
Abstract
Background
Comorbidities of coronavirus disease 2019 (COVID-19)/coronary heart disease (CHD) pose great threats to disease outcomes, yet little is known about their shared pathology. The study aimed to examine whether comorbidities of COVID-19/CHD involved shared genetic pathology, as well as to clarify the shared genetic variants predisposing risks common to COVID-19 severity and CHD risks.
Methods
By leveraging publicly available summary statistics, we assessed the genetically determined causality between COVID-19 and CHD with bidirectional Mendelian randomization. To further quantify the causality contributed by shared genetic variants, we interrogated their genetic correlation with the linkage disequilibrium score regression method. Bayesian colocalization analysis coupled with conditional/conjunctional false discovery rate analysis was applied to decipher the shared causal single nucleotide polymorphisms (SNPs).
Findings
Briefly, we observed that the incident CHD risks post COVID-19 infection were partially determined by shared genetic variants. The shared genetic variants contributed to the causality at a proportion of 0.18 (95% CI 0.18–0.19) to 0.23 (95% CI 0.23–0.24). The SNP (rs10490770) located near LZTFL1 suggested direct causality (SNPs → COVID-19 → CHD), and SNPs in ABO (rs579459, rs495828), ILRUN(rs2744961), and CACFD1(rs4962153, rs3094379) may simultaneously influence COVID-19 severity and CHD risks.
Interpretation
Five SNPs located near LZTFL1 (rs10490770), ABO (rs579459, rs495828), ILRUN (rs2744961), and CACFD1 (rs4962153, rs3094379) may simultaneously influence their risks. The current study suggested that there may be shared mechanisms predisposing to both COVID-19 severity and CHD risks. Genetic predisposition to COVID-19 is a causal risk factor for CHD, supporting that reducing the COVID-19 infection risk or alleviating COVID-19 severity among those with specific genotypes might reduce their subsequent CHD adverse outcomes. Meanwhile, the shared genetic variants identified may be of clinical implications for identifying the target population who are more vulnerable to adverse CHD outcomes post COVID-19 and may also advance treatments of ‘Long COVID-19.’
Similar content being viewed by others

Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for coronavirus disease 2019 (COVID-19) and the current global pandemic. It has soon gone virus across the world, affecting more than 200 countries/territories [1]. To date, the world has registered more than 24 million individuals contaminated, with more than 5 million deaths [2]. While the disease has mild effects in most individuals, severe COVID-19 is more likely to be observed in the those with comorbidities such as cardiovascular diseases [3]. Additionally, why certain populations are at a higher risk of adverse CHD outcomes post COVID-19 infection is still unclear [4].
Accumulating evidence revealed a bidirectional relationship between coronary heart disease (CHD) and COVID-19 [5,6,7,8], yet consensus has not been achieved regarding their causality. Patients of cardiovascular diseases are at higher risk of severe COVID-19 and death [3, 9,10,11]. Meanwhile, CHD complications are observed in patients recovering from COVID-19. However, little is known as to whether COVID-19 infections causally induce CHD that were not in existence prior to the infection, or vice versa [8, 12]. From an ethical perspective, causal inference with a randomization clinical trial (RCT) is almost infeasible, as it is unethical to leave patients with one disease untreated with the aim of observing the occurrence of another disease. Therefore, it is anticipated to assess the causality between COVID-19 and CHD with an alternative method, for example, bidirectional Mendelian randomization (MR) [13]. Through leverage of randomly allocated genetic variants, bidirectional MR is expected to overcome the issues arising from ethical perspectives, as well as the confounders that hinder causal inference from observational studies [14, 15].
Observational studies indicated that COVID-19 and CHD may share common genetic variants [16, 17]. Suggestively, several genome-wide association studies (GWASs) identified certain SNPs responsible for COVID-19 susceptibility, including ACE2 and ABO [19, 20], which were also found to be associated with CHD risks [18,19,20]. However, limited studies to date provided with a comprehensive picture of where these shared genetic variants lied in a genome-wide scale. It is thus anticipated to advance the current knowledge of their underlying mechanism by systematically locating these shared genetic variants.
The current study aimed to assess the causality between COVID-19 and CHD, as well as to clarify their shared genetic SNPs.
Methods
Study pipeline and data sources
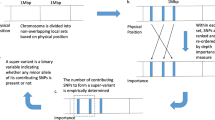
Figure 1 depicts the study pipeline. First, causality and its direction were assessed with a bidirectional MR. Second, to quantify the contribution from the shared genetic variants, we utilized the linkage disequilibrium score regression (LDSC method. Third, we applied the Bayesian colocalization (COLOC) and conditional/conjunctional false discovery rate (cond/conj FDR) to locate shared causal SNPs. For distinguishing SNPs of vertical pleiotropy (SNP → Trait1 → Trait2) with horizontal pleiotropy (SNP simultaneously influence Trait1&2), we searched biological pathway databases to examine whether SNPs were involved in multiple pathways [30].

