
Abstract
Microvillus inclusion disease (MVID) is characterised by onset of intractable life-threatening watery diarrhoea during infancy. Transmission electron microscopy demonstrates shortening or absence of apical microvilli, pathognomonic microvillus inclusions in mature enterocytes and subapical accumulation of periodic acid-Schiff-positive granules or vesicles confirming diagnosis. Mutations in MYO5B have been found to cause MVID. In two patients with MVID, whole-exome sequencing of DNA revealed homozygous truncating mutations in STX3. Mutations in these genes disrupt trafficking between apical cargo vesicles and the apical plasma membrane. Thus, disturbed delivery of certain brush border membrane proteins is a common defect in MVID.
Similar content being viewed by others


The history of microvillus inclusion disease
In 1978, Davidson and colleagues first described five infants with severe diarrhoea from birth and failure to thrive [1]. The disease was identified as a congenital enteropathy marked by villus atrophy, severe diarrhoea with partial sodium loss and malabsorption. Furthermore, jejunal biopsies displayed in electron microscopy (EM) cytoplasmic inclusions with brush border microvilli on their inside. Over the following years, the disease was given several names: Davidson disease [1], congenital familial protracted diarrhoea with enterocyte brush-border abnormalities, congenital microvillus atrophy [2] and microvillus inclusion disease (MVID). The latter one was shaped by Cutz and colleagues in 1989 [3]. This study sets the diagnostic standard, already discussing further possible mechanisms for the formation of microvillus inclusions (ectopic brush border formation at intracellular sites [3] versus engulfment of brush border via autophagy/macropinocytosis [1]; see also below the paragraph on pathophysiology). The following years, an increasing number of patients were diagnosed with MVID and diagnostic criteria were refined [4–6]. In 2008, mutations in MYO5B were identified as causal for MVID [7]. This finding initiated further research trying to unravel the pathophysiology of MVID and the specific function of the motor protein Myo5B in polarised epithelial cells [8–10]. With the identification of mutations in a second gene, STX3, causative for MVID, molecular and genetic analyses gained pace pushing MVID to the ‘centre stage’ of molecular paediatric research.
Clinical features of MVID
MVID patients typically present intractable watery diarrhoea, leading to a severe loss of body weight, and metabolic acidosis due to bicarbonate loss [1, 3, 4, 11]. Early-onset MVID that starts within the first days after birth can be discriminated from late-onset MVID cases [4, 12]. The latter one becomes clinically apparent only at the age of 2 to 3 months. In general, pregnancy was reported to be without complications, but occasionally, maternal polyhydramnios is present. Diarrhoea is the main, often life-threatening symptom of MVID. Stool volumes range between 150 and 300 ml/kg/day and respond only slightly to bowel arrest [4].
In most cases, no additional clinical signs, such as malformations, or other organ manifestations accompany MVID. However, cholestatic liver disease might be present in up to one third of patients [13], and cases of associated renal Fanconi syndrome [14] have been reported.
Treatment aims at supplementing the fluid and nutrient loss. Thus, life-long total parenteral nutrition (TPN) is generally required. No causative cure is available, but small bowel transplantation (intestinal Tx) is able to cure the severe diarrhoea. Both TPN and intestinal Tx come with side effects, such as cholestasis or infections caused by the immunosuppressive regime.
The limited therapeutic options and the severity of the disease often lead to the patients’ death within the first 3 years. However, patients with late-onset MVID sometime tolerate enteral feeding. The requirement of parenteral nutrition can be reduced to once or twice per week [4, 6].
Genetics
Since the identification of mutations in the MYO5B gene in 2008, an increasing number of mutations causing MVID are described in the literature [7, 8, 12], thereby confirming autosomal recessive inheritance of MVID. Homozygous (mostly originating from consanguine parents) and compound-heterozygous missense and nonsense mutations (mostly from non-consanguine parents) were reported. However, in some patients, only one heterozygous mutation and no second mutation could be identified [12] upon sequencing all exons and exon-intron boundaries. With this technique, deep intronic mutations, mutations in regulatory regions, and deletions and duplications involving whole exons cannot be revealed and may be missed. Nowadays, commercial multiplex ligation-dependent probe amplification (MLPA) analysis is available to detect larger intragenic deletions and duplications. Sequencing and MLPA analysis detect biallelic mutations in >90 % of patients referred with a clinical/histopathologic diagnosis of MVID (n = 70, our unpublished data). Van der Velde and colleagues have methodically characterised all currently reported mutations in MYO5B and linked them to potentially impaired functions of the different domains of the actin motor protein MYO5B [12].
