
Abstract
Beyond hemostasis, thrombosis and wound healing, it is becoming increasingly clear that platelets play an integral role in inflammatory response and immune regulation. Platelets recognize pathogenic microorganisms and secrete various immunoregulatory cytokines and chemokines, thus facilitating a variety of immune effects and regulatory functions. In this review, we discuss recent advances in signaling of platelet activation-related biomarkers in inflammatory settings and application prospects to apply for disease diagnosis and treatment.
Similar content being viewed by others


Introduction
Platelets were first reported to be involved in hemostasis, thrombosis and wound healing. However, there is accumulating evidence that platelets also have distinct roles in inflammatory response and immune regulation [1, 2]. This stems from several platelet characteristics, including the ability to bind infectious pathogens, secrete various immunoregulatory cytokines and chemokines, and express receptors for various immune effects and regulatory functions [3]. In addition, although platelets have no nucleus, using messenger RNA (mRNA) as templates, they can synthesize a limited number of proteins under different environmental pressures and transport inflammatory substances to inflammatory cells [4].
Platelets are discoid-shaped fragments generated from megakaryocytes in the bone marrow, which change to a compact sphere with dendritic extensions facilitating adhesion on activation. A steady supply is secured by a continuous production and clearance of 1011 platelets per day to maintain 150–400*109/L of blood level [5]. Thus, numerous platelets may encounter pathogens, which are immediately bound following their intrusion to prevent pathogen dissemination and protect from infections. In fact, adverse clinical outcomes in septic patients are correlated with the decline of platelet count [6]. Observational studies also suggested that antiplatelet therapy was associated with a decrease in mortality from sepsis, without causing an excessive bleeding [7]. In line with this, platelet transfusions improved survival in mouse sepsis models [8, 9]. It is becoming increasingly clear that platelets are multifunctional and critical in many other physiological and pathological processes, such as inflammatory response and immune regulation [1, 10,11,12,13,14].
The association between platelets and inflammation has been evaluated for decades in the study of atherosclerosis [15]. Atherosclerosis is a chronic inflammatory process, and platelets interact with white blood cells and endothelial cells to promote inflammatory response in atherosclerosis. Platelet activation, adhesion to endothelial cells, and secretion of inflammatory molecules support the migration and adhesion of monocytes to the lesion and accelerate atherosclerosis progress. This review highlights these non-hemostatic aspects of platelets in inflammation and immunity, and their application prospect as candidates for targeted therapeutic approaches.
Platelets and pathogens
Platelets can carry pathogens, including viruses, bacteria and parasites, on their plasma membrane and internally [11, 16,17,18]. Platelets are also known to be involved in acute and chronic liver disease associated with hepatitis B virus (HBV) by upregulating migration of virus-specific CD8+ T cells and non-specific inflammatory cells into the liver [19]. In contrast, platelets directly eliminate Escherichia coli (E. coli), particularly when the bacteria are opsonized by IgG [20]. Two groups recently reported that platelets and megakaryocytes, precursor cells of platelets, engulf influenza virus and dengue virus, respectively [21, 22]. Generally, the antimicrobial activity in platelets is dependent on the IgG receptor FcγRIIA and actin rearrangement processes [20]. Furthermore, the activated platelets adhere onto Staphylococcus aureus (S. aureus) by interaction of P-selectin with P-selectin glycoprotein ligand-1 (PSGL-1) of neutrophils in neutrophil extracellular traps (NETs), thus they can inhibit bacterial growth by secreting the antibacterial peptide β-defensins or involving in the high mobility group box 1 protein (HMGB1) signaling pathway [23,24,25,26]. Besides contributing to the elimination of pathogens directly and indirectly, activated platelets can also kill infected red blood cells via platelet factor (PF) 4- and Duffy Ag-dependent manner, thereby controlling the infection of plasmodium falciparum [11]. However, the mechanism of how platelets kill endogenous parasites is unclear. Thus, patients with platelet disorders consistently are more susceptible to infection, which justifies detailed examination of the role of platelets in inflammation and immune responses.
