
Abstract
Background
Chronic myeloid leukemia (CML) comprises ~3 % of pediatric leukemia. Although therapy with tyrosine kinase inhibitors (TKIs) is highly effective for CML, multiple factors have been identified as predictive of treatment failure. Chromosomal abnormalities involving the MECOM locus at 3q26 portend therapy resistant disease in adults, yet have never been described in pediatric patients and have not been associated with T lymphoblastic progression.
Case presentation
We present a case of an 11-year-old boy with CML possessing the unique combination of T lymphoblastic transformation and a subclone harboring inv(3)(q21q26.2) at diagnosis. This is the first reported case of pediatric CML with inv(3)(q21q26.2) and the first case of T lymphoblastic progression associated with this karyotype. The patient was treated with single agent TKI therapy with robust initial response. Marrow histology at one month showed restoration of trilineage hematopoiesis and BCR-ABL RT-PCR at three months showed a 1.4 log reduction in transcript levels.
Conclusions
The karyotypic abnormality of inv(3)(q21q26.2) in CML is not restricted to adult patients. Moreover, while chromosome 3 abnormalities are markers of TKI resistance in adults, our patient showed a robust early response to single agent TKI therapy. This finding suggests pediatric CML with inv(3)(q21q26.2) may have distinct features and more favorable treatment responses than those described in adults.
Similar content being viewed by others


Background
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm characterized morphologically by overproduction of maturing granulocytes and genetically by the BCR-ABL1 fusion oncogene. CML constitutes 15–20 % of adult leukemia [1] yet is uncommon in children, comprising only 2–3 % of all pediatric leukemia [2]. The natural history of CML is either biphasic or triphasic, with progression from an indolent chronic phase (CP) to a terminal blast phase (BP), occasionally through an intermediate or accelerated phase (AP). Advanced disease is infrequent at diagnosis, with only 15 % of adult and 5 % of pediatric patients initially presenting with AP or BP [2, 3]. Morphologically, BP resembles acute leukemia and is not restricted to the myeloid lineage, indicating that very early hematopoietic progenitors harbor the BCR-ABL1 translocation. Between 50–65 % of CML-BP shows myeloid differentiation, while lymphoid and undifferentiated phenotypes comprise 20–25 % and 15–25 %, respectively [4, 5]. The majority of lymphoid BP in CML is B lymphoblastic, while T lymphoblastic transformation is rare.
The hallmark karyotypic abnormality of CML is t(9;22)(q34;q11), yet complex translocations, such as t(6;9;22), are seen in 5–10 % of cases. The resulting BCR-ABL1 fusion protein is sensitive to imatinib and related tyrosine kinase inhibitors (TKIs). Use of these agents has vastly improved prognosis; however, a subset of patients progress to AP or BP despite adequate treatment, and prognosis for CML-BP remains poor [6].
Progression from CP to AP and BP is associated with acquisition of additional chromosomal abnormalities (ACAs). ACAs of trisomy 8, isochromosome 17q, and Philadelphia chromosome amplification, often referred to as “major-route” changes, serve as genetic markers of high-risk disease and are, therefore, sufficient for classifying CML-AP [5, 7]. Less frequent “minor-route” ACAs are more varied and have poorly described treatment implications. One notable exception is abnormalities of 3q26.2 resulting in overexpression of the MECOM locus [8, 9]. Increased expression of MECOM, an oncogenic transcription factor involved in hematopoietic stem cell renewal and differentiation, is associated with resistance to TKIs and progression to myeloid CML-BP [10–13]. Recently, rearrangements involving the 3q26.2 locus have been shown to be an independent predictor of inferior outcomes in adults with CML [14]. As such, 3q26.2 alterations, found either directly by genetic assays or indirectly by determination of MECOM expression levels, may be markers for subclassification of patients with CML. Of note, 3q26.2 locus abnormalities have not been reported in pediatric CML and thus are of uncertain significance in this population.