The second gene in which MVID causing mutations were identified was STX3 [15]. So far, two patients of 2 years of age have been reported, both with nonsense mutations resulting in truncations of the apically targeted N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin3 [15]. Genotype-phenotype correlation with respect to MYO5B and STX3-related MVID has not been reported so far, due to the small number and young age of patients with STX3 mutations.
Diagnosis
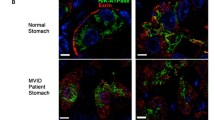
The clinical presentation of MVID is that of severe diarrhoea, most often starting within the first week of life. The diarrhoea has a secretory component, which persists even when giving nil per mouth and does not clinically resolve upon elimination of dietary components. Villus atrophy, brush border reduction and increased periodic acid-Schiff (PAS) staining throughout the subapical cytoplasm are often found in light microscopy. Immunohistochemistry for brush border components CD10 (a neutral membrane-associated peptidase) [6, 16], Villin [17] and Rab11a [18, 19] shows an irregular, broadened apical signal, corresponding to abnormal subapical localization of brush border components, which strongly raises suspicion of MVID. Analysis of small intestinal biopsies by EM remains the best tool for histological diagnosis of MVID. EM reveals shortening or absence of brush border microvilli, so-called microvillus inclusions (vacuoles bearing centripetal microvilli in about 10 % of small gut villus enterocytes), and a subapical accumulation of different kinds of vesicular/tubular structures, referred to as ‘PAS positive secretory granules’ [5]—the three ultrastructural/diagnostic hallmarks of MVID (compare Fig. 1). Differences in the abundance of these features are often observed between villus and crypt enterocytes. While villus enterocytes may display all three hallmarks, crypt enterocytes generally display just secretory granules. Only a minority of enterocytes might display the hallmarks at the time of EM analysis, and the histological presentation of MVID varies from patient to patient.

Electron micrograph of patient’s duodenal enterocytes depicting the three ultrastructural hallmarks of MVID (homozygous c.1323–2A > G splice-site mutation in MYO5B). Black arrow heads mark the shortening or loss of apical microvilli. Subapical accumulations of tubulo-/vesicular structures (secretory granules) are marked by black arrows. MI intracellular microvillus inclusion, Lys lysosomes. Scale bar = 2 μm
Additionally, increasing availability and decreasing costs of modern genetic analysis render it a suitable tool to verify MVID diagnosis. Targeted mutational analysis of the MYO5B and STX3 loci, high-throughput sequencing of genes causing congenital diarrhoeas (‘gene panel’ sequencing) and whole-exome sequencing should be performed.
Pathophysiology
Over the last years, several studies have added to the understanding of MVID’s pathophysiology [7–10, 15, 20–22]. Besides native biopsy material from MVID patients, the human colorectal adenoma cell line CaCo2 has been used as the model system in most of these studies. As this cell line has the ability to establish enterocyte polarity in culture, it has been the basis for loss of Myo5B studies and its impact on epithelial polarity. This approach allowed to gain further insight into the role of Myo5B in intracellular trafficking as down-regulation and knockout recapitulate the loss of apical microvilli, loss of polarity and the accompanying mislocalisation of apical transporters [8, 9, 20]. This might be a crucial step for understanding MVID, as the mislocalisation of the Na+/H+ exchanger NHE3 [20] might account for the sodium loss diarrhoea reported from MVID cases [23]. The identification of a second gene causal for MVID, the t-SNARE STX3, points towards a role of the apical exocytic pathway in epithelial cells. Both Myo5B and Stx3 are involved in apical trafficking [15, 24].
The origin of microvillus inclusions [10, 21] was addressed as well, and hypotheses range from autophagocytosed/endocytosed apical plasma membrane [1, 10, 21, 25] to de novo formed, intracellular apical domains [3, 19]. Conceivably, microvillus inclusions might not add much to the pathophysiology but could rather be a secondary effect [25] of overall disrupted epithelial polarity, since similar structures known as ‘vacuolar apical compartments (VACs)’ also occur in epithelial cancers, or following experimental disruption of the cytoskeleton or of intercellular contacts [26–30].
