
Abstract
Background
Male-pattern baldness (MPB) is the most common cause of hair loss in men. It can be categorized into three types: type 2 (T2), type 3 (T3), and type 4 (T4), with type 1 (T1) being considered normal. Although various MPB-associated genetic variants have been suggested, a comprehensive study for linking these variants to gene expression regulation has not been performed to the best of our knowledge.
Results
In this study, we prioritized MPB-related tissue panels using tissue-specific enrichment analysis and utilized single-tissue panels from genotype-tissue expression version 8, as well as cross-tissue panels from context-specific genetics. Through a transcriptome-wide association study and colocalization analysis, we identified 52, 75, and 144 MPB associations for T2, T3, and T4, respectively. To assess the causality of MPB genes, we performed a conditional and joint analysis, which revealed 10, 11, and 54 putative causality genes for T2, T3, and T4, respectively. Finally, we conducted drug repositioning and identified potential drug candidates that are connected to MPB-associated genes.
Conclusions
Overall, through an integrative analysis of gene expression and genotype data, we have identified robust MPB susceptibility genes that may help uncover the underlying molecular mechanisms and the novel drug candidates that may alleviate MPB.
Similar content being viewed by others

Introduction
Male-pattern baldness (MPB), also known as androgenetic alopecia, is the leading cause of hair loss in men. MPB is a polygenic disease with a high heritability rate (~ 80%), reaching a prevalence of approximately 50% by the age of 50 [1, 2]. MPB is usually caused by testosterone, an androgenic hormone, with the hairline and crown being more susceptible to androgen than the occipital scalp [3, 4]. As a result, MPB typically starts with a receding hairline and hair loss on the crown, eventually leaving only a horseshoe-shaped area of hair on the back of the head [1, 5, 6]. The severity of MPB is assessed using the Hamilton-Norwood scale (HNS) that identifies characteristic patterns of progression [7,8,9,10]. This scale is divided into three main types ranging from type 2 (mild MPB) to type 4 (severe MPB), while type 1 is considered the normal phenotype. MPB has a complex pathophysiology, potentially involving interactions between various tissues including skin, adipose, blood, and other endocrine systems [11]. Currently, finasteride and dutasteride are used to temporarily alleviate MPB by inhibiting or decomposing testosterone, so these drugs are not effective for severe MPB, may have reproductive side effects, and often result in high recurrence rates [12, 13]. Therefore, further research on the genetic basis of MPB and its application to therapeutic investigation is necessary.
Previous studies have suggested that the androgen receptor (AR) on the X chromosome and/or the genes involved with the Wnt/β-catenin signaling pathway are causal genetic factors that promote MPB [2, 4, 14]. However, these genes cannot fully explain the high heritability and mechanisms of MPB [1]. A previous genome-wide association study (GWAS) was performed to investigate the genetic features of MPB and several risk loci with high heritability were identified [1]. Although this study identified risk loci associated with MPB, it is necessary to map these loci to specific genes to gain a deeper understanding of the underlying genomic and transcriptomic mechanisms. As observed in many GWAS, the risk loci are often found in intergenic regions, such as single nucleotide polymorphisms (SNPs) in non-coding regions. These SNPs often regulate the expression of genes located far away from them, making it challenging to directly map them to genes based on their genomic locations [15,16,17,18,19,20]. In addition, GWAS identifies linkage disequilibrium (LD) blocks of associated variants rather than specific variant-trait associations [20]. Therefore, it is difficult to discover the causal variants using GWAS risk loci. To address these issues, transcriptome-wide association study (TWAS) has been proposed as a comprehensive method that takes into account SNP-mediated gene expression based on the correlation between SNPs and gene expression levels [21]. TWAS predicts the gene expression levels associated with complex traits by calculating associations with genetic variants using expression quantitative trait loci (eQTL) panels. Recently, the eQTL panels from context-specific genetics (CONTENT), which combine tissue-shared genetic features in the regulation of gene expression, have shown improved statistical power in TWAS [22]. By utilizing the advantages of TWAS that has proven effective in investigating the pathogenesis of traits and prioritizing causal genes, this study aims to address the limitations of previous studies and provide new insights into the mechanisms of MPB.
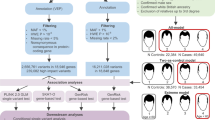
Here, we present a study that examines gene expression to investigate the association between risk loci and genes. We conducted TWAS on three types of MPB (type 2, type 3, and type 4) utilizing three GWAS datasets from the UK Biobank (UKBB), which consists of individuals of European ancestry. Prior to TWAS, the linkage disequilibrium score-specifically expressed genes (LDSC-SEG) method was used to prioritize tissue panels that were significantly associated with each type of MPB [23]. Using the results of the tissue prioritization with LDSC-SEG, we performed TWAS on single-tissue panels from the genotype-tissue expression (GTEx) version 8 and cross-tissue panels from the CONTENT. To verify the robustness of MPB susceptibility genes, we conducted a colocalization (COLOC) and conditional and joint analysis, which identified 10, 11, and 54 significantly associated genes with MPB type 2, type 3, and type 4, respectively. As downstream analyses of these MPB signatures, we conducted a phenome-wide association study (PheWAS) and in silico drug repositioning (Fig. 1).

Workflow of the entire study. The GWAS summary statistics were obtained from GWAS ATLAS and tissue panels were prioritized using LDSC-SEG. We conducted TWAS and COLOC using both single-tissue panels and cross-tissue panels. The biological functions of TWAS results from single-tissue panels were evaluated through functional annotation analysis. Downstream analyses were performed using 10 (type 2), 11 (type 3), and 54 (type 4) genes that showed significance in the conditional and joint analysis
Materials and methods
Description of GWAS summary statistics and data pre-processing
The GWAS summary statistics for TWAS analyses were obtained from GWAS ATLAS (https://atlas.ctglab.nl/). The datasets were categorized into four types based on the disease states of MPB: type 1 (HNS I and II), type 2 (HNS II, III, IIIa, and Iva), type 3 (HNS III vertex-V), and type 4 (HNS IV, V, Va, VI, and VII) [2, 24, 25]. Except for the normal type 1, the three datasets consisted of 176,380 Europeans from the UKBB and additional details can be found in Supplementary Table S1. The datasets were converted into the LD score format using the LD score regression (LDSC) software (version 1.0.1).
The tissue-specific enrichment analysis based on genetic heritability
To prioritize tissue panels associated with MPB for TWAS analyses, we conducted a tissue-specific enrichment analysis using LDSC-SEG, a tool developed by Finucane et al. [23]. for identifying disease-relevant tissues. This method applies stratified LDSC to GWAS summary statistics to evaluate the contribution of the heritable element in multiple tissue types [23]. We tested the tissue-specific enrichments for the three types of MPB using the multi-tissue gene expression and multi-tissue chromatin modification (DNase hypersensitivity, histone acetylation, and histone methylation) datasets. The multi-tissue gene expression dataset consists of the GTEx [26] and Franke lab datasets [27, 28] and is classified into nine major groups for visualization purposes: adipose, blood/immune, cardiovascular, central nervous system, digestive, liver, musculoskeletal/connective, pancreas, and “other”. The multi-tissue chromatin modification dataset includes data from the Roadmap Epigenomics [29] and the Encyclopedia of DNA Elements (ENCODE) projects [30] and is classified as the multi-tissue gene expression dataset. Since chromatin modification can influence gene expression, we used the multi-tissue chromatin modification dataset to validate the results obtained from the multi-tissue gene expression dataset and to account for epigenetic modifications. While MPB is known to be associated with the sex chromosomes, we aimed to identify responses in somatic cells that occur regardless of the sex chromosomes by considering general tissues or cells rather than those specific to sex chromosomes.
