
Abstract
Introduction
BCR-ABL1, resulting from t(9;22), is the oncogenic driver of chronic myeloid leukemia and the therapeutic target of the disease. Molecular studies have been the gold standard modality for patient assessment since the advent of tyrosine kinase inhibitor therapy. In spite of that, there are cytogenetic abnormalities that can render the disease unresponsive to conventional therapy, thus making cytogenetics an important component of patient management guidelines.
Case presentation
We present a case of a Tajik, Afghan patient with chronic myeloid leukemia with del(6)(q23.3q27), t(9;22)(q34;q11.2), monosomy 11, monosomy 12, and marker chromosome who, despite having typical clinical and hematological disease with initial response to therapy, progressed to blast crisis very early and thus required special interventions.
Conclusion
Cytogenetic monitoring is an important pillar in the management of patients with chronic myeloid leukemia that cannot be ignored. It should therefore be a part of patient management not only during diagnosis but also during management. We present an unusual cytogenetic abnormality in a patient with chronic myeloid leukemia that resulted in early disease progression.
Similar content being viewed by others

Introduction
BCR-ABL1, the oncogenic driver of chronic myeloid leukemia (CML), results from a balanced translocation, t(9;22)(q34;q11.2), involving a fusion of the Abelson gene (ABL) from chromosome 9q34 with the breakpoint cluster region (BCR) gene on chromosome 22q11.2 [1]. This fusion gene is the main molecular target in the management of CML, and, since its first introduction in early 2000s, tyrosine kinase inhibitor (TKI) therapy has been very effective in improving outcome and prognosis in affected patients [2]. The first agent with TKI activity that acquired license for treatment of patients with CML was imatinib [3]. Then the new-generation drugs were added, including nilotinib, dasatinib, and ponatinib [4].
The current therapeutic protocols are all based on cytogenetic and molecular genetic predictors of disease [1, 5]. For instance, the first tool to identify patient response with a great level of confidence was quantitative polymerase chain reaction (PCR) for BCR-ABL1, aiming to assess patients for major molecular response, which was the deepest scrutiny into a disease process for its time [6]. Since then, scrutiny of the disease process has grown deeper and deeper, with more agile and accurate methods of identifying abnormal clones with ratio of 1:10,000 versus normal clones, called the deep molecular response (DMR) [7]. Now, patients who have achieved DMR and are able to maintain it for a specific time period are even entitled to be allowed complete discontinuation of TKI therapy [8].
In spite of all the above-mentioned molecular progress in diagnosis and monitoring of CML, the role of cytogenetics is still undeniably very significant because molecular modalities, though very useful, might still be unable to identify extra Philadelphia chromosome (Ph) abnormalities, including the major route cytogenetic abnormalities that can affect management as well as prognosis [9]. We present a case of a patient with CML who was diagnosed in chronic phase (CP) and subsequently started on TKI therapy with initial hematological response; however, within 5 months, the patient experienced a blast crisis due to acquisition of extra Ph abnormalities.
Case presentation
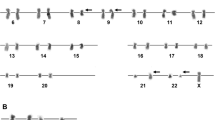
Our patient was a 42-year-old Tajik, Afghan man with no known medical illness who presented to our institution with lethargy, anorexia, pallor, and progressive abdominal distension that had developed over a duration of 4 months. On examination, the patient was pale, not in distress, and had no lymphadenopathy. His abdominal examination revealed gross splenomegaly crossing the midline with the liver just two finger breadths in the subcostal region. His complete blood count revealed moderate anemia, thrombocytosis, and hyperleukocytosis (450,000 white blood cells per microliter), showing predominance of granulocytes with a bimodal peak of mature neutrophils (68%) and myelocytes (33%). The patient’s blast count was 4% with normal basophil count. The clinical impression at that time was CML in CP based on the initial clinical and hematological evaluation. The patient was counseled to proceed with cytogenetic analysis for confirmation and prognostication of disease, but, due to financial constraints, he opted to start on TKI therapy. Accordingly, the patient was started on conventional imatinib therapy 400 mg/day. Initially, he responded well with resolution of hyperleukocytosis and improvement in hemoglobin and platelet count toward normal, but he did not achieve complete hematological remission. A repeated complete blood count analysis revealed moderate anemia, mild thrombocytopenia, and the presence of >90% blast cells in a total white cell count of 62,000/μl. After detailed counseling, the patient agreed to proceed with cytogenetic analysis, in which the analyzed metaphases of all 20 cells revealed 45,XY, presence of Ph chromosome (9q34;22q11.2), and additional complex chromosomal abnormalities, including deletion of chromosomes 6q23.3 to q27, monosomy 11, monosomy 12, insertion 12p13.3, and a marker chromosome, as shown in Fig. 1. Ultimately, the patient and his family were advised to proceed with allogeneic bone marrow transplant, considering the patient’s complex karyotype and nonresponsiveness to TKI therapy.