So far, secretory granules could only rarely be reproduced in in vitro culture [8, 15]. However, a genome-edited CaCo2 cell line was recently published, demonstrating all three ultrastructural hallmarks including an accumulation of subapical vesicles. With this, better understanding of MVID’s pathophysiology was achieved [31].
Only recently, MYO5B knockout mouse models have been published. Despite the different genetic approaches applied, constitutive whole organism knockout [32] and inducible bowel-specific knockout [19, 25]demonstrate all these studies MVID-like phenotypes in mice. Schneeberger and colleagues demonstrated that organoid cultures of murine intestinal stem cells might serve a useful tool to test future therapies for MVID [19]. For this, crypt stem cells obtained by endoscopic biopsies are cultured to form ‘mini-guts’ ex vivo.
Outlook
The increase in understanding the pathophysiology of MVID will eventually allow developing targeted therapies to treat specific symptoms, e.g. diarrhoea, and could overcome the need to transplant the small bowel and perform long-term parenteral nutrition. This could lead to a drastic increase in the quality of life of MVID patients. New therapeutic approaches could be tested on organoid cultures derived from mice or patients. This, of course, remains speculative at the moment, but conceivable targets of therapy could be the disrupted epithelial polarity and the accompanying mislocalisation of apical enzymes and transporters.
In recent years, novel genome editing technologies, such as CRISPR/Cas9, once again moved gene therapy into the spotlights. Enterocyte stem cells reside at the bottom of every crypt in the bowel [33]. Thus, all these stem cell niches have to be targeted in order to achieve therapeutic efficiency. Besides all still unsolved problems of genome editing, e.g. off targets [34], crypt stem cells niches make gene therapy rather unsuitable for MVID.
Abbreviations
- EM:
-
electron microscopy
- MLPA:
-
multiplex ligation-dependent probe amplification
- MVID:
-
microvillus inclusion disease
- PAS:
-
periodic acid-Schiff
- SNARE:
-
N-ethylmaleimide-sensitive factor attachment protein receptor
- TPN:
-
total parenteral nutrition
- Tx:
-
transplantation
References
Davidson GP, Cutz E, Hamilton JR, Gall DG (1978) Familial enteropathy: a syndrome of protracted diarrhea from birth, failure to thrive, and hypoplastic villus atrophy. Gastroenterology 75(5):783–790
Phillips AD, Jenkins P, Raafat F, Walker-Smith JA (1985) Congenital microvillous atrophy: specific diagnostic features. Arch Dis Child 60(2):135–140
Cutz E, Rhoads JM, Drumm B, Sherman PM, Durie PR, Forstner GG (1989) Microvillus inclusion disease: an inherited defect of brush-border assembly and differentiation. N Engl J Med 320(10):646–651. doi:10.1056/nejm198903093201006
Phillips AD, Schmitz J (1992) Familial microvillous atrophy: a clinicopathological survey of 23 cases. J Pediatr Gastroenterol Nutr 14(4):380–396
Phillips AD, Szafranski M, Man LY, Wall WJ (2000) Periodic acid-Schiff staining abnormality in microvillous atrophy: photometric and ultrastructural studies. J Pediatr Gastroenterol Nutr 30(1):34–42
Ruemmele FM, Schmitz J, Goulet O (2006) Microvillous inclusion disease (microvillous atrophy). Orphanet J Rare Dis 1:22. doi:10.1186/1750-1172-1-22
Muller T, Hess MW, Schiefermeier N, Pfaller K, Ebner HL, Heinz-Erian P, Ponstingl H, Partsch J, Rollinghoff B, Kohler H, Berger T, Lenhartz H, Schlenck B, Houwen RJ, Taylor CJ, Zoller H, Lechner S, Goulet O, Utermann G, Ruemmele FM, Huber LA, Janecke AR (2008) MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat Genet 40(10):1163–1165. doi:10.1038/ng.225