The transcriptome-wide association analysis using multi-tissue panels
The TWAS analysis was performed using the functional summary-based imputation (FUSION) software. FUSION integrates GWAS summary statistics and eQTL weights to compute gene expression-mediated associations between genotype and phenotype [21]. We used eQTL panels from GTEx version 8, which were filtered based on tissue-specific enrichment analysis. For multiple comparison adjustment, the Bonferroni correction method was applied for TWAS associations of tissue panels in MPB type 2, type 3, and type 4, respectively. To obtain additional TWAS associations with tissue-shared or tissue-specific effects, we utilized the cross-tissue expression panel from the CONTENT. The CONTENT panel computes based on the admixture of GTEx single-tissue panels and combines tissue-shared and tissue-specific associations through linear regression calculated by Thompson et al. [22] For TWAS results using the CONTENT panel, the Bonferroni correction method was used and only results corresponding to the tissue panels of tissue-specific enrichment analysis were selected.
The colocalization analysis between eQTL and GWAS signals
Since TWAS is based on predictive models that include the genotype, gene expression, and phenotype, there may be hidden confounding factors including LD contamination, which can lead to false correlations between predicted gene expression and phenotype. To address this issue, we performed the COLOC analysis, a Bayesian-based statistical method that estimates the probability of shared causal variants between eQTL and GWAS signals [31]. The posterior probabilities (PP) for hypotheses 0–4 were as follows: H0 (no causal variant), H1 (only causal variants between genotype and phenotype), H2 (only causal variants for eQTL), H3 (phenotype and gene expressions driven by two independent causal variants), and H4 (phenotype and gene expressions share one causal variant). Following previous studies, we set the significance threshold as PP3 + PP4 > 0.8 and PP4 / PP3 > 2 [16, 32].
The conditional and joint analysis
To ensure accuracy in identifying potential LD-contaminated associations, a conditional and joint analysis was conducted. This analysis takes into account the extent to which GWAS signal remains significant when other associations are included as covariates. If a GWAS signal remains significant even after conditioning, it suggests that the signal is conditionally independent, meaning it is not reliant on other associations. However, if the signal becomes less significant after conditioning, it suggests that the signal was influenced by other associations [15, 21]. Genes that still exhibit significance after conditioning were defined as jointly significant genes, while genes showing reduced significance after conditioning are considered as marginally significant genes due to co-regulation. We selected the jointly significant genes as the reliable genetic signature for each type of MPB and named them as MPB signatures.
The functional enrichment analysis in biological processes using GSEA
The functional annotations of TWAS associations were conducted using the gene set enrichment analysis (GSEA) version 4.3.2, a computational method that interprets groups of genes that share a common biological function by using gene expression data and a gene set database [33]. The functional enrichment analysis utilized gene sets derived from the gene ontology (GO) biological process and ranked gene lists with z-scores calculated from TWAS. Biological processes that met a significance threshold (P-value < 0.05) were shown based on the normalized enrichment scores (NES). The NES value reflects the degree of up-regulated and down-regulated gene enrichment through multiple hypothesis testing.
The phenome-wide association study using MPB signatures to identify MPB-associated traits
The PheWAS is a method used to search for phenotypes that share specific SNP associations across thousands of human phenotypes. This approach tests associations between genetic variants and a large number of phenotypes to discover pleiotropic effects, where specific genetic variants may be associated with multiple traits [34, 35]. By utilizing the PheWAS database available at GWAS ATLAS that contains leading SNPs from 4,756 GWAS summary statistics, we examined the phenotype associations of MPB signatures [36]. For each SNP in the MPB signatures, we obtained a list of associations and filtered the results to identify significant traits linked to genetic loci of each type of MPB using a Bonferroni-corrected significance threshold (P-value < 1.05 × 10− 5).
Drug-gene connectivity analysis for drug repositioning
To identify drugs that are associated with genes, we used the connectivity map (CMAP) and library of integrated network-based cellular signatures (LINCS) unified environment (CLUE) (https://clue.io/about) that is a web-based tool for drug repositioning. CLUE generates drug-gene connections by analyzing gene expression and proteomics assays including the LINCS L1000 and CMAP databases [37, 38]. To validate the drug candidates identified by CLUE, we also used the Networkanalyst that can offer comprehensive list of protein-drug interaction networks collected from the DrugBank database (version 5.0) [39]. The networks were visualized using Cytoscape application (version 3.9.1) and only networks that were directly linked to MPB signatures in both results were displayed [40].
Calculating the protein-protein interaction score
The protein-protein interaction (PPI) analysis was conducted between the already known markers and signatures of MPB. For the known MPB genes, we selected 336 genes that appeared when searching for ‘androgenetic alopecia’ in the Open Targets Platform (https://platform.opentargets.org/). The interaction score was calculated using the combined score from the STRING database (https://string-db.org/). As a control, we calculated the interaction score between randomly selected genes and known MPB genes. To determine whether the two groups of interaction scores were significantly different, we analyzed the degree of interaction using a one-tailed t-test with 10,000 repetitions.
Results
Panel prioritization to select MPB-related tissue panels
Since TWAS calculates genetic associations based on tissue panels, it is necessary to select appropriate tissues to obtain accurate results. In particular, to determine the effects of MPB on somatic cells, it is better to select MPB-related tissue panels from general tissue panels rather than focusing on tissue panels related to the sex chromosomes. To examine the genetic contribution in tissue-specific gene expression of MPB and select significantly associated tissue panels, we conducted a tissue-specific enrichment analysis using the LDSC-SEG with the multi-tissue gene expression dataset [23]. The LDSC-SEG analysis with the multi-tissue gene expression dataset suggested that four (adipose, digestive, musculoskeletal/connective, and “other”) out of nine major categories showed significant association with MPB type 2 (coefficient P-value < 0.05) (Fig. 2A). For MPB type 3, we identified significant enrichment in five (adipose, blood/immune, digestive, musculoskeletal/connective, and “other”) out of nine major categories (coefficient P-value < 0.05) (Fig. 2B). In the case of MPB type 4, the analysis revealed significant associations in four major categories (adipose, blood/immune, cardiovascular, and “other”; coefficient P-value < 0.05) (Fig. 2C and Supplementary Data S1).