Nevertheless, the combination of infectious agents with platelets may lead to the spread of infection. During sepsis, platelet activation promotes the development of disseminated intravascular coagulation (DIC), which blocks blood vessels instead and increases ischemia and multiple organ failure [27]. Additionally, septic-induced platelet activation upregulates both pro-inflammatory and anti-inflammatory cytokine networks [28]. Platelets bind to neutrophils and release NETs, which is beneficial in trapping bacteria. Yet it can be harmful to the host as a double-edged sword. For instance, when the neutrophils activated by LPS-bearing platelets, they are significantly activated and release NETs with reactive oxygen species, causing damage to the underlying endothelial cells [25]. In addition, neutrophils can scan activated platelets through the P-selectin ligand signaling pathway, leading to inflammation cascade [14]. At present, there is abundant evidence that platelets can act as pathogen sensors in peripheral circulation because they express several receptors that have no obvious effect on hemostasis [2].
Platelet TLRs
Toll-like receptors (TLRs) are a major family of receptors that recognize pathogen-associated molecular patterns (PAMPs). Ligands of TLRs have been extensively studied, ranging from the secretory components of pathogens to nucleic acids. It has been demonstrated that platelets express TLRs 1–9, and some of them play a role as signal transducer, such as TLR4, which is a classical initiator of innate immune responses [29,30,31]. During sepsis, platelet TLR4 triggers the previously mentioned platelet-neutrophil interaction, which leads to NETs formation and subsequent bacterial capture [13]. It was indicated that platelets may be primarily responsible for reactivity to bacterial products and platelets may act as circulatory sentinels, binding to infectious agents and presenting them to neutrophils and/or cells of the reticuloendothelial system [32,33,34]. Numerous studies have also showed that activated TLRs and endotoxemia can increase the level of thrombopoietin (TPO) induced platelet-neutrophil aggregation in turn [35, 36]. Additionally, platelet TLR2 can recognize PAMPs in G+ bacteria, mycobacteria and fungi [37, 38]. Platelet TLR2 can promote the expression of P-selectin and GPIIb/IIIa integration, producing reactive oxygen species which may act directly on bacteria [39]. Additionally, stimulation of TLR7 leads to the expression of P-selectin and CD154, suggesting select α-granule release [40]. Other TLRs, like TLR9, also have functions in platelet activation and but their role in inflammation remains to be determined [41].
Platelet CD40L (CD154)
Activated platelets also express CD40L (also known as CD154), a member of the tumor necrosis factor family [42]. Platelet CD40L can bind to the endothelial cell membrane and interact with CD40, triggering a variety of inflammatory reactions, leading to the local release of adhesion molecules, such as ICAM1, VCAM1, CCL2, etc. [42] Furthermore, platelets are known as the predominant source of soluble CD40L (sCD40L), which can induce vascular cells to express E-selectin and P-selectin and initiate the release of tissue factor and interleukin- (IL-) 6 [43]. CD40L−/− mice have defect in thrombus formation, but infusion of recombinant sCD40L normalizes this deficiency, demonstrating the prothrombotic activity of sCD40L [42]. Therefore, platelet CD40L-CD40 axis may play a central link between thrombosis and inflammation.
Expression of CD40L on platelets has been shown to affect dendritic cells as well as B and T lymphocytes, suggesting that it provides a communicative link between innate and adaptive immunity [44,45,46]. For example, platelets inhibit dendritic cells differentiation in a CD40L-dependent manner and significantly suppress the proinflammatory cytokines production [47]. In addition, activated platelets enhance lymphocyte adhesion to endothelial cells and promote lymphocyte homing and migration to inflammatory areas [48, 49]. Besides, platelets expressed CD40L can promote B cell differentiation and Ab class switching [50]. Platelets can therefore modulate adaptive immune mechanisms through the CD40L-CD40 axis.