The study pipeline to investigate putative COVID-19-mediated causal pathways to CHD. * X and Y in blue circles are nonspecific designations for either COVID-19 or CHD, depending on previous results from bidirectional Mendelian randomization. G in green circles are putative causal SNPs (either of vertical pleiotropy or horizontal pleiotropy) to be identified
We leveraged publicly available GWAS summary statistics of COVID-19 and CHD to perform the analyses. A detailed description of the GWAS profiles was presented in Additional file 1: Table S1. Specifically, the GWAS summary statistics of COVID-19 infections were drawn from the COVID-19 Host Genetic Initiative (COVID-19 HG) (https://www.covid19hg.org/results/r5/). The COVID-19 Host Genetics Initiative is an international collaboration aimed at uncovering the genetic determinants of COVID-19 susceptibility and severity. To achieve this goal, researchers collected individual-level clinical and genetic data and conducted individual GWAS. All participating cohorts imputed genotypes to the Haplotype Reference Consortium, 1000 Genomes, or TOPMed reference panels. Each cohort categorized ancestry through self-report or genetic data and performed single-variant association testing while adjusting for covariates such as age, age2, sex, age × sex, genetic ancestry principal components, and study-specific factors. Its round 5 release provided summary statistics of COVID-19 genetic susceptibility of differentiated severity among Europeans. COVID-19 genetic susceptibility was investigated in 3 different datasets, namely, COVID-19_A defined as the very severe respiratory confirmed COVID-19 versus the general population (5101 cases/1,383,241 controls), COVID-19_B defined as the hospitalized versus the general population (9986 cases/1,877,672 controls), and COVID-19_C defined as a positive COVID-19 diagnosis versus the general population (38,984 cases/1,644,784 controls). Normally, these 3 datasets assessed the genetic susceptibility to COVID-19 of differentiated severity [21].
The CHD GWAS summary statistic was acquired from the CARDIoGRAMplusC4D consortium (http://www.cardiogramplusc4d.org/), contributed by CARDIoGRAMplusC4D investigators, which currently possesses the largest publicly available CHD GWAS meta-analysis results for Europeans [22]. The CHD dataset (CARDIoGRAM GWAS) comprised 22,233 CHD cases and 64,762 controls from 22 case–control studies [23]. Case status was defined by an inclusive CHD diagnosis (e.g., myocardial infarction, acute coronary syndrome, chronic stable angina, or coronary stenosis > 50%). Each study was analyzed separately under additive logistic regression, and the results were merged by meta-analysis using an inverse-variance weighted fixed-effects model [23]. We applied the LiftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver) to flip CHD GWAS build to GRCh37, so as to align with the COVID-19 coordinate.
Bidirectional mendelian randomization
We applied bidirectional MR to test whether COVID-19 causally affected CHD risks or vice versa, where either COVID-19-associated SNPs or CHD-associated SNPs (P < 5 × 10–5) were used as instrumental variables (IVs). We further clumped the IVs at a linkage disequilibrium threshold of 0.2 (r2 < 0.2) within a distance of 5000 kb. Finally, the SNPs for being palindromic with intermediate allele frequencies were removed when harmonized with the variants.
Of the four MR methods applied in this study, the inverse-variance-weighted (IVW) method was performed in the primary analysis [24]. Additional sensitivity analyses, namely, the MR-Egger, the weighted mode and the simple mode method, were performed to test for robustness of the causality. The MR-Egger method was applied to control potential bias in cases of invalid or weak IVs, which was reported to be capable of controlling the pleiotropic effect of genetic variants that is not mediated via exposure [25]. Similarly, the weighted mode and simple mode were both applied to reduce invalid or weak instrument bias [26, 27]. To account for the potential influence of BMI and T2D on the association between COVID-19 and CHD, we conducted multivariable mendelian randomization analyses using GWAS summary statistics from the IEU open GWAS project (https://gwas.mrcieu.ac.uk/datasets/). GWAS summary statistics for BMI were derived from the UK Biobank (N = 461,460, GWAS id: ukb-b-19953), and those for T2D were obtained from another European study with a sample size of N = 655,666 (GWAS id: ebi-a-GCST006867).