To validate the results of the tissue-specific enrichment analysis using the multi-tissue gene expression dataset, we also utilized the multi-tissue chromatin modification dataset [23]. For MPB type 2, the analysis revealed significant enrichment in four (digestive, musculoskeletal/connective, pancreas, and “other”) out of nine major categories, of which three categories showed significance in both the multi-tissue gene expression and multi-tissue chromatin modification datasets (coefficient P-value < 0.05) (Fig. 2D). The results for MPB type 3 showed significant enrichment in four major categories (adipose, digestive, musculoskeletal/connective, and “other”; coefficient P-value < 0.05) (Fig. 2E). These four categories were also significantly associated with MPB type 3 using the multi-tissue gene expression dataset. MPB type 4 showed significant enrichment in the adipose, blood/immune, digestive, musculoskeletal/connective, and “other” categories, three of which showed significance in both the multi-tissue gene expression and multi-tissue chromatin modification datasets (coefficient P-value < 0.05) (Fig. 2F). The specific tissue categories and statistics for MPB type 2, type 3, and type 4 are presented in Supplementary Data S1 and S2. Finally, we selected five, six, and five tissue eQTL panels from GTEx, based on their significant enrichment in both the multi-tissue gene expression and multi-tissue chromatin modification datasets (Table 1). These selected tissue eQTL panels were used for subsequent TWAS analyses.

Results of tissue prioritization using the multi-tissue gene expression and chromatin modification datasets. Scatter plots showing the tissue prioritization of MPB type 2, 3, and 4 using both multi-tissue gene expression dataset (A–C) and multi-tissue chromatin modification dataset (D–F). These tissue prioritization results were categorized into nine major groups and are plotted on the X-axis (scarlet: adipose; mustard: blood/immune; yellow green: cardiovascular; green: central nervous system; blue green: digestive; turquoise: liver; blue: musculoskeletal/connective; pink: pancreas; and lilac: “other”). The Y-axis represents –log(P-value) and the dashed lines indicate the significant thresholds (coefficient P-value < 0.05). Any tissues above the threshold are considered significant
The transcriptome-wide association study to identify associations with MPB
To investigate the susceptibility genes for MPB, we conducted TWAS using the FUSION method, which identifies associations between GWAS phenotype and gene expressions. We used three GWAS summary statistics and relevant tissue eQTL panels from the GTEx selected based on tissue-specific enrichment analysis (Table 1 and Supplementary Table S1). Among a total of associations, 89, 156, and 331 associations in 20, 25, and 101 loci were significantly associated with MPB type 2, type 3, and type 4, respectively (Bonferroni-corrected P-value < 0.05) (Fig. 3A, Supplementary Fig. 1, and Supplementary Data S3). The significant associations can be divided into positive associations and negative associations based on the direction of the z-score, indicating that increased/decreased expression leads to increased/decreased risk of MPB. In MPB type 2, there were 18 positive associations and 71 negative associations. In case of MPB type 3, it had 122 positive associations and 34 negative associations, while MPB type 4 showed 182 positive associations and 149 negative associations. The number of significant associations according to tissue panels is shown in Fig. 3B. For MPB type 2 and type 3, the sun-exposed and not-sun-exposed skin panels had the largest number of associations. In the case of MPB type 4, subcutaneous adipose tissue had the largest number of associations, surpassing both skin tissue panels (Fig. 3B). Among the results, we found that 7, 10, 16 genes were significant in all tissue panels (Supplementary Fig. 2D–F). We then examined the Chi-squared TWAS z-scores for each tissue and observed that the tissue panels within individual types of MPB had similarly grouped means of effect sizes (type 2: 5.39 to 5.84, type 3: 7.70 to 8.47, and type 4: 8.57 to 9.21) (Supplementary Fig. 2A–C) [41]. This indicates that the associations of MPB may evenly affect gene expression in the tissue panels.
To examine the tissue-specific or tissue-shared genetic effects in TWAS associations at the pathway level, we performed the functional annotation for each tissue panel using GSEA. We found that all the 79, 111, and 116 pathways that were statistically significant were enriched in multiple tissue panels (Supplementary Fig. 3 and Supplementary Data S4). The majority of tissue panels showed significant enrichments in pathways previously implicated in MPB pathogenesis, including apoptosis, signaling pathway, and immune response (Supplementary Table S2) [42,43,44,45]. This suggests that the cumulative functional characteristics of TWAS associations for MPB are consistent across tissue panels and have shared effects across tissues.
Although TWAS using GTEx eQTL panels can identify associations for each tissue, it does not account for all intra-individual correlations found in multiple contexts [22]. To analyze the tissue-shared effects of MPB-associated genetic variants, we performed additional TWAS analyses using cross-tissue panels called CONTENT. Since there was no cultured fibroblast panel in CONTENT, we conducted the analysis using the remaining panels. After Bonferroni correction, we found 27, 56, and 149 associations in 6, 10, and 33 loci that were significantly associated with MPB type 2, type 3, and type 4. Among these associations, 12, 17, and 71 associations were not found in the results using GTEx panels (Bonferroni-corrected P-value < 0.05) (Supplementary Data S5, Fig. 3A and G-I). The majority of TWAS associations for GTEx and CONTENT were located on chromosome 17 that is known to be related to various human genetic diseases, including cancers, DNA damage response, and MPB (Supplementary Fig. 1) [46, 47]. Detailed results on the number of associations for each tissue panel are presented in Fig. 3C. The number of significant associations across all tissue panels was two, five, and six (Supplementary Fig. 4D–F). We also examined Pearson’s correlation between the results from the GTEx and CONTENT panels, which showed moderate to high levels of consistency with minor differences (0.62 to 0.75 of Pearson’s R). These results indicate that our analysis accounted for both tissue-specific and tissue-shared features of MPB (Supplementary Fig. 4A–C).
To filter out LD-contaminated associations and ensure rigorousness, we additionally conducted the COLOC analysis. The COLOC analysis evaluates the consistency between GWAS signals and eQTL to determine if the signals are driven by a shared causal variant by calculating PPs for hypotheses of colocalized patterns between GWAS and eQTL signals. Out of the significant associations identified in the TWAS analyses, 52, 75, and 144 associations met the COLOC threshold (PP3 + PP4 > 0.8 and PP4 / PP3 > 2) (Fig. 3D–I and Supplementary Table S3).

Results of TWAS and COLOC analyses.(A) A bar graph displaying the number of associations obtained using TWAS (GTEx and CONTENT) and COLOC, which passed the threshold for each analysis. The results are represented by different colors: green (type 2), orange (type 3), and blue (type 4). (B) Bar graphs showing the results of TWAS using individual tissue panels of GTEx. (C) Bar graphs displaying the results of TWAS using individual tissue panels of CONTENT. The bars are color-coded according to MPB types. Ternary plots showing TWAS and COLOC results for (D) MPB type 2, (E) type 3, and (F) type 4, respectively. PP0 + PP1 + PP2 indicates underpowered signals; PP3 indicates independent variants between phenotype and gene expression; and PP4 indicates colocalized variants where the causal SNP is associated with both phenotype and gene expression. Non-significant TWAS associations are represented with gray points, while significant associations are marked in blue, of which with a high posterior probability of colocalized signals are highlighted in red. Venn diagrams represent the number of associations from GTEx, CONTENT, and COLOC for (G) MPB type 2, (H) type 3, and (I) type 4, respectively. Genes used for further analysis are indicated in bold
The conditional and joint analysis to identify conditionally independent genes
To thoroughly eliminate any remaining potential LD contaminations, the conditional and joint analysis were performed to determine the conditional or joint associations of significant levels [21]. We tested the joint significance of 52, 75, and 144 associations by conditioning on the GWAS signals and identified 13, 14, and 82 jointly significant associations in 9, 8, and 44 loci for MPB type 2, type 3, and type 4, respectively (joint P-value < 0.05) (Fig. 4 and Supplementary Table S4).