We report a case of pediatric CML with the unique combination of T lymphoblastic progression and a subclone harboring inv(3)(q21q26.2) at diagnosis. This is the first reported case of CML in which inv(3)(q21q26.2) occurs simultaneously with T lymphoblastic progression, as well as the first reported instance of inv(3)(q21q26.2) in pediatric CML. Notably, immunohistochemical staining (IHC) of the bone marrow using a novel antibody against MECOM demonstrated the T lymphoblastic population to be independent of the subpopulation with inv(3)(q21q26.2). Despite these adverse features, the patient initially responded well to TKI monotherapy. We discuss the diagnostic, prognostic, and therapeutic implications for this patient and for patients with CML with inv(3)(q21q26.2) in general.
Case presentation
The patient is an 11-year-old male with attention deficit hyperactivity disorder (ADHD) who presented with an unintentional 20 lb. weight loss that was initially attributed to ADHD medication. Exam was notable for cervical lymphadenopathy. Laboratory workup showed leukocytosis with left-shifted granulocytes (WBC 210,000 cells/μL; 65 % neutrophils, 6 % bands, 5 % lymphocytes, 3 % monocytes, 2 % eosinophils, 3 % metamyelocytes, 13 % myelocytes, 2 % promyelocytes, 1 % blasts), normocytic anemia (Hgb 10.8 g/dL, MCV 92 fL), elevated LDH (1,858 U/L), and elevated uric acid (7.0 mg/dL). Abdominal CT scan showed splenomegaly.
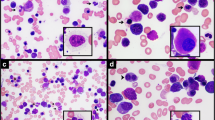
A bone marrow study revealed hypercellular marrow (>95 %) with increased myeloid cells at all stages of maturation, increased eosinophils, scattered basophils, few erythroid progenitors, so-called dwarf megakaryocytes, and sea-blue histiocytes (Fig. 1a–d). Blasts were <5 % morphologically. Flow cytometry identified a population of atypical CD3+ MPO- TDT+ T lymphoblasts (9 % of total events). IHC of the bone marrow core biopsy confirmed these latter findings, showing individual and clusters of CD3+ TDT+ T lymphoblasts comprising ~10 % of overall cellularity and 20–30 % of cells in restricted areas (Fig. 1d–g).

Morphologic and immunohistochemical evaluation of diagnostic bone marrow aspirate and core biopsy. Diagnostic bone marrow aspirate (a and b, Wright-Giemsa, 600x) and biopsy (c, h & e, 200x) showing myeloid elements at all stages of maturation. The aspirate shows that blasts are not markedly increased and cells with cytology consistent with lymphoblasts are not readily apparent. Eosinophils (green arrows), basophils (arrowhead) and dwarf megakaryocytes (black arrows) are identified. Immunohistochemical staining of the bone marrow biopsy (100x) shows an expansion of CD34(subset) + TDT+ blasts (d and e, respectively) that form clusters. An expansion of CD3+ T-cells (f) without an expansion of CD19+ B-cells (g) is seen. In panel h, double staining of the biopsy (400x) shows that MECOM expressing cells (brown nuclear stain) are distinct from CD3+ T-cells (red membranous stain)
RT-PCR from peripheral blood was positive for the BCR-ABL1 p210 transcript. Cytogenetic analysis of the bone marrow showed 46,XY,t(6;9;22)(p22;q34;q11.2)[9]/46,sl,inv(3)(q21q26.2)[11], confirming the presence of a variant three-way translocation generating the BCR-ABL1 fusion. The presence of a subclone (11 of 20 cells analyzed) with inv(3)(q21q26.2) suggested disease progression. Following identification of this inversion and validation of a novel MECOM antibody, combined IHC for MECOM and CD3 was performed on the bone marrow core biopsy. Interestingly, the CD3+ population and the MECOM-overexpressing population were non-overlapping (Fig. 1h), indicating the T lymphoblastic transformation was independent of the acquisition of inv(3)(q21q26.2).
Based on these findings, a diagnosis of CML with T lymphoblastic transformation was rendered. The patient was started on hydroxyurea and allopurinol, followed by single-agent treatment with imatinib (500 mg daily). Repeat bone marrow studies on day 25 of treatment showed restoration of trilineage hematopoiesis and normal cellular morphology, with abnormal T lymphoblasts comprising 1 % of total cellularity by flow cytometry. Subsequent to this study, he was transitioned from imatinib to dasatinib (100 mg daily) due to the development of oral ulcers, and continued to improve clinically on TKI alone. A third bone marrow biopsy and aspirate at day 54 showed 0.02 % T lymphoblasts. Peripheral blood quantitative RT-PCR analysis at three months showed a 1.4 log reduction of BCR-ABL1 transcripts (4.3 % IS units) (Table 1). Despite this response to TKI monotherapy, a matched unrelated stem cell donor was identified and transplantation is scheduled given the high-risk features of his disease.
