Wu L, Chu Y, Yi J, Wang X, Sun Y, Jeong LS, Liu J, Jia L (2012) The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res 72(13):3360–3371
Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA, Milhollen M, Blakemore S, Thomas M, Berger A, Carew JS (2013) Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res 19(13):3577–3590
Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, Oberheu K, Padmanabhan S, O’Dwyer M, Nawrocki ST, Giles FJ, Carew JS (2010) Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood 115(18):3796–3800
Wang X, Li L, Liang Y, Li C, Zhao H, Ye D, Sun M, Jeong LS, Feng Y, Fu S, Jia L, Guo X (2014) Targeting the neddylation pathway to suppress the growth of prostate cancer cells: therapeutic implication for the men’s cancer. Biomed Res Int 2014:974309
Yang D, Tan M, Wang G, Sun Y (2012) The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. PLoS One 7(3):e34079
Milhollen MA, Traore T, Adams-Duffy J, Thomas MP, Berger AJ, Dang L, Dick LR, Garnsey JJ, Koenig E, Langston SP, Manfredi M, Narayanan U, Rolfe M, Staudt LM, Soucy TA, Yu J, Zhang J, Bolen JB, Smith PG (2010) MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-kappaB-dependent lymphoma. Blood 116(9):1515–1523
Smith MA, Maris JM, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Morton CL, Wu J, Smith PG, Yu J, Houghton PJ (2012) Initial testing of the investigational NEDD8-activating enzyme inhibitor MLN4924 by the pediatric preclinical testing program. Pediatr Blood Cancer 59(2):246–253
Jia L, Li H, Sun Y (2011) Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia 13(6):561–569
Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B (2011) Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res 71(8):3042–3051
Luo Z, Pan Y, Jeong LS, Liu J, Jia L (2012) Inactivation of the Cullin (CUL)-RING E3 ligase by the NEDD8-activating enzyme inhibitor MLN4924 triggers protective autophagy in cancer cells. Autophagy 8(11):1677–1679
Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, Tayber O, Hather G, Liu R, Narayanan U, Milhollen MA, Lightcap ES (2013) Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res 73(1):225–234
Liao H, Liu XJ, Blank JL, Bouck DC, Bernard H, Garcia K, Lightcap ES (2011) Quantitative proteomic analysis of cellular protein modulation upon inhibition of the NEDD8-activating enzyme by MLN4924. Mol Cell Proteomics 10(11):M111
Tan M, Li H, Sun Y (2014) Endothelial deletion of Sag/Rbx2/Roc2 E3 ubiquitin ligase causes embryonic lethality and blocks tumor angiogenesis. Oncogene 33(44):5211–5220
Tentler JJ, Micel LN, Selby HM, Tan AC, Brunkow KL, Pitts TM, Blakemore S, Smith PG, Eckhardt SG (2011) Molecular determinants of response to the investigational small molecule inhibitor of Nedd8-activating enzyme (NAE) MLN4924 in melanoma cell lines and patient-derived tumor explant models (Abstract). Mol Cancer Ther 10:B178
Traore T, Milhollen M, Garnsey JJ, Berger A, Manfredi M, Cosmopoulos K, Donelan J, Smith PG (2011) Antitumor activity of MLN4924, an investigational inhibitor of NEDD8-activating enzyme (NAE), in preclinical models of melanoma (Abstract). J Clin Oncol 29:8594
Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ, Cleary JM, Mahalingam D, Pickard MD, Faessel HM, Berger AJ, Burke K, Mulligan G, Dezube BJ, Harvey RD (2016) Phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res 22(4):847–857
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC (2005) Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352(24):2487–2498
de Vos S, Goy A, Dakhil SR, Saleh MN, McLaughlin P, Belt R, Flowers CR, Knapp M, Hart L, Patel-Donnelly D, Glenn M, Gregory SA, Holladay C, Zhang T, Boral AL (2009) Multicenter randomized phase II study of weekly or twice-weekly bortezomib plus rituximab in patients with relapsed or refractory follicular or marginal-zone B-cell lymphoma. J Clin Oncol 27(30):5023–5030
Reece DE, Hegenbart U, Sanchorawala V, Merlini G, Palladini G, Blade J, Fermand JP, Hassoun H, Heffner L, Vescio RA, Liu K, Enny C, Esseltine DL, van d, V, Cakana A, Comenzo RL (2011) Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase 1/2 study. Blood 118(4):865-873
Shah JJ, Jakubowiak AJ, O’Connor OA, Orlowski RZ, Harvey RD, Smith MR, Lebovic D, Diefenbach C, Kelly K, Hua Z, Berger AJ, Mulligan G, Faessel HM, Tirrell S, Dezube BJ, Lonial S (2016) Phase I study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res 22(1):34–43
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, Hua Z, Blakemore SJ, Faessel H, Sedarati F, Dezube BJ, Giles FJ, Medeiros BC (2015) Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol 169(4):534–543
Swords RT, Savona MR, Maris MB, Erba HP, Hua Z, Faessel H, Blakemore S, Sederati F, Dezube BJ, Medeiros BC (2014) First-in-class NAE inhibitor MLN4924 in combination with azacitidine for acute myeloid leukemia (AML) patients considered unfit for conventional chemotherapy: results from the C15009 trial (Abstract). Haematologica 99:223–224
DeAngelo DJ, Erba HP, Maris MB, Swords RT, Anwer F, Altman JK, Hua Z, Blakemore S, Faessel H, Dezube BJ, Medeiros BC (2013) MLN4924, a novel investigational inhibitor of NEDD8-activating enzyme (NAE), in adult patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS): results from multiple dosing schedules in a phase 1 study (Abstract). Blood 122:1443