There were 10, 11, and 54 genes found in jointly significant associations, which were denoted as MPB signatures below, containing one, four, and nine non-coding RNAs, as well as 9, 7, and 45 protein-coding genes. Especially, among the protein coding genes, we identified two, two, and four novel genes that had not been recognized as MPB risk genes in previous studies and gene databases: cluster of differentiation 59 (CD59) and zinc finger DHHC-type palmitoyltransferase 5 (ZDHHC5) in MPB type 2; chromodomain helicase DNA binding protein 6 (CHD6) and zic family member 2 (ZIC2) in MPB type 3; and ADAM metallopeptidase with thrombospondin type 1 motif 18 (ADAMTS18), ATPase H + transporting V0 subunit D1 (ATP6V0D1), transmembrane protein 178B (TMEM178B), and ZDHHC5 in MPB type 4 (Table 2).

Results of the conditional and joint analysis. Dot plots showing joint genes that remained significant after conditioning in (A) MPB type 2, (B) type 3, and (C) type 4. The X-axis represents –log(joint P-value) and the dashed line indicates the significance threshold (joint P-value < 0.05). The dot color corresponds to the tissue panels (scarlet: subcutaneous adipose; mustard: visceral adipose; yellow green: cultured fibroblasts; green: sigmoid colon; light blue: transverse colon; blue: not-sun-exposed skin; pink: sun-exposed skin; and lilac: whole blood)
Phenome-wide association study of MPB signatures to identify potential pleiotropic effects
To explore the potential pleiotropic effects of our MPB signatures and identify shared genetic factors with other diseases or traits, we conducted a PheWAS analysis using GWAS ATLAS, which can reveal traits associated with MPB-signatures. We identified 223, 244, and 490 traits that were categorized into 19, 19, and 25 domains, respectively, under the Bonferroni-corrected significance threshold (Fig. 5A–C and Supplementary Data S6). The psychiatric domain showed the highest number of traits associated with MPB type 2 and type 3, while the metabolic domain had the highest number of traits in MPB type 4. The analysis revealed that variants from MPB signatures were highly enriched in five domains across all types of MPB: activities, immunological, metabolic, neurological, and psychiatric domains. It is well known that these domains are associated with an increased risk of MPB [1, 44, 48,49,50,51].
Among the variants that had pleiotropic effects, there were 6, 6, and 33 variants associated with five domains for MPB type 2, type 3, and type 4, respectively (Fig. 5D–F). Among the six variants in MPB type 2, rs4277389, rs199535, and rs199451 showed a high number of associations with psychiatric and other domains, regulating genes such as microtubule-associated protein tau (MAPT), RP11-259G18.3, and KAT8 regulatory NSL complex subunit 1 (KANSL1), which are known to be involved in neuropsychiatric functions (Fig. 5D) [52,53,54]. In MPB type 3, rs8072451 and rs199456 were particularly associated with the psychiatric domain, regulating RP11-798G7.8 and RP11-259G18.3 (Fig. 5E). Among the 33 variants associated with MPB type 4, rs174574, which regulates fatty acid desaturase 1 (FADS1) known to play a crucial role in the metabolic response of fatty acids, showed the largest number of associations with the metabolism domain (Fig. 5F) [55, 56]. We sought that these variants may cause pleiotropic effects by regulating gene expression in multiple tissues. By examining the traits associated with these variants, the traits that may be related to MPB can be confirmed.

PheWAS results of SNP-trait associations. Bar graphs indicating the number of traits per each domain in (A) MPB type 2, (B) type 3, and (C) type 4. Heatmaps showing the number of traits associated with each MPB signature variant for the top five domains in (D) MPB type 2, (E) type 3, and (F) type 4
Identification of potential drug candidates related to MPB signatures
In order to identify potential drug candidates for treating MPB, we analyzed the connectivity between drugs and MPB signatures using CLUE and Drugbank database. We manually excluded drugs known to cause hair loss. Among the connected drugs, curcumin was directly connected to type 2 MPB signatures (Fig. 6A). For MPB type 4, five drug molecules showed a direct connection with type 4 MPB signatures: alpha-linolenic-acid, genistein, diethylstilbestrol, afimoxifene, and succinic-acid (Fig. 6B). Previous studies on curcumin and succinic-acid have suggested their potential use in treating MPB, supporting the reliability of our potential drug candidates [57, 58]. Unfortunately, there were no drug molecules connected with type 3 MPB signatures.
Since we identified potential drug candidates based on the MPB signatures, it is important to assess the functional connectivity between MPB signatures and known MPB marker genes in order to establish credibility for our results. To address this issue, we compared the connectivity between MPB signatures and known MPB markers to the connectivity between random genes and known MPB markers (Supplementary Data S7). We calculated the interaction scores using protein interaction datasets from the STRING database. The results showed that the mean connectivity of type 2 and type 4 MPB signatures was significantly higher than that of random genes in nearly half of the cases in our simulation repeated up to 10,000 times (Supplementary Fig. 5). This indicates that MPB signatures may exhibit functional connectivity with known MPB markers, suggesting that drug candidates potentially affect the known MPB markers via MPB signatures.

Drug-gene interaction network using CLUE and Drugbank database. The drug-gene interactions for (A) MPB type 2 and (B) type 4 were identified using CLUE and Drugbank database and only drugs common to both results were visualized using Cytoscape. The solid line represents drug-gene interaction from CLUE, dashed line indicates drug-gene interaction from Drugbank database, and parallel lines indicate drug-gene interaction from both CLUE and Drugbank database
Discussion
MPB is a type of hair loss in men with specific patterns, which is caused by various genetic variants. Although previous studies have attempted to identify the MPB-associated genetic variants, the associations between genetically regulated gene expression and MPB have not been studied yet. To investigate gene expression changes influenced by genetics in MPB, we utilized TWAS, a method that calculates the expected changes in gene expression modulated by genetic variants and identifies associations between genes and the trait [16].
Since MPB is a complex disease that may involve multiple tissues in its development, it is crucial to prioritize tissue panels based on gene expression patterns. We conducted tissue-specific enrichment analysis using the LDSC-SEG method, utilizing multi-tissue gene expression and chromatin modification datasets. As a result, we selected five, six, and five tissue panels for MPB type 2, type 3, and type 4, respectively (Fig. 2; Table 1). These tissue panels consisted of adipose, colon, fibroblasts, skin, and whole blood. It has been reported that these tissues are associated with biological processes involved in the pathology of MPB, including inflammatory response and growth factors [59,60,61,62,63,64].