Linkage disequilibrium score regression
To quantify the contribution from shared genetic variants of COVID-19 and CHD, we used the LDSC method (https://github.com/bulik/ldsc) [28, 29]. The genetic overlap laid basis for further locating causal SNPs. To examine the genetic overlap across the whole genome, we considered the effects of all SNPs, with uncorrected P values [30]. European ancestry information from the 1000 Genomes Project was used as the linkage disequilibrium reference panel, aligning with the European origin of GWAS samples.
Colocalization and false discovery rate
We performed the following analyses to locate shared causal SNPs in a genome-wide scale, and mutually verify IVs in previous MR.
First, we applied Bayesian colocalization (COLOC) analyses to identify SNPs of pleiotropy. Briefly, colocalization analysis examines whether associations detected by MR methods are driven by the same causal variants [31]. And the shared causal variants identified from COLOC is assumed of ‘causality’ exempted from confounding like linkage disequilibrium, where IVs in MR may confer [30]. Given that COLOC itself could not distinguish vertical or horizontal pleiotropy [30], we searched biological pathway databases (KEGG https://www.kegg.jp) to examine whether the SNPs identified were involved in multiple biological pathways [30]. Specifically, if genes were mapped to one certain biological pathway instead of multiple pathways, the SNPs identified by COLOC are suspected to function via vertical pleiotropy and are thus more likely to be valid IVs in MR. The candidate SNPs identified from COLOC were verified with IVs used in previous MR to finally prioritize SNPs of vertical pleiotropy. In essence, the Bayesian approach COLOC assumes that (1) in each test region, there exists at most one causal SNP for either trait; (2) the probability that a SNP is causal is independent of the probability that any other SNP in the genome is causal; and (3) all causal SNPs are genotyped or imputed and included in the analysis. According to these assumptions, there are five mutually exclusive hypotheses for each test region: (1) there is no causal SNP for either trait (H0); (2) there is one causal SNP for trait 1 only (H1); (3) there is one causal SNP for trait 2 only (H2); (4) there are two distinct causal SNPs, one for each trait (H3); and (5) there is a causal SNP common to both traits (H4) [31]. Our primary interest lied in the last hypothesis, H4 colocalization. Support for each of the hypotheses was quantified by the posterior probability (PP), denoted by PP0, PP1, PP2, PP3 and PP4 accordingly. These PPs were calculated from the priors and the approximate Bayes factors. We set the prior probability of each SNP that is causal to either of the traits to 1 × 10−4 (i.e., one in 10,000 SNPs in the genome are causal to either trait) and causal to both traits to 1 × 10−6 (i.e., one in 100 SNPs in the genome causal to one trait are causal to both traits). We used the GWAS summary statistics of COVID-19 and CHD to approximate the Bayesian factors. Testing for colocalization was performed with the R package coloc (http://cran.r-project.org/web/packages/coloc).
To further identify SNPs of horizontal pleiotropy that were excluded by MR previously, and also to rigorously provide a genome-wide view of SNPs showing horizontal pleiotropy, we performed cond/conjFDR [32]. We denoted the conjFDR as FDRtrait1&trait2, which is defined as the posterior probability that a SNP is null for either phenotype or both simultaneously, given that its P values for associations with both phenotypes are as small as or smaller than the observed ones. A conservative estimate of conjFDR was given by the maximum between FDRtrait1|trait2 and FDRtrait2|trait1, which required that loci exceeded a conjFDR significance threshold for both phenotypes jointly. SNPs with a conjFDR value less than 0.05 were considered shared loci [32].
Role of the funding source
The study was supported by the Special Fund for Health Scientific Research in Public Welfare (Grant No. 201502006), the Key Project of Natural Science Funds of China (Grant No. 8123066), the National Natural Science Foundation of China (Grant No. 81872695), the Fujian Provincial Health Technology Project (Grant No. 2020CXB009), the Natural Science Foundation of Fujian Province, China (Grant No. 2021J01352) and the China Postdoctoral Science Foundation (Grant No. BX2021021). These funding sources supported the study as a not-for-profit endeavor.
Results
The causal effect of COVID-19 on CHD risks
The bidirectional MR suggested a one-way causal relationship of COVID-19 with CHD (COVID-19 → CHD), with higher COVID-19 susceptibility increasing CHD risks. Each per unit increase in liability to very severe COVID-19 corresponded to an increased risk of CHD (MRIVW: OR = 1.01, 95% CI 1.01–1.02, P = 2.2E−14). Sensitivity analyses generated consistent effect estimates (Table 1). MR Egger intercept tests did not detect any horizontal pleiotropy outliers. Generally, the four MR methods yielded rather consistent results. However, we found no evidence of reversed causal effects from CHD to COVID-19 (Additional file 1: Table S1).