Platelet MHC class I
Platelets always express major histocompatibility complex (MHC) class I, and MHC class I significantly increases during infection both on their plasma membrane and intracellularly [51, 52]. On the platelet plasma membrane, MHC class I appears unstable because molecules passively separated from platelets while stored in the blood bank, or eluted from the surface by chloroquine diphosphate or acid washing without affecting the integrity of platelet membranes [53]. At the allogeneic level, through transfusion, denatured platelet MHC class I interact erratically with CD8+ T cells (also known as CTLs) [54]. For example, the platelet MHC class I molecules themselves do not stimulate CTL-mediated cytotoxicity, but mediate “transfusion effect” known as the immunosuppressant response to the transfusion product [53]. Compared with non-transfused recipients, CBA mice transfused with allogeneic BALB/c platelets are easier to receive donor-specific skin grafts [55]. This observation supports the concept that allogeneic platelets may interfere with T cell-mediated cytotoxicity responses. Instead, recent study suggest that, platelet intracellular MHC class I molecules are connected with α granules and are mainly integral membrane proteins co-localized with β2-macroglobulin [56]. Now it seems that, activated platelets can express nascent MHC class I molecules, which have the ability to present antigens to CD8+ T cells. Thus, platelets can mediate T cell inhibition or activation, depending on the source of the MHC (on their plasma membrane or intracellularly).
Platelet cytokines/chemokines
Platelets carry a number of chemokines and cytokines, which play substantial roles in various processes including hemostasis and wound repair, as well as pro-inflammatory and anti-inflammatory processes [57]. Platelets release various chemokines and cytokine upon activation such as CXCL1, PF4 (CXCL4), CXCL5, CXCL7, IL-8 (CXCL8), CXCL12, macrophage inflammatory protein- (MIP-) 1 α (CCL3), and RANTES (regulated on activation, normal T cell expressed and secreted, also called as CCL5) [58,59,60]. The major effect of these cytokines is to regulate inflammatory functions, like leukocyte migration, phagocytosis and reactive oxygen species (ROS) generation [61]. The most abundant chemokine is PF4, a positively charged protein that binds to glycosaminoglycans. PF4 not only has a role in haemostasis/thrombosis, but also is a chemotactic protein for monocytes and neutrophils, with immunoregulatory activity [62]. Interestingly, Guo et al. identify that PF4, a vital immunoregulatory chemokine, is essential for protecting mice against influenza A virus infection, especially as it affects the development of lung injury and neutrophil mobilization to the inflamed lung [63]. Moreover, PF4 prevents monocyte apoptosis, promotes monocyte differentiation into macrophages and induces phagocytosis and generation of ROS [64]. RANTES/CCL5 from platelets also play an important role in leukocyte recruitment, due to their potency to attract monocytes to inflamed endothelium [65], an effect that was found to be dependent on CCL5-receptor CCR1 [66]. Secreted RANTES form heterodimers to promote monocyte recruitment to the endothelium [67] by engaging its receptors CCR1 and CCR5, respectively [68,69,70].
In addition, platelets also synthesize and release IL-1β, IL-6, IL-8 and TNF-α in both micro vesicle and soluble form [71, 72]. When released upon platelet activation, IL-1β has been reported to mediate monocyte and leukocyte mobilization by binding to IL-1 receptors [73]. Furthermore, dengue virus infection could trigger IL-1β through the action of the nucleotide-binding domain, leucine-rich repeat containing protein 3 (NLRP3) inflammasomes in platelets [74]. Though without nucleus, platelets were recently reported to be competent for dengue virus replication, which may explain the thrombocytopenia associated with clinical dengue infection [75].
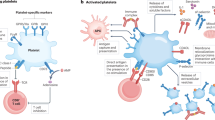
A summary of all the above-mentioned characteristics is shown in Table 1 and illustrated in Fig. 1.