As indicated by our gene prioritization analyses, SNP-mediated gene expression associated with MPB exhibits both tissue-specific and tissue-shared aspects (Supplementary Fig. 3 and Fig. 3). By rigorously assessing the initial TWAS results throughout the conditional and joint analysis, we identified 10, 11, and 54 robust MPB signatures, of which two, two, and four protein-coding genes were novel, respectively (Fig. 4; Table 2). Among the type 2 MPB signatures, the CD59 gene plays a role in defending host cells from the complement system during inflammation response, which is considered one of the causes of MPB [65, 66]. ZDHHC5, significant in both MPB type 2 and type 4, is involved in various biological processes, including cell adhesion, neuronal activity, and innate immune response [67,68,69]. In particular, reduced hair follicle cell adhesion can cause hair loss by stimulating stem cell exhaustion [70]. In MPB type 3, both the CHD6 and ZIC2 genes are involved with transcriptional processes by activating or repressing transcription [71, 72]. Especially, the ZIC2 gene can inhibit Wnt/β-catenin signaling, a known cause of MPB [14, 73]. In MPB type 4, the gene product of ADAMTS18 is a member of the ADAMTS protein family with proteolytic function [74]. Proteolytic activity affects the process of hair shedding driven by enzymatic mechanisms, which can lead to baldness with insufficient replacement of hair [75, 76]. ATP6V0D1 is a gene encoding a component of vacuolar ATPase that utilizes ATP energy to produce a proton gradient [77]. Previous studies have shown that ATP-related processes play an important role in MPB and a previously known treatment, minoxidil, also acts on ATP processes [78,79,80]. TMEM178B is a gene that encodes a transmembrane protein and its function may be attributed to the diffusion or transport of steroid hormones including androgen [81]. Therefore, based on previous studies, these novel genes may be involved in mechanisms related to MPB.
Using these MPB signatures, we conducted PheWAS and drug repositioning as downstream analyses. The results of the PheWAS revealed that all types of MPB were commonly associated with activities, immunological, metabolic, neurological, and psychiatric domains (Fig. 5). The traits involved in the metabolic and psychiatric domains were previously reported in a GWAS study by Pirastu et al. [1] and those of activities, immunological, and neurological domains were also known to be associated with MPB [1, 2, 44, 48,49,50,51, 82,83,84]. One of the significantly associated traits among the activities domain was UV exposure that can cause hair damage and hair loss [82]. In the immunological domain, several immune-related cells play a significant role in hair follicle regulation and regeneration [44]. Diabetes, overweight, and high cholesterol levels were shown in the metabolism domain, which have been previously reported to be related to MPB [49, 83]. Parkinson’s disease, known as an MPB-related, has been reported to share genetic factors with MPB in previous studies, particularly in the neurological domain [2, 50]. In addition, traits related to drinking, smoking and stress in the psychiatric domain may affect the mechanisms of MPB such as hair follicle inflammation [51].
We analyzed the connections between genes and drug molecules to identify potential drug candidates for MPB (Fig. 6). We suggested curcumin as a potential drug candidate for MPB type 2, which have already been mentioned as potential molecules for treating baldness. Curcumin is a plant-derived substance that exhibits anti-inflammatory, anti-bacterial, and antioxidant properties. Previous studies using curcumin for the treatment of MPB have shown significant improvements in hair growth and hair loss without any side-effects [57, 85]. Additionally, curcumin analogs can block the activity of AR that plays a crucial role in MPB pathogenesis [86, 87]. For MPB type 4, five drug candidates were estimated to be effective in treating MPB. The lack of alpha-linolenic acid, an omega-3 fatty acid, can lead to scalp hemorrhagic folliculitis and maintaining adequate levels of alpha-linolenic acid can help strengthen the skin barrier [88,89,90,91]. Treatment with linolenic acid has been found to increase hair cell growth by antagonizing Wnt/β-catenin signaling, making it effective for treating baldness [92]. Estrogen-related drugs such as afimoxifene, diethylstilbestrol, and genistein have been reported to alleviate hair loss by controlling abnormal estrogen levels [93,94,95,96]. Succinic-acid, used in ATP generation reaction in the mitochondria, can promote hair growth by increasing ATP concentration in the scalp and regulating the inflammatory response [58, 97,98,99]. In MPB type 3, the MPB signatures were involved in cellular functions such as transcriptional processes and chromosomal remodeling, which may cause drug-induced cell death when targeted [100].
While our study has provided insights into the complex underlying mechanisms of MPB, there are still some limitations within the study design. First, there was no scalp skin panel in GTEx, so we had to use available skin panels obtained from the suprapubic or lower leg instead of scalp skin. However, it is important to note that the scalp belongs to the ectoderm-oriented epidermis, thus choosing skin panels was the best alternative for this study. Although MPB is associated with sex chromosomes, the GWAS datasets we used only include autosomal chromosomes. This was done intentionally to observe the effects of MPB on autosomal chromosomes only in this study. Additionally, since this study was performed in silico approach, our findings need to be validated with additional experimental studies. Especially, some drug candidates for treating MPB have not been studied yet, so the efficacy and delivery method of these drug candidates need to be further tested using in vivo and in vitro models. Despite these limitations, we believe that our study will contribute to the understanding of the genetic mechanisms of MPB and provide new insights for treatment of MPB.
Data availability
The GWAS summary statistics used in this study can be found in GWAS Atlas (https://atlas.ctglab.nl/) with the accession ID 3501, 3502, and 3503. Multi-tissue gene expression and chromatin modification datasets for LDSC-SEG can be downloaded in following github page (https://github.com/bulik/ldsc/wiki/Cell-type-specific-analyses). Tissue-specific eQTL panels from GTEx can be downloaded from FUSION (http://gusevlab.org/projects/fusion/). Tissue-shared eQTL panels from CONTETN can be found in following github page (https://github.com/cozygene/CONTENT). Functional gene sets can be retrieved from MsigDB (http://software.broadinstitute.org/gsea/msigdb). Previously reported MPB markers were searched on Open Targets Platform (https://platform.opentargets.org/). All data generated during this study are included in this published article and its supplementary files.