Conclusions
We describe a case of pediatric CML with variant translocation t(6;9;22)(p22;q34q11.2) and two identifiable subclonal populations at presentation, one of which harbors inv(3)(q21q26.2) while the other is comprised of abnormal T lymphoblasts. CML is rare in the pediatric population, and only 5 % of patients have evidence of advanced disease at presentation [2]. Given the T lymphoblasts identified at diagnosis, a primary lymphoblastic process was considered; however, many of the features of this case favor a diagnosis of CML with T lymphoblastic progression, including: splenomegaly; peripheral blood with left-shifted granulocytosis and a marked increase in myelocytes; myeloid hyperplasia, dwarf megakaryocytes, and sea-blue histiocytes in the marrow; and t(6;9;22) producing the p210 BCR-ABL1 transcript. Although the possibility of two independent processes (CML and T lymphoblastic lymphoma) was not formally disproven, a single disease entity (CML with progression) is more likely.
T lymphoblastic progression of CML is particularly rare [5]. Our patient did not meet formal WHO morphologic or genetic criteria for AP or BP [7]; however, recent studies in adult patients suggested flow cytometric detection of abnormal lymphoid populations at diagnosis to be highly predictive of rapid progression despite adequate treatment with TKIs [15]. Surprisingly, an expansion of lymphoblasts was not identified in the marrow aspirate smears, which is likely due to the non-uniform distribution of the lymphoblasts in the bone marrow. This finding indicates that extensive immunophenotypic and genetic evaluation of the marrow should be undertaken to rule out AP or BP even if the bone marrow aspirate smear demonstrate CP only.
Given the coincident T lymphoblastic transformation and inv(3)(q21q26.2), we performed IHC for both MECOM and CD3 and showed that the T lymphoblasts likely did not harbor the chromosome 3 inversion. The finding that these populations were independent was not altogether unexpected, as most cases of CML with inv(3)(q21q26.2) are associated with thrombocytosis, thrombosis, and dysmegakaryopoiesis rather than T lymphoblastic transformation [16]. Indeed, the most recent and comprehensive analysis of CML with chromosome 3 alterations [14] contained no cases with T lymphoblastic transformation.
MECOM is a zinc finger transcription factor whose overexpression promotes leukemogenesis via several mechanisms, including apoptotic resistance through direct transcriptional modulation [17] and epigenetic changes via promoter DNA methylation [18]. MECOM has been extensively studied in acute myeloid leukemia and myelodysplastic syndromes, in which increased expression is associated with poor prognosis [7, 19–21]. High levels of MECOM have also been detected in CML-BP [10] as well as a subset of patients with CML-CP with resistance to TKIs [14, 22]. Currently, the customary method for detecting MECOM overexpression is restricted to RT-PCR from tumor mRNA and is not standard practice in the clinical setting. Our detection of MECOM protein expression in the bone marrow core biopsy suggests that IHC may be a useful clinical method of evaluating MECOM expression and may potentially serve as a marker for high-risk disease.
ACAs are common at diagnosis in CML in any phase of disease, and “major-route” cytogenetic changes such as trisomy 8, isochromosome 17q, and amplification of the Philadelphia chromosome suggest both clonal evolution and adverse prognosis. While MECOM abnormalities are not formal “major-route” alterations, recent analysis identified abnormalities involving 3q26.2 in roughly 3 % of adult patients with CML and found them to be at particular risk for disease progression with near uniform unresponsiveness to TKIs [14]. In this study, none of the patients with inv(3)(q21q26.2) or t(3;3;)(q21;q26.2) showed sustained major molecular response (MMR) or complete cytogenetic response (CCyR). This finding was consistent with earlier studies in animal models demonstrating TKI resistance in Evi1-overexpressing cells in both CML-CP and CML-BP cells [12]. Additionally, studies in adult CML have found MECOM rearrangements to be associated with evolution of TKI resistance and progression to myeloid blast crisis despite adequate therapy [11, 13]. Given these associations, genetic detections of 3q26.2 abnormalities or immunohistochemical detection of MECOM overexpression in CML are potentially valuable markers for high-risk disease.