The key roles of platelets in modulating inflammatory processes. (1) Platelets are activated by invading pathogens (or their products) that have already been targeted by IgG receptor FcγRIIA (via IgG production). (2) Platelets can carry and eliminate pathogens, and via the expression of TLRs they can bind bacterial LPS and activate neutrophils, inducing NETs formation. (3) Platelet CD40L expression allows them to interact with different immune cells and either activate (arrow) and/or suppress (T bar) them. Furthermore, CD40L may be cleaved into a soluble form (sCD40L) that enhances platelet activation, aggregation, and platelet-leukocyte conjugation. (4) Intact platelet MHC class I molecules are located intracellularly but upon activation are expressed and can activate antigen-specific CD8+ T cells. In contrast, the MHC class I molecules on the surface of resting platelets are denatured and lead to CD8+ T cell inhibition. (5) Platelets contain many proinflammatory and anti-inflammatory cytokines and chemokines and, upon activation, can release them to the extracellular space. The culmination of these events makes platelets a formidable immunomodulatory host
Clinical applications
In recent years, platelets have become important markers for various diseases. They are multifunctional blood particles, which may be candidates for innovative targeted therapeutic approaches. Besides playing a major role in physiological hemostasis, thrombosis and wound healing, platelets can also make great contributions to host inflammation and immune responses to infection and injury. Under uncontrolled pathological conditions, platelets play critical roles in acute coronary syndrome [76], central nervous system diseases [77], autoimmune diseases [78], and rheumatoid arthritis [79].
After activation, platelets secret numerous cytokines into peripheral circulation. Several of these cytokines, such as IL-1β, P-selectin, CD40L, PF4, and RANTES, are currently under consideration as molecular targets against inflammation and atherosclerosis [71, 80, 81]. Recently, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial demonstrated a beneficial effect of the therapeutic monoclonal IL-1β antibody canakinumab on reducing the risk for recurrent cardiovascular events in high risk patients who have sustained a prior myocardial infarction [82]. The JAK/STAT pathway is recognized as one of the major mechanisms by which cytokine receptors transduce intracellular signals [83]. Numerous inflammatory immune-related diseases are driven by inflammatory mediators, which rely on JAK-STAT signaling. Therefore, inhibition of this pathway using JAK inhibitors might be a useful therapeutic strategy for these diseases. Tofacitinib, the first rheumatologic JAK inhibitor, is US Food and Drug Administration (FDA) approved for rheumatoid arthritis and is currently under investigation for other autoimmune diseases. TD-1473, a JAK inhibitor in phase II clinical trial of Crohn’s disease, exhibits inhibitory potencies in cellular assays similar to Tofacitinib [84]. Preclinical studies demonstrate that JAK inhibitor have potential suppression effects on platelet-derived growth factor in vascular smooth muscle [85] and antiplatelet effect [86], suggesting that JAK inhibition might be a viable strategy to treat multiple inflammatory response induced by platelets.
Moreover, due to short half-life of platelets, platelet-monocyte aggregates have emerged as new markers of platelet activation. Platelet-monocyte aggregates persist longer in peripheral blood and more sensitive in vivo than other platelet surface markers [80, 87].
Microparticles are extracellular vesicles produced by cytoplasmic vesiculation and division of cells ranging in diameter from 100 to 1000 nm. Platelets and megakaryocytes are the primary source of microparticles in circulation [88]. They can carry the nucleus and cytoplasmic components including proteins, lipids, and RNA from their precursor cells and transmit inflammatory signals to nearby or distant cells. Furthermore, given that they are induced in several inflammatory pathologies, such as atherosclerosis, stroke and autoimmune diseases, clinical applications of platelet-derived microparticles have been widely investigated [89,90,91]. Some medications, such as statins and aspirin, have been shown to reduce microparticles levels in patients and thus can be used to assess therapeutic efficacy [90, 92, 93]. Although important efforts in standardizing the preanalytical and analytical variables have been developed, standardization of microparticles detection and quantification methods is highly required to further confirm and generalize the results.
Conclusion
Platelets are best known as primary mediators of hemostasis and thrombin generation; however, emerging evidence demonstrates that platelets are far more complex than previously regarded, equipped with elaborate intracellular machinery. Now recognized as key players in inflammation and immune responses, these anucleate cellular fragments express and secrete several pro- and anti-inflammatory molecules that serve to initiate and modulate immune functions. Understanding the signals of platelet biomarkers in inflammatory settings and how they modulate the immune system could expand new diagnostic and therapeutic methods to monitor and confront inflammatory immune-related diseases.
Availability of data and materials
Not applicable.