Abbreviations
- ADAMTS18 :
-
ADAM Metallopeptidase with Thrombospondin Type 1 Motif 18
- AR :
-
Androgen Receptor
- ATP6V0D1 :
-
ATPase H + Transporting V0 Subunit D1
- CD59 :
-
Cluster of Differentiation 59
- CHD6 :
-
Chromodomain Helicase DNA Binding Protein 6
- CLUE:
-
Connectivity Map and Library of Integrated Network-based Cellular Signatures Unified Environment
- CMAP:
-
Connectivity Map
- COLOC:
-
Colocalization
- CONTENT:
-
Context-specific Genetics
- ENCODE:
-
Encyclopedia of DNA Elements
- eQTL:
-
Expression Quantitative Trait Loci
- FADS1 :
-
Fatty Acid Desaturase 1
- FUSION:
-
Functional Summary-based Imputation
- GO:
-
Gene Ontology
- GSEA:
-
Gene Set Enrichment Analysis
- GTEx:
-
Genotype-tissue Expression
- GWAS:
-
Genome-wide Association Study
- HNS:
-
Hamilton-Norwood Scale
- KANSL1 :
-
KAT8 Regulatory NSL Complex Subunit 1
- LD:
-
Linkage Disequilibrium
- LDSC:
-
LD Score Regression
- LDSC-SEG:
-
Linkage Disequilibrium Score-specifically Expressed Genes
- LINCS:
-
Library of Integrated Network-based Cellular Signatures
- MAPT :
-
Microtubule-associated Protein tau
- MPB:
-
Male-pattern baldness
- NES:
-
Normalized Enrichment Scores
- PheWAS:
-
Phenome-wide Association Study
- PP:
-
Posterior Probabilities
- PPI:
-
Protein-protein Interaction
- SNPs:
-
Single Nucleotide Polymorphisms
- TMEM178B :
-
Transmembrane Protein 178B
- TWAS:
-
Transcriptome-wide Association Studies
- UKBB:
-
UK biobank
- ZDHHC5 :
-
Zinc Finger DHHC-type Palmitoyltransferase 5
- ZIC2 :
-
Zic Family Member 2
References
Pirastu N, Joshi PK, de Vries PS, Cornelis MC, McKeigue PM, Keum N, et al. GWAS for male-pattern baldness identifies 71 susceptibility loci explaining 38% of the risk. Nat Commun. 2017;8(1):1584.
Hagenaars SP, Hill WD, Harris SE, Ritchie SJ, Davies G, Liewald DC, et al. Genetic prediction of male pattern baldness. PLoS Genet. 2017;13(2):e1006594.
Ong J-S, Seviiri M, Dusingize JC, Wu Y, Han X, Shi J, et al. Uncovering the complex relationship between balding, testosterone and skin cancers in men. Nat Commun. 2023;14(1):5962.
Brown CJ, Goss SJ, Lubahn DB, Joseph DR, Wilson EM, French FS, et al. Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet. 1989;44(2):264–9.
Itami S, Sonoda T, Kurata S, Takayasu S. Mechanism of action of androgen in hair follicles. J Dermatol Sci. 1994;7:98–S103.
Ceruti JM, Leirós GJ, Balañá ME. Androgens and androgen receptor action in skin and hair follicles. Mol Cell Endocrinol. 2018;465:122–33.
Gupta M, Mysore V. Classifications of patterned hair loss: a review. J Cutan Aesthet Surg. 2016;9(1):3–12.
Hamilton JB. Patterned loss of hair in man; types and incidence. Ann N Y Acad Sci. 1951;53(3):708–28.
Norwood OT. Male pattern baldness: classification and incidence. South Med J. 1975;68(11):1359–65.
Guarrera M, Cardo P, Arrigo P, Rebora A. Reliability of hamilton-norwood classification. Int J Trichology. 2009;1(2):120–2.
Randall VA. Androgens and hair growth. Dermatol Ther. 2008;21(5):314–28.
Irwig MS, Kolukula S. Persistent sexual side effects of finasteride for male pattern hair loss. J Sex Med. 2011;8(6):1747–53.
Arif T, Dorjay K, Adil M, Sami M. Dutasteride in Androgenetic Alopecia: an update. Curr Clin Pharmacol. 2017;12(1):31–5.
Choi BY. Targeting Wnt/β-Catenin pathway for developing therapies for hair loss. Int J Mol Sci 2020, 21(14).
Kapoor M, Chao MJ, Johnson EC, Novikova G, Lai D, Meyers JL, Schulman J, Nurnberger JI, Porjesz B, Liu Y, et al. Multi-omics integration analysis identifies novel genes for alcoholism with potential overlap with neurodegenerative diseases. Nat Commun. 2021;12(1):5071.
Song J, Kim D, Lee S, Jung J, Joo JWJ, Jang W. Integrative transcriptome-wide analysis of atopic dermatitis for drug repositioning. Commun Biology. 2022;5(1):615.
Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci. 2017;20(8):1052–61.
Huckins LM, Dobbyn A, Ruderfer DM, Hoffman G, Wang W, Pardiñas AF, Rajagopal VM, Als TD, Girdhar HTN. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat Genet. 2019;51(4):659–74.
Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–5.
Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, Ermel R, Ruusalepp A, Quertermous T, Hao K, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51(4):592–9.
Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, Jansen R, de Geus EJ, Boomsma DI, Wright FA, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48(3):245–52.
Thompson M, Gordon MG, Lu A, Tandon A, Halperin E, Gusev A, Ye CJ, Balliu B, Zaitlen N. Multi-context genetic modeling of transcriptional regulation resolves novel disease loci. Nat Commun. 2022;13(1):5704.
Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh PR, Lareau C, Shoresh N, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50(4):621–9.
Chen Y, Hysi P, Maj C, Heilmann-Heimbach S, Spector TD, Liu F, Kayser M. Genetic prediction of male pattern baldness based on large independent datasets. Eur J Hum Genet. 2023;31(3):321–8.
Giles GG, Severi G, Sinclair R, English DR, McCredie MRE, Johnson W, Boyle P, Hopper JL. Androgenetic Alopecia and prostate Cancer: findings from an Australian case-control Study1. Cancer Epidemiol Biomarkers Prev. 2002;11(6):549–53.
Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–60.
Fehrmann RS, Karjalainen JM, Krajewska M, Westra HJ, Maloney D, Simeonov A, Pers TH, Hirschhorn JN, Jansen RC, Schultes EA, et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat Genet. 2015;47(2):115–25.
Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, Lui JC, Vedantam S, Gustafsson S, Esko T, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2015;6:5890.
Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28(10):1045–8.
The ENCODE. (ENCyclopedia of DNA elements) project. Science. 2004;306(5696):636–40.
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between Pairs of Genetic Association studies using Summary statistics. PLoS Genet. 2014;10(5):e1004383.
Li YI, Wong G, Humphrey J, Raj T. Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat Commun. 2019;10(1):994.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences 2005, 102(43):15545–15550.
Bastarache L, Denny JC, Roden DM. Phenome-wide Association studies. JAMA. 2022;327(1):75–6.
Safarova MS, Satterfield BA, Fan X, Austin EE, Ye Z, Bastarache L, Zheng N, Ritchie MD, Borthwick KM, Williams MS, et al. A phenome-wide association study to discover pleiotropic effects of PCSK9, APOB, and LDLR. npj Genomic Med. 2019;4(1):3.
Watanabe K, Stringer S, Frei O, Umićević Mirkov M, de Leeuw C, Polderman TJC, van der Sluis S, Andreassen OA, Neale BM, Posthuma D. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51(9):1339–48.
Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, Gould J, Davis JF, Tubelli AA, Asiedu JK, et al. A Next Generation Connectivity Map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171(6):1437–1452e1417.
Lamb J. The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer. 2007;7(1):54–60.
Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47(W1):W234–41.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, Eeles R, Freedman M, Haiman C, Pasaniuc B. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat Commun. 2018;9(1):4079.
Botchkareva NV, Ahluwalia G, Shander D. Apoptosis in the hair follicle. J Invest Dermatology. 2006;126(2):258–64.
Paus R, Peters EM, Eichmüller S, Botchkarev VA. Neural mechanisms of hair growth control. J Investig Dermatol Symp Proc. 1997;2(1):61–8.
Rahmani W, Sinha S, Biernaskie J. Immune modulation of hair follicle regeneration. Npj Regenerative Med. 2020;5(1):9.
Michel L, Reygagne P, Benech P, Jean-Louis F, Scalvino S, Ly Ka So S, Hamidou Z, Bianovici S, Pouch J, Ducos B, et al. Study of gene expression alteration in male androgenetic alopecia: evidence of predominant molecular signalling pathways. Br J Dermatol. 2017;177(5):1322–36.
Zody MC, Garber M, Adams DJ, Sharpe T, Harrow J, Lupski JR, Nicholson C, Searle SM, Wilming L, Young SK, et al. DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature. 2006;440(7087):1045–9.
Heilmann-Heimbach S, Hochfeld LM, Paus R, Nöthen MM. Hunting the genes in male-pattern alopecia: how important are they, how close are we and what will they tell us? Exp Dermatol. 2016;25(4):251–7.
Piérard-Franchimont C, Quatresooz P, Piérard GE. Effect of UV Radiation on Scalp and Hair Growth. In: Aging Hair Edited by Trüeb RM, Tobin DJ. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010: 113–121.
Braun N, Heinrich U. What Can Complex Dietary Supplements Do for Hair Loss and How Can It Be Validly Measured—A Review. In: Applied Sciences vol. 10; 2020.
Li R, Brockschmidt FF, Kiefer AK, Stefansson H, Nyholt DR, Song K, Vermeulen SH, Kanoni S, Glass D, Medland SE, et al. Six Novel susceptibility loci for early-onset Androgenetic Alopecia and their unexpected Association with Common diseases. PLoS Genet. 2012;8(5):e1002746.
Kavadya Y, Mysore V. Role of smoking in Androgenetic Alopecia: a systematic review. Int J Trichology. 2022;14(2):41–8.
Rittman T, Rubinov M, Vértes PE, Patel AX, Ginestet CE, Ghosh BCP, Barker RA, Spillantini MG, Bullmore ET, Rowe JB. Regional expression of the MAPT gene is associated with loss of hubs in brain networks and cognitive impairment in Parkinson disease and progressive supranuclear palsy. Neurobiol Aging. 2016;48:153–60.
Katrin L, Lewerissa EII, Anouk HAV, Michele G, Monica F, Teun MKG, Lynn D, Edda U, Astrid O, Chantal S et al. KANSL1 Deficiency causes neuronal dysfunction by oxidative stress-Induced Autophagy. bioRxiv 2020:2020.2008.2007.241257.
Duncan LE, Cooper BN, Shen H. Robust findings from 25 years of PTSD Genetics Research. Curr Psychiatry Rep. 2018;20(12):115.
Lankinen MA, de Mello VD, Meuronen T, Sallinen T, Ågren J, Virtanen KA, Laakso M, Pihlajamäki J, Schwab U. The FADS1 genotype modifies metabolic responses to the Linoleic Acid and Alpha-linolenic acid containing plant oils-genotype based Randomized Trial FADSDIET2. Mol Nutr Food Res. 2021;65(7):e2001004.
He Z, Zhang R, Jiang F, Zhang H, Zhao A, Xu B, Jin L, Wang T, Jia W, Jia W, et al. FADS1-FADS2 genetic polymorphisms are associated with fatty acid metabolism through changes in DNA methylation and gene expression. Clin Epigenetics. 2018;10(1):113.
Pumthong G, Asawanonda P, Varothai S, Jariyasethavong V, Triwongwaranat D, Suthipinittharm P, Ingkaninan K, Leelapornpisit P, Waranuch N. Curcuma Aeruginosa, a novel botanically derived 5α-reductase inhibitor in the treatment of male-pattern baldness: a multicenter, randomized, double-blind, placebo-controlled study. J Dermatolog Treat. 2012;23(5):385–92.
Adachi K, Yokoyama D, Tamai H, Sadai M, Oba K. Effect of the glyceride of pentadecanoic acid on energy metabolism in hair follicles. Int J Cosmet Sci. 1993;15(3):125–31.
Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV Jr., Sheppard D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol. 1996;133(4):921–8.
Nepal S, Venkataram A, Mysore V. The role of adipose tissue in hair regeneration: a potential Tool for Management? J Cutan Aesthet Surg. 2021;14(3):295–304.
Liu Y, Wang H, Wang J. Exosomes as a novel pathway for regulating development and diseases of the skin. Biomed Rep. 2018;8(3):207–14.
Heilmann-Heimbach S, Herold C, Hochfeld LM, Hillmer AM, Nyholt DR, Hecker J, Javed A, Chew EGY, Pechlivanis S, Drichel D, et al. Meta-analysis identifies novel risk loci and yields systematic insights into the biology of male-pattern baldness. Nat Commun. 2017;8(1):14694.
Keum N, Cao Y, Lee DH, Park SM, Rosner B, Fuchs CS, Wu K, Giovannucci EL. Male pattern baldness and risk of colorectal neoplasia. Br J Cancer. 2016;114(1):110–7.
Tsuboi R, Yamazaki M, Matsuda Y, Uchida K, Ueki R, Ogawa H. Antisense oligonucleotide targeting fibroblast growth factor receptor (FGFR)-1 stimulates cellular activity of hair follicles in an in vitro organ culture system. Int J Dermatol. 2007;46(3):259–63.
Peyravian N, Deo S, Daunert S, Jimenez JJ. The inflammatory aspect of male and female pattern hair loss. J Inflamm Res. 2020;13:879–81.
Sampaziotis F, Kokotas S, Gorgoulis VG. P53 possibly upregulates the expression of CD58 (LFA-3) and CD59 (MIRL). Med Hypotheses. 2002;58(2):136–40.
Woodley KT, Collins MO. S-acylated Golga7b stabilises DHHC5 at the plasma membrane to regulate cell adhesion. EMBO Rep. 2019;20(10):e47472.
Brigidi GS, Santyr B, Shimell J, Jovellar B, Bamji SX. Activity-regulated trafficking of the palmitoyl-acyl transferase DHHC5. Nat Commun. 2015;6:8200.
Lu Y, Zheng Y, Coyaud É, Zhang C, Selvabaskaran A, Yu Y, Xu Z, Weng X, Chen JS, Meng Y, et al. Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science. 2019;366(6464):460–7.
Zhang C, Wang D, Wang J, Wang L, Qiu W, Kume T, Dowell R, Yi R. Escape of hair follicle stem cells causes stem cell exhaustion during aging. Nat Aging. 2021;1(10):889–903.