Based on the adult literature of CML with 3q26.2 rearrangements, we were unsure if our patient would respond to TKI monotherapy; however, after three months of treatment, BCR-ABL1 transcript level had fallen to 4.3 % (IS). Multiple studies of adult (and a few adolescent) patients with CML receiving TKI therapy have found early molecular response (EMR), defined as BCR-ABL1 transcript levels ≤10 % (IS) at 3 months, to be associated with significantly higher rates of overall survival, event- and progression-free survival, CCyR and MMR [23–25]: in fact, BCR-ABL1 transcript level >10 % (IS) at three months was found to be the strongest predictor of poor clinical outcome [25]. Of note, the association between EMR and improved outcomes held true for patients who required transition to second-generation TKIs within the first three months due to imatinib failure or intolerance [24]. Our patient’s robust response to TKI monotherapy at three months suggests that clinical and biologic features of CML with inv(3)(q21q26.2) may be distinct in adult and pediatric patients.
Since the introduction of TKIs, hematopoietic stem cell transplant (HSCT) in CML has been largely regarded as salvage therapy for refractory disease. However, a recent large-scale prospective randomized comparison of TKIs and early HSCT found HSCT improved both long-term survival and sustained molecular remission in patients with high-risk disease and low transplant risk [26]. Given these results, combined with the lack of data in pediatric CML with inv(3)(q21q26.2), the high risk of TKI resistance in adult patients with this genetic abnormality, and the concomitant T lymphoblastic progression, we considered allogeneic stem cell transplant to be the best treatment option.
Abbreviations
ACA, additional chromosomal abnormality; CCyR, complete cytogenetic response; CML, chronic myeloid leukemia; CML-AP, accelerated phase CML; CML-BP, blast phase CML; CML-CP, chronic phase CML; EMR, early molecular response; EVI-1, ecotropic virus integration site 1; HSCT, hematopoietic stem cell transplant; IHC, immunohistochemistry; IS, international standard units; MECOM, MDS1 and EVI1 complex locus; MMR, major molecular response; TKI, tyrosine kinase inhibitor
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
Millot F, Traore P, Guilhot J, Nelken B, Leblanc T, Leverger G, et al. Clinical and biological features at diagnosis in 40 children with chronic myeloid leukemia. Pediatrics. 2005;116:140–3.
Faderl S, Talpaz M, Estrov Z, O’Brien S, Kurzrock R, Kantarjian HM. The biology of chronic myeloid leukemia. N Engl J Med. 1999;341:164–72.
Derderian PM, Kantarjian HM, Talpaz M, O'Brien S, Cork A, Estey E, et al. Chronic myelogenous leukemia in the lymphoid blastic phase: characteristics, treatment response, and prognosis. Am J Med. 1993;94:69–74.
Kantarjian HM, Keating MJ, Talpaz M, Walters RS, Smith TL, Cork A, et al. Chronic myelogenous leukemia in blast crisis. Analysis of 242 patients. Am J Med. 1987;83:445–54.
Helhmann R. How I, treat CML blast crisis. Blood. 2012;120:737–47.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissue. 4th ed. Lyon: International Agency for Research on Cancer; 2008.
Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–81.
Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, Yamamoto M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25:415–27.
Ogawa S, Kurokawa M, Tanaka T, Tanaka K, Hangaishi A, Mitani K, et al. Increased Evi-1 expression is frequently observed in blastic crisis of chronic myelocytic leukemia. Leukemia. 1996;10:788–94.
Paquette RL, Nicoll J, Chalukya M, Elashoff D, Shah NP, Sawyers C, et al. Frequent EVI1 translocations in myeloid blast crisis CML that evolves through tyrosine kinase inhibitors. Cancer Genet. 2011;204:392–7.
Sato T, Goyama S, Kataoka K, Nasu R, Tsuruta-Kishino T, Kagoya Y, et al. Evi1 defines leukemia-initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene. 2014;33:5028–38.
Theil KS, Cotta CV. The prognostic significance of an inv(3)(q21q26.2) in addition to a t(9;22)(q34;q11.2) in patients treated with tyrosine kinase inhibitors. Cancer Genet. 2014;207:171–6.