Karyogram of one of the two cells assessed, showing 45,XY with del(6)(q23.3q27), t(9;22)(q34;q11.2), monosomy 11, monosomy 12, and marker chromosome labeled as (mar)
Discussion
CML, once an indolent but indefinitely progressive disease of the hematopoietic system, has now become a success story in the field of hemato-oncology [10]. Almost all patients now achieve complete remission in a matter of months, and this success is attributable to the discovery of TKI agents and progress in the field of molecular genetics [11, 12].
The field of molecular genetics has enabled physicians not only to identify the disease burden but also to tailor management in accordance with measurable residual disease [1, 13]. All the guidelines since the advent of quantitative real-time PCR for BCR-ABL1, just two decades ago, include close patient monitoring with consideration of molecular targets [9, 14]. Quantitation of disease burden with consideration of established guidelines not only has enabled identification of patients for whom TKI therapy needs to be switched from one agent to a more potent one during therapy but also has made it possible to identify patients for whom a higher class of TKI must be considered right from the start [15].
Targeting deep molecular response has enabled identification of individuals who can successfully achieve complete molecular remission with successful discontinuation of therapy [8]. Novel, highly sensitive quantitative PCR analysis methods are now implemented to monitor patients who discontinue TKI therapy after successful achievement of molecular targets [16].
Mutational studies have now become a commonplace practice in the management of patients with CML [10]. The presence of mutation(s) of bad prognostic value implies that the patient needs commencement with expensive but more specific tyrosine kinase domain targeting agents, such as dasatinib and ponatinib [12, 17]. In such cases, it has been demonstrated that ponatinib, the newest agent in the market, is the agent that is effective in almost all the mutant forms of BCR-ABL1 demonstrated so far [18].
Despite all the mentioned progression in molecular studies, cytogenetics plays a very important role and thus has always been incorporated in CML management guidelines [19]. Chromosomal abnormalities that are acquired during either the pathogenesis or the treatment of CML have a deep impact on disease progression, prognosis, and thus shift the paradigm of management. Although even patients with the mutations of worst prognostic significance can benefit from third-generation TKI therapy, such as ponatinib, patients who harbor cytogenetic markers of prognostic significance have to be considered for alternative therapeutic modalities, especially allogeneic bone marrow stem cell transplant [20]. This is because it has been demonstrated that such clones that acquire additional cytogenetic abnormalities are resistant to conventional TKI inhibitor therapy [19].
Our patient, despite having the characteristic clinical and hematological profile of CML, progressed to blast crisis early during therapy for harboring complex chromosomal abnormalities that were in addition to the pathognomonic Ph. Thus, the role played by cytogenetic analysis in the management of patients with CML along with the modern molecular modalities is absolutely not replaceable. Therefore, when feasible, cytogenetic studies must be included in the analysis of patients for whom molecular tests are being done.
Conclusion
We present a case of a patient with CML who had an unusual “complex” cytogenetic abnormality in addition to Ph that resulted in early blast crisis. To our knowledge, this is the first case report of a patient with CML harboring del(6)(q23.3q27), t(9;22)(q34;q11.2), monosomy 11, monosomy 12, and marker chromosome in addition to Ph.
Availability of data and materials
All the generated data are included in this article.
Abbreviations
- ABL1:
-
Abelson gene 1
- BCR:
-
Break point cluster region
- CML:
-
Chronic myeloid leukemia
- CP:
-
Chronic phase
- PCR:
-
Polymerase chain reaction
- Ph:
-
Philadelphia chromosome
- TKI:
-
Tyrosine kinase inhibitor
References
Haidary AM, Azma Z, Ithnin A, Alauddin H, Rafeah Tumian N, Tamil AM, et al. FISH versus real-time quantitative PCR for monitoring of minimal residual disease in chronic myeloid leukaemia patients on tyrosine kinase inhibitor therapy. Malays J Pathol. 2019;41(2):149–60.
Van Etten RA. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leuk Res. 2004;28(Suppl 1):S21–8.