Ruemmele FM, Muller T, Schiefermeier N, Ebner HL, Lechner S, Pfaller K, Thoni CE, Goulet O, Lacaille F, Schmitz J, Colomb V, Sauvat F, Revillon Y, Canioni D, Brousse N, de Saint-Basile G, Lefebvre J, Heinz-Erian P, Enninger A, Utermann G, Hess MW, Janecke AR, Huber LA (2010) Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum Mutat 31(5):544–551. doi:10.1002/humu.21224
Thoeni CE, Vogel GF, Tancevski I, Geley S, Lechner S, Pfaller K, Hess MW, Müller T, Janecke AR, Avitzur Y, Muise A, Cutz E, Huber LA (2014) Microvillus inclusion disease: loss of myosin Vb disrupts intracellular traffic and cell polarity. Traffic 15(1):22–42. doi:10.1111/tra.12131
Knowles BC, Roland JT, Krishnan M, Tyska MJ, Lapierre LA, Dickman PS, Goldenring JR, Shub MD (2014) Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J Clin Invest 124(7):2947–2962. doi:10.1172/jci71651
Erickson RP, Larson-Thome K, Valenzuela RK, Whitaker SE, Shub MD (2008) Navajo microvillous inclusion disease is due to a mutation in MYO5B. Am J Med Genet A 146a(24):117–3119. doi:10.1002/ajmg.a.32605
van der Velde KJ, Dhekne HS, Swertz MA, Sirigu S, Ropars V, Vinke PC, Rengaw T, van den Akker PC, Rings EH, Houdusse A, van Ijzendoorn SC (2013) An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations. Hum Mutat 34(12):1597–1605. doi:10.1002/humu.22440
Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, Sauvat F, Irtan S, Davit-Spraul A, Jacquemin E, Ruemmele F, Rainteau D, Goulet O, Colomb V, Chardot C, Henrion-Caude A, Debray D (2014) MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology 60(1):301–310. doi:10.1002/hep.26974
Golachowska MR, van Dael CM, Keuning H, Karrenbeld A, Hoekstra D, Gijsbers CF, Benninga MA, Rings EH, van Ijzendoorn SC (2012) MYO5B mutations in patients with microvillus inclusion disease presenting with transient renal Fanconi syndrome. J Pediatr Gastroenterol Nutr 54(4):491–498. doi:10.1097/MPG.0b013e3182353773
Wiegerinck CL, Janecke AR, Schneeberger K, Vogel GF, van Haaften-Visser DY, Escher JC, Adam R, Thoni CE, Pfaller K, Jordan AJ, Weis CA, Nijman IJ, Monroe GR, van Hasselt PM, Cutz E, Klumperman J, Clevers H, Nieuwenhuis EE, Houwen RH, van Haaften G, Hess MW, Huber LA, Stapelbroek JM, Muller T, Middendorp S (2014) Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology 147(1):65–68.e10. doi:10.1053/j.gastro.2014.04.002
Youssef N, MR F, Goulet O, Patey N (2004) CD10 expression in a case of microvillous inclusion disease. Ann Pathol 24(6):624–627
Shillingford NM, Calicchio ML, Teot LA, Boyd T, Kurek KC, Goldsmith JD, Bousvaros A, Perez-Atayde AR, Kozakewich HP (2015) Villin immunohistochemistry is a reliable method for diagnosing microvillus inclusion disease. Am J Surg Pathol 39(2):245–250. doi:10.1097/pas.0000000000000355
Talmon G, Holzapfel M, DiMaio DJ, Muirhead D (2012) Rab11 is a useful tool for the diagnosis of microvillous inclusion disease. Int J Surg Pathol 20(3):252–256. doi:10.1177/1066896911430959
Schneeberger K, Vogel GF, Teunissen H, van Ommen DD, Begthel H, El Bouazzaoui L, van Vugt AH, Beekman JM, Klumperman J, Muller T, Janecke A, Gerner P, Huber LA, Hess MW, Clevers H, van Es JH, Nieuwenhuis EE, Middendorp S (2015) An inducible mouse model for microvillus inclusion disease reveals a role for myosin Vb in apical and basolateral trafficking. Proc Natl Acad Sci U S A 112(40):12408–12413. doi:10.1073/pnas.1516672112
Ameen NA, Salas PJ (2000) Microvillus inclusion disease: a genetic defect affecting apical membrane protein traffic in intestinal epithelium. Traffic 1(1):76–83
Reinshagen K, Naim HY, Zimmer KP (2002) Autophagocytosis of the apical membrane in microvillus inclusion disease. Gut 51(4):514–521