Wang W, Cortes JE, Lin P, Beaty MW, Ai D, Amin HM, et al. Clinical and prognostic significance of 3q26.2 and other chromosome 3 abnormalities in CML in the era of tyrosine kinase inhibitors. Blood. 2015;126:1699–706.
El Rassi F, Bergsagel JD, Arellano M, Gaddh M, Jillella A, Kota V, et al. Predicting early blast transformation in chronic-phase chronic myeloid leukemia: is immunophenotyping the missing link? Cancer. 2015;121:872–5.
Bernstein R, Bagg A, Pinto M, Lewis D, Mendelow B. Chromosome 3q21 abnormalities associated with hyperactive thrombopoiesis in acute blastic transformation of chronic myeloid leukemia. Blood. 1986;68:652–7.
Konantz M, Andre MC, Ebinger M, Grauer M, Wang H, Grzywna S, et al. EVI-1 modulates leukemogenic potential and apoptosis sensitivity in human acute lymphoblastic leukemia. Leukemia. 2013;27:56–65.
Lugthart S, Figueroa ME, Bindels E, Skrabanek L, Valk PJ, Li Y, et al. Aberrant DNA hypermethylation signature in acute myeloid leukemia directed by EVI1. Blood. 2011;117:234–41.
Barjesteh van Wallwijk van Doorn-Khosrovani S, Erpelinck C, van Putten WL, Valk PJ, van der Poel-van de Luytgaarde S, Hack R, et al. High EVI1 expression predicts poor survival in acute myeloid leukemia: a study of 319 de novo AML patients. Blood. 2003;101:837–45.
Lugthart S, Groschel S, Beverloo HB, Kayser S, Valk PJ, van Zelderen-Bhola SL, et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J Clin Oncol. 2010;28:3890–8.
Thol F, Yun H, Sonntag AK, Damm F, Weissinger EM, Krauter J, et al. Prognostic significance of combined MN1, ERG, BAALC, and EVI1 (MEBE) expression in patients with myelodysplastic syndromes. Ann Hematol. 2012;91:1221–33.
Daghistani M, Marin D, Khorashad JS, Wang L, May PC, Paliompeis C, et al. EVI-1 oncogene expression predicts survival in chronic-phase CML patients resistant to imatinib treated with second-generation tyrosine kinase inhibitors. Blood. 2010;116:6014–7.
Yeung DT, Mauro MJ. Prognostic significance of early molecular response in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Hematology Am Soc Hematol Educ Program. 2014;2014:240–3.
Branford S, Kim DW, Soverini S, Hague A, Shou Y, Woodman RC, et al. Initial molecular response at 3 months may predict both response and event-free survival at 24 months in imatinib-resistant or -intolerant patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase treated with nilotinib. J Clin Oncol. 2012;30:4323–9.
Marin D, Ibrahim AR, Lucas C, Gerrard G, Wang L, Szydlo RM, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012;30:232–8.
Gratwohl A, Pfirrmann M, Zander A, Kroger N, Beelen D, Novotny J, et al. Long-term outcome of patients with newly diagnosed chronic myeloid leukemia: a randomized comparison of stem cell transplantation with drug treatment. Leukemia. 2016;30:562–9.
Acknowledgements
None.
Funding
None.
Availability of data and materials
N/A.
Authors’ contributions
ML reviewed all case material and wrote the manuscript. RG, EC and RA are the doctors in charge of the present case, reviewed guidelines and studies regarding treatment of CML, and assisted with manuscript preparation. GW is the hematopathologist in charge of the initial diagnostic material and wrote the manuscript. MQ, VP, MP, AB and DF are consulting pathologists and assisted in manuscript preparation. EH is a consulting hematologist/oncologist and assisted in manuscript preparation. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Written informed consent was obtained from the patient’s parent for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Ethics approval and consent to participate
N/A.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lewen, M., Gresh, R., Queenan, M. et al. Pediatric chronic myeloid leukemia with inv(3)(q21q26.2) and T lymphoblastic transformation: a case report. Biomark Res 4, 14 (2016). https://doi.org/10.1186/s40364-016-0069-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-016-0069-0