Abbreviations
- mRNA:
-
Messenger RNA
- HBV:
-
Hepatitis B virus
- E. coli :
-
Escherichia coli
- S. aureus :
-
Staphylococcus aureus
- PSGL-1:
-
P-selectin glycoprotein ligand-1
- NETs:
-
Neutrophil extracellular traps
- HMGB1:
-
High mobility group box 1 protein
- PF:
-
Platelet factor
- DIC:
-
Disseminated intravascular coagulation
- TLRs:
-
Toll-like receptors
- PAMPs:
-
Pathogen-associated molecular patterns
- TPO:
-
Thrombopoietin
- sCD40L:
-
Soluble CD40L
- MHC:
-
Major histocompatibility complex
- MIP:
-
Macrophage inflammatory protein
- RANTES:
-
Regulated on activation, normal T cell expressed and secreted
- ROS:
-
Reactive oxygen species
- NLRP3:
-
Nucleotide-binding domain, leucine-rich repeat containing protein 3
References
Semple JW, Italiano JJ, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. 2011;11(4):264–74.
Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123(18):2759–67.
Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115(12):3339–47.
Italiano JJ, Shivdasani RA. Megakaryocytes and beyond: the birth of platelets. J Thromb Haemost. 2003;1(6):1174–82.
Machlus KR, Italiano JJ. The incredible journey: from megakaryocyte development to platelet formation. J Cell Biol. 2013;201(6):785–96.
Moreau D, Timsit JF, Vesin A, Garrouste-Orgeas M, de Lassence A, Zahar JR, Adrie C, Vincent F, Cohen Y, Schlemmer B, et al. Platelet count decline: an early prognostic marker in critically ill patients with prolonged ICU stays. Chest. 2007;131(6):1735–41.
Akinosoglou K, Alexopoulos D. Use of antiplatelet agents in sepsis: a glimpse into the future. Thromb Res. 2014;133(2):131–8.
Wuescher LM, Takashima A, Worth RG. A novel conditional platelet depletion mouse model reveals the importance of platelets in protection against Staphylococcus aureus bacteremia. J Thromb Haemost. 2015;13(2):303–13.
Xiang B, Zhang G, Guo L, Li XA, Morris AJ, Daugherty A, Whiteheart SW, Smyth SS, Li Z. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat Commun. 2013;4:2657.
Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. 2002;99(11):4021–9.
Mcmorran BJ, Marshall VM, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, Corbin JE, Alexander WS, Foote SJ. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science. 2009;323(5915):797–800.
Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. 2013;14(8):785–92.
Clark SR, Ma AC, Tavener SA, Mcdonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, Mcavoy E, Sinclair GD, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–9.
Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nacher M, Pitaval C, Radovanovic I, Fukui Y, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234–8.
Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482–94.
Assinger A. Platelets and infection - an emerging role of platelets in viral infection. Front Immunol. 2014;5:649.
Yeaman MR. Bacterial-platelet interactions: virulence meets host defense. Future Microbiol. 2010;5(3):471–506.
Kerrigan SW, Cox D. Platelet-bacterial interactions. Cell Mol Life Sci. 2010;67(4):513–23.
Aiolfi R, Sitia G. Chronic hepatitis B: role of anti-platelet therapy in inflammation control. Cell Mol Immunol. 2015;12(3):264–8.
Riaz AH, Tasma BE, Woodman ME, Wooten RM, Worth RG. Human platelets efficiently kill IgG-opsonized E. coli. FEMS Immunol Med Microbiol. 2012;65(1):78–83.
Koupenova M, Corkrey HA, Vitseva O, Manni G, Pang CJ, Clancy L, Yao C, Rade J, Levy D, Wang JP, et al. The role of platelets in mediating a response to human influenza infection. Nat Commun. 2019;10(1):1780.
Vogt MB, Lahon A, Arya RP, Spencer CJ, Rico-Hesse R. Dengue viruses infect human megakaryocytes, with probable clinical consequences. PLoS Negl Trop Dis. 2019;13(11):e7837.
Flaujac C, Boukour S, Cramer-Borde E. Platelets and viruses: an ambivalent relationship. Cell Mol Life Sci. 2010;67(4):545–56.
Kraemer BF, Campbell RA, Schwertz H, Franks ZG, Vieira DAA, Grundler K, Kile BT, Dhakal BK, Rondina MT, Kahr WH, et al. Bacteria differentially induce degradation of Bcl-xL, a survival protein, by human platelets. Blood. 2012;120(25):5014–20.
Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126(2):242–6.
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D'Angelo A, Bianchi ME, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074–88.
Greco E, Lupia E, Bosco O, Vizio B, Montrucchio G. Platelets and multi-organ failure in sepsis. Int J Mol Sci. 2017;18(10):2200.
Vardon-Bounes F, Ruiz S, Gratacap MP, Garcia C, Payrastre B, Minville V. Platelets are critical key players in sepsis. Int J Mol Sci. 2019;20(14):3494.
Andonegui G, Kerfoot SM, Mcnagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional toll-like receptor-4. Blood. 2005;106(7):2417–23.
Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83(2):196–8.
Damien P, Cognasse F, Eyraud MA, Arthaud CA, Pozzetto B, Garraud O, Hamzeh-Cognasse H. LPS stimulation of purified human platelets is partly dependent on plasma soluble CD14 to secrete their main secreted product, soluble-CD40-ligand. BMC Immunol. 2015;16:3.
Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood. 2007;109(11):4803–5.
Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006;108(1):167–76.
Koessler J, Niklaus M, Weber K, Koessler A, Kuhn S, Boeck M, Kobsar A. The role of human platelet preparation for toll-like receptors 2 and 4 related platelet responsiveness. TH Open. 2019;3(2):e94–102.
D'Atri LP, Rodriguez CS, Miguel CP, Pozner RG, Ortiz WJ, Negrotto S, Carrera SE, Heller P, Schattner M. Activation of Toll-like receptors 2 and 4 on CD34+ cells increases human megakaryo/thrombopoiesis induced by thrombopoietin. J Thromb Haemost. 2019;17(12):2196–210.
Undi RB, Sarvothaman S, Narasaiah K, Gutti U, Gutti RK. Toll-like receptor 2 signalling: significance in megakaryocyte development through wnt signalling cross-talk and cytokine induction. Cytokine. 2016;83:245–9.
Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE. Stimulation of toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009;104(3):346–54.
Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. 2019;10(1):2402.
Amirkhosravi A, Mousa SA, Amaya M, Blaydes S, Desai H, Meyer T, Francis JL. Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454. Thromb Haemost. 2003;90(3):549–54.
Koupenova M, Vitseva O, Mackay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood. 2014;124(5):791–802.
Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, Byzova TV, Podrez EA. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ Res. 2013;112(1):103–12.
Andre P, Nannizzi-Alaimo L, Prasad SK, Phillips DR. Platelet-derived CD40L: the switch-hitting player of cardiovascular disease. Circulation. 2002;106(8):896–9.
Mobarrez F, Sjovik C, Soop A, Hallstrom L, Frostell C, Pisetsky DS, Wallen H. CD40L expression in plasma of volunteers following LPS administration: a comparison between assay of CD40L on platelet microvesicles and soluble CD40L. Platelets. 2015;26(5):486–90.
Nishat S, Wuescher LM, Worth RG. Platelets enhance dendritic cell responses against Staphylococcus aureus through CD40-CD40L. Infect Immun. 2018;86(9):e00186–18.
Hamzeh-Cognasse H, Cognasse F, Palle S, Chavarin P, Olivier T, Delezay O, Pozzetto B, Garraud O. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol. 2008;9:54.
Aloui C, Prigent A, Sut C, Tariket S, Hamzeh-Cognasse H, Pozzetto B, Richard Y, Cognasse F, Laradi S, Garraud O. The signaling role of CD40 ligand in platelet biology and in platelet component transfusion. Int J Mol Sci. 2014;15(12):22342–64.
Kissel K, Berber S, Nockher A, Santoso S, Bein G, Hackstein H. Human platelets target dendritic cell differentiation and production of proinflammatory cytokines. Transfusion. 2006;46(5):818–27.
Stoll G, Nieswandt B. Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol. 2019;15(8):473–81.
Diacovo TG, Catalina MD, Siegelman MH, von Andrian UH. Circulating activated platelets reconstitute lymphocyte homing and immunity in L-selectin-deficient mice. J Exp Med. 1998;187(2):197–204.
von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100(1):27–40.