Michaux G, Massey-Harroche D, Nicolle O, Rabant M, Brousse N, Goulet O, Bivic AL, Ruemmele FM (2015) The localisation of the apical Par/Cdc42 polarity module is specifically affected in microvillus inclusion disease. Biology of the cell/under the auspices of the European Cell Biology Organization. doi:10.1111/boc.201500034
Salomon J, Goulet O, Canioni D, Brousse N, Lemale J, Tounian P, Coulomb A, Marinier E, Hugot JP, Ruemmele F, Dufier JL, Roche O, Bodemer C, Colomb V, Talbotec C, Lacaille F, Campeotto F, Cerf-Bensussan N, Janecke AR, Mueller T, Koletzko S, Bonnefont JP, Lyonnet S, Munnich A, Poirier F, Smahi A (2014) Genetic characterization of congenital tufting enteropathy: epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum Genet 133(3):299–310. doi:10.1007/s00439-013-1380-6
Roland JT, Bryant DM, Datta A, Itzen A, Mostov KE, Goldenring JR (2011) Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc Natl Acad Sci U S A 108(7):2789–2794. doi:10.1073/pnas.1010754108
Weis V, G, Knowles BC, Choi E, Goldstein AE, Williams JA, Manning EH, et al. (2015) Loss of MYO5B in mice recapitulates microvillus inclusion disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cellular and Molecular Gastroenterology and Hepatology. (In press). doi:10.1016/j.jcmgh.2015.11.009
Remy L (1986) The intracellular lumen: origin, role and implications of a cytoplasmic neostructure. Biol Cell 56(2):97–105
Vega-Salas DE, Salas PJ, Rodriguez-Boulan E (1987) Modulation of the expression of an apical plasma membrane protein of Madin-Darby canine kidney epithelial cells: cell-cell interactions control the appearance of a novel intracellular storage compartment. J Cell Biol 104(5):1249–1259
Achler C, Filmer D, Merte C, Drenckhahn D (1989) Role of microtubules in polarized delivery of apical membrane proteins to the brush border of the intestinal epithelium. J Cell Biol 109(1):179–189
Gilbert T, Rodriguez-Boulan E (1991) Induction of vacuolar apical compartments in the Caco-2 intestinal epithelial cell line. J Cell Sci 100(Pt 3):451–458
Vega-Salas DE, San Martino JA, Salas PJ, Baldi A (1993) Vacuolar apical compartment (VAC) in breast carcinoma cell lines (MCF-7 and T47D): failure of the cell-cell regulated exocytosis mechanism of apical membrane. Differentiation 54(2):131–141
Vogel GF, Klee KM, Janecke AR, Muller T, Hess MW, Huber LA (2015) Cargo-selective apical exocytosis in epithelial cells is conducted by Myo5B, Slp4a, Vamp7, and Syntaxin 3. J Cell Biol 211(3):587–604. doi:10.1083/jcb.201506112
Carton-Garcia F, Overeem AW, Nieto R, Bazzocco S, Dopeso H, Macaya I, Bilic J, Landolfi S, Hernandez-Losa J, Schwartz S, Ramon YCS, van Ijzendoorn SC, Arango D (2015) Myo5b knockout mice as a model of microvillus inclusion disease. Sci Rep 5:12312. doi:10.1038/srep12312
Koo BK, Clevers H (2014) Stem cells marked by the R-spondin receptor LGR5. Gastroenterology 147(2):289–302. doi:10.1053/j.gastro.2014.05.007
Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z, Lv J, Xie X, Chen Y, Li Y, Sun Y, Bai Y, Songyang Z, Ma W, Zhou C, Huang J (2015) CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6(5):363–372. doi:10.1007/s13238-015-0153-5
Acknowledgements
The authors thank the Austrian Science Funds (FWF) within the SFB021 special research programme and the MCBO PhD programme which supported this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors conceived and wrote the manuscript. HMW provided the figure. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vogel, G.F., Hess, M.W., Pfaller, K. et al. Towards understanding microvillus inclusion disease. Mol Cell Pediatr 3, 3 (2016). https://doi.org/10.1186/s40348-016-0031-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-016-0031-0