Branford S, Rudzki Z, Harper A, Grigg A, Taylor K, Durrant S, et al. Imatinib produces significantly superior molecular responses compared to interferon alfa plus cytarabine in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Leukemia. 2003;17(12):2401–9.
Loren CP, Aslan JE, Rigg RA, Nowak MS, Healy LD, Gruber A, et al. The BCR-ABL inhibitor ponatinib inhibits platelet immunoreceptor tyrosine-based activation motif (ITAM) signaling, platelet activation and aggregate formation under shear. Thromb Res. 2015;135(1):155–60.
Hehlmann R, Saußele S, Voskanyan A, Silver RT. Management of CML-blast crisis. Best Pract Res Clin Haematol. 2016;29(3):295–307.
Cross NC, White HE, Müller MC, Saglio G, Hochhaus A. Standardized definitions of molecular response in chronic myeloid leukemia. Leukemia. 2012;26(10):2172–5.
Hughes TP, Ross DM. Moving treatment-free remission into mainstream clinical practice in CML. Blood. 2016;128(1):17–23.
Kumagai T, Nakaseko C, Nishiwaki K, Yoshida C, Ohashi K, Takezako N, et al. Dasatinib cessation after deep molecular response exceeding 2 years and natural killer cell transition during dasatinib consolidation. Cancer Sci. 2018;109(1):182–92.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Arber DA, Hasserjian RP, Le Beau MM, Orazi ASR. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press; 2017.
Yeung CC, Egan D, Radich J. New methodologies in the molecular monitoring of CML. Curr Hematol Malig Rep. 2016;11(2):94–101.
Mensink E, van de Locht A, Schattenberg A, Linders E, Schaap N, Geurts van Kessel A, et al. Quantitation of minimal residual disease in Philadelphia chromosome positive chronic myeloid leukaemia patients using real-time quantitative RT-PCR. Br J Haematol. 1998;102(3):768–74.
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am J Hematol. 2016;91(2):252–65.
Soverini S, De Benedittis C, Mancini M, Martinelli G. Present and future of molecular monitoring in chronic myeloid leukaemia. Br J Haematol. 2016;173(3):337–49.
Usui N. Updated European LeukemiaNet recommendations for the management of CML. In: Kizaki M, editor. Molecular pathogenesis and treatment of chronic myelogenous leukemia. Tokyo: Springer; 2016. p. 81–100.
Shamroe CL, Comeau JM. Ponatinib: a new tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Ann Pharmacother. 2013;47(11):1540–6.
Kitamura H, Tabe Y, Ai T, Tsuchiya K, Yuri M, Misawa S, Horii T, Kawaguchi A, Ohsaka A, Kimura S. A new highly sensitive real-time quantitative-PCR method for detection of BCR-ABL1 to monitor minimal residual disease in chronic myeloid leukemia after discontinuation of imatinib. PLoS One. 2019;14(3):e0207170.
Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–84.
Hoy SM. Ponatinib: a review of its use in adults with chronic myeloid leukaemia or Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs. 2014;74(7):793–806.
Fabarius A, Kalmanti L, Dietz CT, Lauseker M, Rinaldetti S, Haferlach C, et al. Impact of unbalanced minor route versus major route karyotypes at diagnosis on prognosis of CML. Ann Hematol. 2015;94(12):2015–24.
Kim YJ, Kim DW, Lee S, Kim HJ, Kim YL, Hwang JY, et al. Comprehensive comparison of FISH, RT-PCR, and RQ-PCR for monitoring the BCR-ABL gene after hematopoietic stem cell transplantation in CML. Eur J Haematol. 2002;68(5):272–80.
Acknowledgements
We acknowledge the role played by Agha Khan Development Network (AKDN) in establishing the country’s first cytogenetic laboratory.
Funding
The authors received no funding for the writing.
Author information
Authors and Affiliations
Contributions
AMH and HAM conceived the case report. HAM and AMH were the major contributors to the writing of the manuscript. AMH and SN diagnosed the patient. SR, MA, and SS performed cytogenetic studies. AMH, SI, and HAM were the major contributors to critical revision of the manuscript for important intellectual content. SN and AMH provided expert opinion and final approval of the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Malakzai, H.A., Rahmani, S., Haidary, A.M. et al. Complex cytogenetic abnormalities in chronic myeloid leukemia resulting in early progression to blast crisis: a case report. J Med Case Reports 14, 231 (2020). https://doi.org/10.1186/s13256-020-02539-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-020-02539-x