The genetic correlation between COVID-19 and CHD
As listed in Table 2, the correlation attributable to shared genetic variants accounted for 0.18 (95% CI 0.18–0.19) to 0.23 (95% CI 0.23–0.24). Generally, phenotypic correction of more severe COVID-19 and CHD can be explained by shared genetic variants to a larger extent.
Bayesian colocalization analysis
We first included 2,508,363 SNPs that were associated with either COVID-19 or CHD We mapped all the overlapped SNPs between COVID19/CHD to the genomic regions with SNP to Gene (S2G) [33], which could be available at https://alkesgroup.broadinstitute.org/cS2G/. With Bonferroni correction for the multiple testing in inflation of Type I error, the significance threshold was set as 0.05/890 = 5.0E−05. Each of the SNPs and their neighboring SNPs (distance within 200 kb) were then utilized to define a test region. After merging overlapping regions, we tested for colocalization in their respective unique regions. Among these unique test regions, one region on chromosome 3 near LZTFL1/LOC107986083 displayed suggestive causality shared by both COVID-19 and CHD, with a posterior PP4 exceeding 0.70 (Table 3). Within this region, the SNP Chr3:45859561 (rs10490770, nearest gene LZTFL1) exhibited the highest maximum PP4 and was considered the putative causal SNP. Additionally, rs17713054, also located in this region, demonstrated suggestive causality concerning the genetic correlation between COVID-19 and CHD. Notably, rs10490770 was previously identified as an IV in MR analyses. Furthermore, as indicated in the KEGG pathway analysis, LZTFL1 was not found to be involved in multiple pathways. Considering the LD relationship between rs10490770 with rs10490770, rs17713054 was not selected as an IV. Assuming that at most one causal SNP exists in the gene/region, the presence of two SNPs with high LD and high PP4 suggests that these two SNPs either both play a causal role in the outcome or are in strong LD with a single causal variant. Interestingly, both rs10490770 and rs17713054 exhibited associations with COVID-19 (P < 5 × 10−8) but not with CHD in their original GWAS (P > 0.05). Consequently, the SNPs identified in this study displayed vertical pleiotropy, impacting CHD solely through their influence on COVID-19 risk.
The cond/conjFDR analysis
To mutually verify the IV validity in MR, and to compensate for the inefficiency in identifying SNPs of horizontal pleiotropy in MR, we further provided a comprehensive, unselected map of shared loci between COVID-19 and CHD with a cond/conjFDR analysis (Fig. 2). With a predefined threshold at conjFDR < 0.05, we identified 5 distinct genetic loci shared between CHD and COVID-19 (Table 4). We observed that 2 SNPs mapped in the ABO gene (rs579459 Chr9:136154168 and rs495828 Chr9:136154867) showed consistent signals across varied COVID-19 severities, which implied the value of the ABO gene for jointly influencing CHD and COVID-19 (P < 5 × 10–4).

Conjunctional false discovery rate (conjFDR) Manhattan plots of conjunctional − log10(FDR) values
To observe increments of SNP enrichment for CHD as a function of the significance of COVID-19, a conditional QQ plot was presented (Additional file 1: Fig. S2). Gradual leftward curves indicated that the proportion of nonnull SNPs varies considerably across different levels of association with CHD, which supports the polygenic overlap between these phenotypes.
Discussion
In the current study, we observed a single-way causal effect of COVID-19 exerted on CHD. Shared genetic variants contributed to the causality, where rs10490770 in LZTFL1 suggested direct causality (SNPs → COVID-19 → CHD), and SNPs in ABO (rs579459, rs495828), ILRUN (rs2744961), and CACFD1(rs4962153, rs3094379) may simultaneously influence their risks.
To date, limited evidence is available in terms of the causality between COVID-19 and CHD. The current study indicated that the genetically determined risk of COVID-19 infection contributed to higher CHD complication risk. Several observational studies reported adverse CHD outcomes after COVID-19 infection [12, 34], which was consistent with our findings. A recent review also reported a considerable proportion of patients who recovered from COVID-19 continued to experience complications including CHD, even in the absence of a detectable viral infection [8]. This condition, which is often referred to as ‘post-acute COVID-19’ or ‘long COVID’, has been the major concern of clinical care for COVID-19 patients [35]. However, results of most observational studies might be confounded and/or influenced by reverse causality [36]. We thus attempted to infer causality with an alternative method, i.e., bidirectional MR [25,26,27]. For the reliability of causality observed in our study, we made multiple attempts to provide a rather robust estimate that was less likely to be false positive, for example, multiple sensitivity analyses, and IV validity check in an integrated framework. To be specific, the SNP (rs10490770) identified in colocalization analysis was suspected of vertical pleiotropy with its biological role validated from KEGG. It is suggested that LZTFL1, where rs10490770 was mapped, was involved in merely one pathway known to date, namely ‘BBSome-mediated cargo-targeting to cilium’ pathway. No additional evidence was shown regarding its direct association with CHD. Meanwhile, we noted that rs10490770 functioned exactly as IV, which strengthened our beliefs regarding its direct causality of SNP → COVID-19 → CHD. Regarding the relatively moderate effect size, the causality estimate was assumed to be attenuated compared to the true causal effect, for the limited number of genetic variants that overlapped between the COVID-19 HG meta-analysis and the datasets that were used for this study. Results should be interpreted with caution in terms of the genetically determined causality between the CHD incidence and COVID-19. In essence, the merit of MR limited its estimation of causality to the genetically determined risk of COVID-19 and CHD risks. Given that the genetic susceptibility of COVID-19 accounted for only a certain proportion of its total phenotypic variation, true causal estimates between these two traits were supposed to be much larger.