Manning BJ, Yusufzai T. The ATP-dependent chromatin remodeling enzymes CHD6, CHD7, and CHD8 exhibit distinct nucleosome binding and remodeling activities. J Biol Chem. 2017;292(28):11927–36.
Lv Z, Qi L, Hu X, Mo M, Jiang H, Fan B, Li Y. Zic Family Member 2 (ZIC2): a potential diagnostic and prognostic biomarker for Pan-cancer. Front Mol Biosci 2021, 8.
Pourebrahim R, Houtmeyers R, Ghogomu S, Janssens S, Thelie A, Tran HT, Langenberg T, Vleminckx K, Bellefroid E, Cassiman JJ, et al. Transcription factor Zic2 inhibits Wnt/β-catenin protein signaling. J Biol Chem. 2011;286(43):37732–40.
Wei X, Prickett TD, Viloria CG, Molinolo A, Lin JC, Cardenas-Navia I, Cruz P, Rosenberg SA, Davies MA, Gershenwald JE, et al. Mutational and functional analysis reveals ADAMTS18 metalloproteinase as a Novel driver in Melanoma. Mol Cancer Res. 2010;8(11):1513–25.
Bhogal RK, Mouser PE, Higgins CA, Turner GA. Protease activity, localization and inhibition in the human hair follicle. Int J Cosmet Sci. 2014;36(1):46–53.
Higgins CA, Westgate GE, Jahoda CA. Modulation in proteolytic activity is identified as a hallmark of exogen by transcriptional profiling of hair follicles. J Invest Dermatol. 2011;131(12):2349–57.
Futai M, Sun-Wada GH, Wada Y, Matsumoto N, Nakanishi-Matsui M. Vacuolar-type ATPase: a proton pump to lysosomal trafficking. Proc Jpn Acad Ser B Phys Biol Sci. 2019;95(6):261–77.
Haslam IS, El-Chami C, Faruqi H, Shahmalak A, O’Neill CA, Paus R. Differential expression and functionality of ATP-binding cassette transporters in the human hair follicle. Br J Dermatol. 2015;172(6):1562–72.
Mikhalchik EV, Morozova OV, Tsimbalenko TV, Kharaeva ZF, Balabushevich NG, Lipatova VA, Gadzhigoroeva AG. Analysis of cytokines and ATP in plucked hair follicles. Bull Exp Biol Med. 2021;170(3):299–302.
Shorter K, Farjo NP, Picksley SM, Randall VA. Human hair follicles contain two forms of ATP-sensitive potassium channels, only one of which is sensitive to minoxidil. Faseb j. 2008;22(6):1725–36.
Trüeb RM. Understanding pattern hair loss-hair Biology impacted by genes, androgens, prostaglandins and epigenetic factors. Indian J Plast Surg. 2021;54(4):385–92.
Sebetić K, Sjerobabski Masnec I, Cavka V, Biljan D, Krolo I. UV damage of the hair. Coll Antropol. 2008;32(Suppl 2):163–5.
Sadgrove NJ. The ‘bald’ phenotype (androgenetic alopecia) is caused by the high glycaemic, high cholesterol and low mineral ‘western diet’. Trends Food Sci Technol. 2021;116:1170–8.
Sawant N, Chikhalkar S, Mehta V, Ravi M, Madke B, Khopkar U. Androgenetic Alopecia: quality-of-life and Associated Lifestyle patterns. Int J Trichology. 2010;2(2):81–5.
Vaughn AR, Branum A, Sivamani RK. Effects of Turmeric (Curcuma longa) on skin health: a systematic review of the clinical evidence. Phytother Res. 2016;30(8):1243–64.
Ellis JA, Stebbing M, Harrap SB. Polymorphism of the androgen receptor gene is associated with male pattern baldness. J Invest Dermatol. 2001;116(3):452–5.
Shi Q, Shih CC, Lee KH. Novel anti-prostate cancer curcumin analogues that enhance androgen receptor degradation activity. Anticancer Agents Med Chem. 2009;9(8):904–12.
Naghshi S, Aune D, Beyene J, Mobarak S, Asadi M, Sadeghi O. Dietary intake and biomarkers of alpha linolenic acid and risk of all cause, cardiovascular, and cancer mortality: systematic review and dose-response meta-analysis of cohort studies. BMJ. 2021;375:n2213.
Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis. 2006;189(1):19–30.
Bjerve KS. n-3 fatty acid deficiency in man. J Intern Med Suppl. 1989;731:171–5.
Simard M, Julien P, Fradette J, Pouliot R. Modulation of the lipid Profile of reconstructed skin substitutes after essential fatty acid supplementation affects testosterone permeability. Cells 2019, 8(10).
Ryu HS, Jeong J, Lee CM, Lee KS, Lee JN, Park SM, Lee YM. Activation of Hair Cell Growth factors by Linoleic Acid in Malva verticillata seed. Molecules 2021, 26(8).
Ohnemus U, Uenalan M, Inzunza J, Gustafsson J-A, Paus R. The hair follicle as an Estrogen Target and source. Endocr Rev. 2006;27(6):677–706.
Duchnik E, Kruk J, Baranowska-Bosiacka I, Pilutin A, Maleszka R, Marchlewicz M. Effects of the soy isoflavones, genistein and daidzein, on male rats’ skin. Postepy Dermatol Alergol. 2019;36(6):760–6.
Hu H-m, Zhang S-b, Lei X-h, Deng Z-l, Guo W-x, Qiu Z-f, Liu S, Wang X-y, Zhang H. Duan E-k: Estrogen leads to reversible hair cycle retardation through inducing premature Catagen and maintaining Telogen. PLoS ONE. 2012;7(7):e40124.
Kische H, Arnold A, Gross S, Wallaschofski H, Völzke H, Nauck M, Haring R. Sex hormones and hair loss in men from the General Population of Northeastern Germany. JAMA Dermatol. 2017;153(9):935–7.
Cheng KK, Wang GY, Zeng J, Zhang JA. Improved succinate production by metabolic engineering. Biomed Res Int. 2013;2013:538790.
Mills E, O’Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol. 2014;24(5):313–20.
Grimolizzi F, Arranz L. Multiple faces of succinate beyond metabolism in blood. Haematologica. 2018;103(10):1586–92.
Hickman JA. Apoptosis induced by anticancer drugs. Cancer Metastasis Rev. 1992;11(2):121–39.
Acknowledgements
This work was a part of the fulfillment of the requirements for Eunyoung Choi’s M.S. degree.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. NRF-2021R1A2C1008804).
Author information
Authors and Affiliations
Contributions
E.C. and J.S. conceptualized and designed the study. E.C. collected the data. E.C analyzed the data. E.C., J.S., Y.L., and Y.J. conducted data interpretation and visualization. E.C. wrote the manuscript. W.J. edited and revised the manuscript. W.J. gave final approval of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Choi, E., Song, J., Lee, Y. et al. Prioritizing susceptibility genes for the prognosis of male-pattern baldness with transcriptome-wide association study. Hum Genomics 18, 34 (2024). https://doi.org/10.1186/s40246-024-00591-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-024-00591-y