Garraud O, Cognasse F. Are platelets cells? And if yes, are they immune cells? Front Immunol. 2015;6:70.
Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, Topham DJ, Baldwin WR, Morrell CN. Platelets present antigen in the context of MHC class I. J Immunol. 2012;189(2):916–23.
Gouttefangeas C, Diehl M, Keilholz W, Hornlein RF, Stevanovic S, Rammensee HG. Thrombocyte HLA molecules retain nonrenewable endogenous peptides of megakaryocyte lineage and do not stimulate direct allocytotoxicity in vitro. Blood. 2000;95(10):3168–75.
Zufferey A, Speck ER, Machlus KR, Aslam R, Guo L, Mcvey MJ, Kim M, Kapur R, Boilard E, Italiano JJ, et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. 2017;1(20):1773–85.
Aslam R, Speck ER, Kim M, Freedman J, Semple JW. Transfusion-related immunomodulation by platelets is dependent on their expression of MHC class I molecules and is independent of white cells. Transfusion. 2008;48(9):1778–86.
Zufferey A, Schvartz D, Nolli S, Reny JL, Sanchez JC, Fontana P. Characterization of the platelet granule proteome: evidence of the presence of MHC1 in alpha-granules. J Proteome. 2014;101:130–40.
Mazzucco L, Borzini P, Gope R. Platelet-derived factors involved in tissue repair-from signal to function. Transfus Med Rev. 2010;24(3):218–34.
Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost. 2014;12(11):1764–75.
Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, and immunity. Int J Lab Hematol. 2013;35(3):254–61.
Cognasse F, Laradi S, Berthelot P, Bourlet T, Marotte H, Mismetti P, Garraud O, Hamzeh-Cognasse H. Platelet inflammatory response to stress. Front Immunol. 2019;10:1478.
Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013;17(3):638–50.
Kowalska MA, Rauova L, Poncz M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb Res. 2010;125(4):292–6.
Guo L, Feng K, Wang YC, Mei JJ, Ning RT, Zheng HW, Wang JJ, Worthen GS, Wang X, Song J, et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. 2017;10(6):1529–41.
Woller G, Brandt E, Mittelstadt J, Rybakowski C, Petersen F. Platelet factor 4/CXCL4-stimulated human monocytes induce apoptosis in endothelial cells by the release of oxygen radicals. J Leukoc Biol. 2008;83(4):936–45.
von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, Hackeng TM, Weber C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. 2005;105(3):924–30.
Kramp BK, Megens RT, Sarabi A, Winkler S, Projahn D, Weber C, Koenen RR, von Hundelshausen P. Exchange of extracellular domains of CCR1 and CCR5 reveals confined functions in CCL5-mediated cell recruitment. Thromb Haemost. 2013;110(4):795–806.
Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. 2005;25(7):1512–8.
Raport CJ, Gosling J, Schweickart VL, Gray PW, Charo IF. Molecular cloning and functional characterization of a novel human CC chemokine receptor (CCR5) for RANTES, MIP-1beta, and MIP-1alpha. J Biol Chem. 1996;271(29):17161–6.
Pakianathan DR, Kuta EG, Artis DR, Skelton NJ, Hebert CA. Distinct but overlapping epitopes for the interaction of a CC-chemokine with CCR1, CCR3 and CCR5. Biochemistry. 1997;36(32):9642–8.
Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, Nelson PJ. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood. 2001;97(4):1144–6.
Weyrich AS, Lindemann S, Zimmerman GA. The evolving role of platelets in inflammation. J Thromb Haemost. 2003;1(9):1897–905.
Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122(3):379–91.
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87(6):2095–147.
Hottz ED, Lopes JF, Freitas C, Valls-De-Souza R, Oliveira MF, Bozza MT, Da PA, Weyrich AS, Zimmerman GA, Bozza FA, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. 2013;122(20):3405–14.
Simon AY, Sutherland MR, Pryzdial EL. Dengue virus binding and replication by platelets. Blood. 2015;126(3):378–85.