As the causality inferred previously may involve an interplay of both genetic and environmental factors, one cannot decide whether the causality inferred previously is contributed from their shared genetic variants. In this context, quantifying the genetic correlation can be thought as a prerequisite for subsequent identification of shared causal SNPs. In the current study, the genetic correlation was estimated at 0.18(COVID-19_C) to 0.23(COVID-19_A), suggesting an increasing trend between CHD and more severe COVID-19. However, it should be noted that the strength of their genetic correlation might be overestimated, as this analysis covers the entire genome. Still, it motivated us to further locate where these shared causal SNPs lied precisely in the genome. Generally, the genetic correlation is thought to be attributable to either vertical pleiotropy or horizontal pleiotropy [37]. Therefore, we applied COLOC coupled with cond/conj FDR to provide a comprehensive view of shared causal SNPs in a genome-wide scale. By integrating corresponding epidemiologic findings from populations with understanding of their biological functions, these SNPs identified may advance current knowledge of underlying mechanisms and may also facilitate clinical care of ‘Long COVID-19’ [35].
Commonly, COVID-19 and CHD are hypothesized to be linked by several biological mechanisms, including immune response, and endothelial dysfunction [21, 38, 39]. Of note, most SNPs identified in the current study were mapped in these pathways. Two SNPs (rs579459 and rs495828) in the ABO gene were assumed to be promising putative causal loci, as they showed consistent signals of pleiotropy when assessing CHD and COVID-19 of the three severity types. Recent studies showed that the ABO gene, located in 9q34.2, which determines blood type, may affect COVID-19 disease severity [40]. Several observational studies further reported a relationship between ABO blood groups and adverse CHD complications post COVID-19 [19, 20, 38]. In a recent GWAS meta-analysis, where investigators sampled 1980 patients with COVID-19-related respiratory failure and analyzed 8582968 SNPs, further cross-replicated the association of rs657152 at locus 9q34 with the COVID-19 severity [41]. In the current study, it is noticed that rs657152 was in linkage disequilibrium with rs579459 (D′ = 0.99), and they both were found to be expression qualitative trait loci (eQTLs) responsible for immune stimulation upon regulatory variant activity (http://pubs.broadinstitute.org/mammals/haploreg/haploreg.php). The activation of the immune response was assumed to be the key for both CHD risks and viral clearance [42]. On the one hand, both rs579459 and rs495828 were repetitively underscored to be associated with CHD risks, where immune response was suspected in the pathology [43,44,45]. On the other hand, both rs579459 and rs495828 functioned as eQTL [46] responsible for immune response activation and thus were hypothesized to influence the COVID-19 outcomes [47]. An alternative hypothesis is Renin-Angiotensin-System (RAS) unbalancing, where RAS-pathway genes, including rs495828 in ABO, was suspected predictive of CHD complications of COVID-19 [48]. Moreover, both rs579459 and rs495828 were found to be associated with Motifs change in Nkx2, where autoimmune mechanism underlying the acute respiratory distress syndrome in SARS-COV-2 was undergirded [47]. Considering Motifs play important regulatory roles, and may bind SARS-CoV-2 spike protein [49], rs579459 and rs495828 identified in the current study are worthy of further replication to fully clarify their roles.