Wolenski FS, Fisher CD, Sano T, Wyllie SD, Cicia LA, Gallacher MJ, Baker RA, Kirby PJ, Senn JJ (2015) The NAE inhibitor pevonedistat (MLN4924) synergizes with TNF-α to activate apoptosis. Cell Death Discov. doi:10.1038/cddiscovery.2015.34
Azijli K, Stelloo E, Peters GJ, VAN DEN Eertwegh AJ (2014) New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer Res 34(4):1493–1505
Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O’Day SJ, Hoos A, Humphrey R, Berman DM, Lonberg N, Korman AJ (2013) Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 1291:1–13
Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, Mateus C, Postow MA, Gergich K, Elassaiss-Schaap J, Li XN, Iannone R, Ebbinghaus SW, Kang SP, Daud A (2014) Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet 384(9948):1109–1117
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A (2013) Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369(2):134–144
Shaw HM, Nathan PD (2013) Vemurafenib in melanoma. Expert Rev Anticancer Ther 13(5):513–522
Kainthla R, Kim KB, Falchook GS (2014) Dabrafenib. Recent Results Cancer Res 201:227–240
Salama AK, Kim KB (2013) Trametinib (GSK1120212) in the treatment of melanoma. Expert Opin Pharmacother 14(5):619–627
Acknowledgments
The authors would like to thank all the patients who participated in this study and their families, as well as staff at all study centers. The authors would also like to thank Steve Hill and Hannah Finnigan of FireKite, an Ashfield company, part of UDG Healthcare plc, for writing support during the development of this manuscript, which was funded by Millennium Pharmaceuticals, Inc., and complied with Good Publication Practice 3 ethical guidelines (Battisti WP, et al. Ann Intern Med 2015; 163:461-4).
Authors’ contributions
SB, ACP, GM, HF, BJD, and OH designed the research. SB, ACP, PB, JAT, GM, and OH performed the research. SB, ACP, PB, JAT, GM, HF, BJD, and OH collected data. SB, GM, MDP, HF, BJD, and OH analyzed and interpreted data (pharmacokinetics: HF; pharmacodynamics: GM). MDP performed statistical analysis. SB, MDP, HF, BJD, and OH wrote the draft manuscript. All authors contributed to the writing and reviewing of the manuscript, and approved the final manuscript for submission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SB: Nothing to disclose. ACP: Nothing to disclose. JAT: Nothing to disclose. PB: Nothing to disclose. GM, MDP, HF, BJD: employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. OH: Nothing to disclose.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all individual participants included in the study.
Funding
This study was funded by Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Role of funding source
Employees of Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, participated in trial design, data collection, data analysis, data interpretation, and writing of the report. The Sponsor of the study was involved in the design of the trial and provided grants to trial sites and had no other involvement in conduct of trial. The Sponsor also provided funding for writing support during the development of this manuscript. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table S1
(DOC 32 kb)
Supplementary Table S2
(DOC 51 kb)
Supplementary Fig. S1
Dose-escalation schema, using a Bayesian continual reassessment method. CRM, continual reassessment method; DLT, dose-limiting toxicity; Midhigh, midpoint between current dose level and next dose level; Midlow, midpoint between previous dose level and current dose level; MTD, maximum tolerated dose; PMTD, predicted maximum tolerated dose. Starting dose presented (50 mg/m2) was for schedule A. Starting dose for schedule B was 157 mg/m2. aDosing intervals were to be 1.33-fold over the previous dose level and were not determined by the CRM algorithm. (GIF 24 kb)
Supplementary Fig. S2
Percent change from baseline over time of NAE-regulated gene transcripts in whole blood during Cycle 1 of pevonedistat dosing at the MTD of 209 mg/m2 on schedule A or at 157 mg/m2 on schedule B. Increases in gene transcript levels can be seen across the genes after pevonedistat dosing on days 1 and 11 in schedule A and days 1 and 15 in schedule B. Dotted lines represent individual patients; symbols represent data obtained at specified times. MTD, maximum tolerated dose. (GIF 116 kb)
(GIF 132 kb)
Supplementary Fig. S3
Tumor reduction in a 61-year-old woman with malignant melanoma who progressed through multiple (>6) prior therapies before receiving pevonedistat 209 mg/m2 on schedule A, achieving a partial response after 4 treatment cycles prior to an assessment of progressive disease in Cycle 6. (GIF 58 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bhatia, S., Pavlick, A.C., Boasberg, P. et al. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Invest New Drugs 34, 439–449 (2016). https://doi.org/10.1007/s10637-016-0348-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-016-0348-5