Navas-Carrillo D, Marin F, Valdes M, Orenes-Pinero E. Deciphering acute coronary syndrome biomarkers: high-resolution proteomics in platelets, thrombi and microparticles. Crit Rev Clin Lab Sci. 2017;54(1):49–58.
Pluta R, Ulamek-Koziol M. Lymphocytes, platelets, erythrocytes, and exosomes as possible biomarkers for alzheimer’s disease clinical diagnosis. Adv Exp Med Biol. 2019;1118:71–82.
Liu X, Gorzelanny C, Schneider SW. Platelets in skin autoimmune diseases. Front Immunol. 2019;10:1453.
Gasparyan AY, Ayvazyan L, Mukanova U, Yessirkepov M, Kitas GD. The platelet-to-lymphocyte ratio as an inflammatory marker in rheumatic diseases. Ann Lab Med. 2019;39(4):345–57.
Gremmel T, Ay C, Riedl J, Kopp CW, Eichelberger B, Koppensteiner R, Panzer S. Platelet-specific markers are associated with monocyte-platelet aggregate formation and thrombin generation potential in advanced atherosclerosis. Thromb Haemost. 2016;115(3):615–21.
Pfluecke C, Berndt K, Wydra S, Tarnowski D, Barthel P, Quick S, Ulbrich S, Christoph M, Waessnig N, Speiser U, et al. Atrial fibrillation is associated with high levels of monocyte-platelet-aggregates and increased CD11b expression in patients with aortic stenosis. Thromb Haemost. 2016;115(5):993–1000.
Ridker PM, Everett BM, Thuren T, Macfadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31.
O'Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(Suppl):S121–31.
D'Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs. 2018;27(7):595–9.
Yellaturu CR, Rao GN. Cytosolic phospholipase A2 is an effector of Jak/STAT signaling and is involved in platelet-derived growth factor BB-induced growth in vascular smooth muscle cells. J Biol Chem. 2003;278(11):9986–92.
Lu WJ, Lin KC, Huang SY, Thomas PA, Wu YH, Wu HC, Lin KH, Sheu JR. Role of a Janus kinase 2-dependent signaling pathway in platelet activation. Thromb Res. 2014;133(6):1088–96.
Shih L, Kaplan D, Kraiss LW, Casper TC, Pendleton RC, Peters CL, Supiano MA, Zimmerman GA, Weyrich AS, Rondina MT. Platelet-monocyte aggregates and c-reactive protein are associated with VTE in older surgical patients. Sci Rep. 2016;6:27478.
Burnouf T, Goubran HA, Chou ML, Devos D, Radosevic M. Platelet microparticles: detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014;28(4):155–66.
Lacroix R, Dubois C, Leroyer AS, Sabatier F, Dignat-George F. Revisited role of microparticles in arterial and venous thrombosis. J Thromb Haemost. 2013;11(Suppl 1):24–35.
El-Gamal H, Parray AS, Mir FA, Shuaib A, Agouni A. Circulating microparticles as biomarkers of stroke: a focus on the value of endothelial- and platelet-derived microparticles. J Cell Physiol. 2019;234(10):16739–54.
Saluk-Bijak J, Dziedzic A, Bijak M. Pro-thrombotic activity of blood platelets in multiple sclerosis. Cells. 2019;8(2):E110.
Chen Y, Li G, Liu ML. Microvesicles as emerging biomarkers and therapeutic targets in cardiometabolic diseases. Genomics Proteomics Bioinformatics. 2018;16(1):50–62.
Suades R, Padro T, Alonso R, Mata P, Badimon L. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb Haemost. 2013;110(2):366–77.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81770348), (No. 81800305).
Funding
This work was supported by the National Natural Science Foundation of China (No. 81770348), (No. 81800305).
Author information
Authors and Affiliations
Contributions
Yufei Chen had the idea for the article; Yufei Chen, Haoxuan Zhong, and Yikai Zhao performed the literature search and data analysis; Yufei Chen drafted the work; Xinping Luo, and Wen Gao critically revised the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Zhong, H., Zhao, Y. et al. Role of platelet biomarkers in inflammatory response. Biomark Res 8, 28 (2020). https://doi.org/10.1186/s40364-020-00207-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-020-00207-2