For rs10490770 located near LZTFL1 in the 3p21.31 region, it was in high LD with rs17713054 (D′ = 1), which was identified in COVID-19 GWASs as conferring a twofold increased risk of respiratory failure [5, 41, 50, 51] and an over twofold increased risk of mortality for individuals under 60 years old [52]. Also, rs10490770 was in LD with rs11385942 (D′ = 0.98) which was cross-validated in a recent COVID-19 GWAS meta-analysis [41]. However, despite repetitive statistical significance reported in the 3p21.31 region, its specific role in COVID-19 infection remained unexplained [50, 51]. We coincidentally observed rs17713054 in this region colocalized between COVID-19 and CHD, which might offer insights for further uncovering the biological role in this region by simultaneously taking COVID-19 and its CHD complication into considerations. Recent studies reported that rs17713054-affected enhancer upregulates LZTFL1 [16, 17], which is currently known to be actively involved in the epithelial–mesenchymal transition in the viral response pathway of COVID-19 [50]. We further supplemented eQTL colocalization analysis in the 3p21.31 region (data not shown in the main text). We confirmed that rs10490770 colocalized with eQTL and was highly expressed in the lung tissue (Additional file 1: Figure S2). Meanwhile, no known evidence showed rs10490770 was directly associated with CHD, nor with its belonging pathway ‘BBSome-mediated cargo-targeting to cilium’ pathway. Evidence gathered thus far consistently supported our findings that rs10490770 function only through COVID-19 to CHD. Although statistical signals are not necessarily validated biological evidence, we provided preliminary hints for further studies to uncover the underlying molecular mechanism.
The current study has several clinical implications. First, this study suggested that genetic predisposition to COVID-19 is a causal risk factor for CHD, leading to the hypothesis that reducing the COVID-19 infection risk or alleviating COVID-19 severity among those with specific genotypes might reduce their subsequent CHD adverse outcomes. Initially recognized as a respiratory system disease, COVID-19 has been found to interact with and affect the cardiovascular system leading to myocardial damage and cardiac and endothelial dysfunction [53]. In fact, cardiac damage has been noted even without clinical features of respiratory disease [54], with new-onset cardiac dysfunction common in this subgroup [55, 56]. Specifically, the CHD incidence secondary to COVID-19 infection might be characterized with massive damage in the vascular endothelium and cardiac myocytes due to the systemic inflammatory response in severe COVID-19, which includes the release of high levels of cytokines (known as cytokine release syndrome) [57,58,59]. These patients with subsequent myocardial involvement could suffer from several potentially life-threatening symptoms [1]. Second, these SNPs identified may be of clinical implications for identifying the target population who are more vulnerable to adverse CHD outcomes post COVID-19 and may also advance treatments of ‘Long COVID-19’ [35].
This study also had limitations worthy of noting. First, the validity of the genetic instruments was not fully understood. It is possible that the genetic instruments may have an indirect effect on the outcome via a currently unknown pathway that does not involve the risk factor for interest. Nevertheless, we addressed this issue by adopting the MR-Egger intercept, although it cannot be ruled out unequivocally. Second, as GWAS summary datasets were extracted from Europeans, the generalizability of the study results was limited to Europeans only. Third, study participants included in the COVID-19 were not screened for CHD at baseline and vice versa. The presence of outcomes in the exposure dataset may bias the causal estimates in MR analyses. However, this is a general limitation of two-sample MR analyses and is inevitable without individual-level data. Third, we acknowledge the potential bias introduced by environmental factors, which could not be completely mitigated, despite our efforts to adjust for age and gender in the GWAS summary statistics and our multivariate Mendelian randomization analysis. However, our results indicate that our findings remain robust in the presence of some established environmental factors. Fourth, the methods we adopted that were built upon GWAS summary statistics, including the LDSC method, along with the Mendelian randomization and Bayesian colocalization, required larger sample sizes than methods that use individual genotypes to achieve equivalent standard error. Of note, our analyses were limited by the number of genetic variants that overlapped between the COVID-19 HG meta-analysis and the datasets that were used for this study. Thus, we could not test some genes that may be of importance. It is also possible that, with larger sample sizes, the genetic association of COVID-19 severity and CHD could become more significant, and confidence intervals would narrow around true estimates.
Conclusion
In summary, this study indicated the putative causality of COVID-19 genetic susceptibility on incident CHD. It underlined rs10490770 located near LZTFL1 and SNPs in ABO (rs579459, rs495828), ILRUN (rs2744961), and CACFD1 (rs4962153, rs3094379) may simultaneously influence their risks. Further studies are warranted to clarify their underlying mechanism.
Data sharing
Anonymized research data could be obtained the COVID-19 Host Genetic Initiative (COVID-19 HG) (https://www.covid19hg.org/results/r5/) and the CARDIoGRAMplusC4D consortium (http://www.cardiogramplusc4d.org/).
References
Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. 2020;17:543–58.
Hodgson SH, Mansatta K, Mallett G, Harris V, Emary KRW, Pollard AJ. What defines an efficacious COVID-19 vaccine? A review of the challenges assessing the clinical efficacy of vaccines against SARS-CoV-2. Lancet Infect Dis. 2021;21:e26-35.
Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054.
Weiss P, Murdoch DR. Clinical course and mortality risk of severe COVID-19. Lancet. 2020;395:1014–5.
Siddiqi HK, Libby P, Ridker PM. COVID-19: a vascular disease. Trends Cardiovasc Med. 2021;31:1–5.
Guzik TJ, Mohiddin SA, Dimarco A, et al. COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020;116:1666.
Azevedo RB, Botelho BG, de Hollanda JVG, et al. Covid-19 and the cardiovascular system: a comprehensive review. J Hum Hypertens. 2021;35:1.
Cenko E, Badimon L, Bugiardini R, et al. Cardiovascular disease and COVID-19: a consensus paper from the ESC Working Group on Coronary Pathophysiology & Microcirculation, ESC Working Group on Thrombosis and the Association for Acute CardioVascular Care (ACVC), in collaboration with the European Heart Rhythm Association (EHRA). Cardiovasc Res 2021; published online Sept 16. https://doi.org/10.1093/CVR/CVAB298.
Wei JF, Huang FY, Xiong TY, et al. Acute myocardial injury is common in patients with COVID-19 and impairs their prognosis. Heart. 2020;106:1154–9.
Shi S, Qin M, Shen B, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 2020;5:802–10.
Guo T, Fan Y, Chen M, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:811–8.
McGonagle D, Plein S, O’Donnell JS, Sharif K, Bridgewood C. Increased cardiovascular mortality in African Americans with COVID-19. Lancet Respir Med. 2020;8:649–51.
Burgess S, Davey Smith G, Davies NM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 2020. https://doi.org/10.12688/WELLCOMEOPENRES.15555.2.
Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5:1205–10.
Ponsford MJ, Gkatzionis A, Walker VM, et al. Cardiometabolic traits, sepsis, and severe COVID-19: a Mendelian randomization investigation. Circulation. 2020;142:1791.
Giudicessi JR, Roden DM, Wilde AAM, Ackerman MJ. Genetic susceptibility for COVID-19-associated sudden cardiac death in African Americans. Heart Rhythm. 2020;17:1487–92.
SeyedAlinaghi SA, Mehrtak M, MohsseniPour M, et al. Genetic susceptibility of COVID-19: a systematic review of current evidence. Eur J Med Res. 2021. https://doi.org/10.1186/s40001-021-00516-8.
Chirinos JA, Cohen JB, Zhao L, et al. Clinical and proteomic correlates of plasma ACE2 (angiotensin-converting enzyme 2) in human heart failure. Hypertension. 2020;76:1526–36.
Dahabreh IJ, Kitsios GD, Trikalinos TA, Kent DM. The complexity of ABO in coronary heart disease. Lancet. 2011;377:1493.
Reilly MP, Li M, He J, et al. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377:383–92.
D’Antonio M, Nguyen JP, Arthur TD, Matsui H, D’Antonio-Chronowska A, Frazer KA. SARS-CoV-2 susceptibility and COVID-19 disease severity are associated with genetic variants affecting gene expression in a variety of tissues. Cell Rep. 2021;37: 110020.
Nikpay M, Goel A, Won HH, et al. A comprehensive 1000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30.
Schunkert H, König IR, Kathiresan S, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–40.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65.
Bowden J, Smith GD, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14.
Hartwig FP, Smith GD, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–98.
Bulik-Sullivan B, Loh PR, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Ni G, Moser G, Ripke S, et al. Estimation of genetic correlation via linkage disequilibrium score regression and genomic restricted maximum likelihood. Am J Hum Genet. 2018;102:1185.
Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014. https://doi.org/10.1371/JOURNAL.PGEN.1004383.
Andreassen OA, Thompson WK, Schork AJ, et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet. 2013. https://doi.org/10.1371/JOURNAL.PGEN.1003455.
Gazal S, Weissbrod O, Hormozdiari F, et al. Combining SNP-to-gene linking strategies to identify disease genes and assess disease omnigenicity. Nat Genet. 2022;54:827.
Driggin E, Madhavan MV, Bikdeli B, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J Am Coll Cardiol. 2020;75:2352–71.
Sudre CH, Murray B, Varsavsky T, et al. Attributes and predictors of long COVID. Nat Med. 2021;27:626–31.
Burgess S, Foley CN, Zuber V. Inferring causal relationships between risk factors and outcomes from genome-wide association study data. Annu Rev Genom Hum Genet. 2018;19:303–27.
Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14:483–95.
Hernández Cordero AI, Li X, Milne S, et al. Multi-omics highlights ABO plasma protein as a causal risk factor for COVID-19. Hum Genet. 2021;140:969–79.
Wang L, Balmat TJ, Antonia AL, et al. An atlas connecting shared genetic architecture of human diseases and molecular phenotypes provides insight into COVID-19 susceptibility. Genome Med. 2021. https://doi.org/10.1186/S13073-021-00904-Z.
Lehrer S, Rheinstein PH. ABO blood groups, COVID-19 infection and mortality. Blood Cells Mol Dis. 2021. https://doi.org/10.1016/J.BCMD.2021.102571.
Ellinghaus D, Degenhardt F, Bujanda L, et al. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med. 2020;383:1522–34.
Tamayo-Velasco Á, Jiménez García MT, Sánchez Rodríguez A, Hijas Villaizan M, Carretero Gómez J, Miramontes-González JP. Association of blood group A with hospital comorbidity in patients infected by SARS-CoV-2. Med Clin. 2021. https://doi.org/10.1016/J.MEDCLI.2021.06.017.
Zhao C, Zhu P, Shen Q, Jin L. Prospective association of a genetic risk score with major adverse cardiovascular events in patients with coronary artery disease. Medicine. 2017. https://doi.org/10.1097/MD.0000000000009473.
Webb TR, Erdmann J, Stirrups KE, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69:823–36.
Winther-Larsen A, Christiansen MK, Larsen SB, et al. The ABO locus is associated with increased fibrin network formation in patients with stable coronary artery disease. Thromb Haemost. 2020;120:1246–56.
Ardlie KG, DeLuca DS, Segrè AV, et al. Human genomics. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60.
Morsy S, Morsy A. Epitope mimicry analysis of SARS-COV-2 surface proteins and human lung proteins. J Mol Graph Model. 2021. https://doi.org/10.1016/J.JMGM.2021.107836.
Gemmati D, Tisato V. Genetic hypothesis and pharmacogenetics side of renin-angiotensin-system in COVID-19. Genes (Basel). 2020;11:1–17.
Beaudoin CA, Hamaia SW, Huang CLH, Blundell TL, Jackson AP. Can the SARS-CoV-2 spike protein bind integrins independent of the RGD sequence? Front Cell Infect Microbiol. 2021. https://doi.org/10.3389/FCIMB.2021.765300.
Downes DJ, Cross AR, Hua P, et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat Genet. 2021;53:1606–15.
Pairo-Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–8.
Nakanishi T, Pigazzini S, Degenhardt F, et al. Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J Clin Investig. 2021. https://doi.org/10.1172/jci152386.
Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-280.e8.
Basu-Ray I, Almaddah N k., Adeboye A, Soos MP. Cardiac Manifestations of Coronavirus (COVID-19). StatPearls 2023; published online Jan 9. https://www.ncbi.nlm.nih.gov/books/NBK556152/ (accessed Oct 2, 2023).
Inciardi RM, Lupi L, Zaccone G, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;5:819–24.
Badawi A, Ryoo SG. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS-CoV): a systematic review and meta-analysis. Int J Infect Dis. 2016;49:129–33.
Shi Y, Wang Y, Shao C, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ. 2020;27:1451–4.
Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol. 2020;17:259–60.
Xiong TY, Redwood S, Prendergast B, Chen M. Coronaviruses and the cardiovascular system: acute and long-term implications. Eur Heart J. 2020;41:1798–800.
Funding
The study was supported by the Special Fund for Health Scientific Research in Public Welfare (Grant No. 201502006), the Key Project of Natural Science Funds of China (Grant No. 8123066), the National Natural Science Foundation of China (Grant No. 81872695), the Fujian Provincial Health Technology Project (Grant No. 2020CXB009), the Natural Science Foundation of Fujian Province, China (Grant No. 2021J01352) and the China Postdoctoral Science Foundation (Grant No. BX2021021).
Author information
Authors and Affiliations
Contributions
SW, TW, YH conceived the study design; SW, FC, CL, YH, JW, MW, assisted with data acquisition; SW, QZ, DC, YW, XQ, YY, JL, supervised statistical analyses and drafting; all authors assisted with data interpretation, manuscript revisions and final manuscript review. All authors reviewed and edited the manuscript. All authors had access to all data in the study and the corresponding author had final responsibility for the decision to submit for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Data used for this study were collected from publicly available GWAS summary statistics, without any individual identifiers. Respective ethical and study protocol approval for original studies can be found elsewhere [21, 23].
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary Tables and Figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, S., Peng, H., Chen, F. et al. Identification of genetic loci jointly influencing COVID-19 and coronary heart diseases. Hum Genomics 17, 101 (2023). https://doi.org/10.1186/s40246-023-00547-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-023-00547-8