
Abstract
Background
Cardiomyopathies are a leading cause of progressive heart failure and sudden cardiac death; however, their genetic aetiology remains poorly understood. We hypothesised that variants in noncoding regulatory regions and oligogenic inheritance mechanisms may help close the diagnostic gap.
Methods
We first analysed whole-genome sequencing data of 143 parent–offspring trios from Genomics England 100,000 Genomes Project. We used gene panel testing and a phenotype-based, variant prioritisation framework called Exomiser to identify candidate genes in trios. To assess the contribution of noncoding DNVs to cardiomyopathies, we intersected DNVs with open chromatin sequences from single-cell ATAC-seq data of cardiomyocytes. We also performed a case–control analysis in an exome-negative cohort, including 843 probands and 19,467 controls, to assess the association between noncoding variants in known cardiomyopathy genes and disease.
Results
In the trio analysis, a definite or probable genetic diagnosis was identified in 21 probands according to the American College of Medical Genetics guidelines. We identified novel DNVs in diagnostic-grade genes (RYR2, TNNT2, PTPN11, MYH7, LZR1, NKX2-5), and five cases harbouring a combination of prioritised variants, suggesting that oligogenic inheritance and genetic modifiers contribute to cardiomyopathies. Phenotype-based ranking of candidate genes identified in noncoding DNV analysis revealed JPH2 as the top candidate. Moreover, a case–control analysis revealed an enrichment of rare noncoding variants in regulatory elements of cardiomyopathy genes (p = .035, OR = 1.43, 95% Cl = 1.095–1.767) versus controls. Of the 25 variants associated with disease (p< 0.5), 23 are novel and nine are predicted to disrupt transcription factor binding motifs.
Conclusion
Our results highlight complex genetic mechanisms in cardiomyopathies and reveal novel genes for future investigations.
Similar content being viewed by others

Background
The cardiomyopathies, herein divided into dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and left ventricular non-compaction cardiomyopathy (LVNC), are leading causes of heart failure [1].
Although there is considerable overlap between different cardiomyopathy subtypes, each has a signature genetic aetiology. HCM and ARVC are largely explained by alterations in sarcomere or desmosome proteins, respectively. Around half of HCM cases are caused by mutations in the genes MYH7 and MYBPC3 [2]. However, the genetic architecture of DCM is far more complex. To date, more than 250 genes have been implicated in DCM causation or risk, including genes encoding for cytoskeletal, sarcolemmal, mitochondrial, calcium cycling, costameric and sarcomeric proteins [3].
Although the aetiological basis of cardiomyopathy is incomplete, recent genetic studies suggest that a large proportion of cases may be explained by alterations in the noncoding genome [4].
Data from the ENCODE project suggest that biochemical functionality could be assigned to 80 per cent of the human genome, affecting regulatory and tissue-specific expression patterns [5]. Furthermore, genome-wide association studies (GWAS) show that over 90% of disease-associated SNPs are in noncoding regions of the genome, including regulatory regions, such as promoters and enhancers [6]. Thus, a key priority in the cardiomyopathy gene discovery pipeline is the identification of regulatory elements controlling genes associated with these disorders.
High-throughput epigenomic profiling methods such as ATAC-seq and ChIP-seq have enabled profiling of chromatin accessibility across samples in a tissue-wide manner, providing the opportunity to identify millions of context-specific regulatory elements. However, these bulk measurements of chromatin accessibility limit the precise understanding of how tissue heterogeneity and multiple cell types in the population contribute to overall disease aetiology [7]. Recent advances in single-cell ‘omics technologies have enabled an unbiased identification of cell-type populations and regulatory elements in a heterogeneous biological sample. By mapping the chromatin-regulatory landscape at a single-cell resolution, studies have demonstrated the potential to link regulatory elements to their target genes, and map regulatory dynamics during complex cellular differentiation processes [7,8,9].
We first performed parent–offspring trio analysis to assess the impact of rare inherited recessive and dominant variants, and of DNVs on cardiomyopathy. We hypothesised that variants in regulatory regions that are specifically active in the adult heart could provide an aetiological basis for cases with unexplained genetics. We further supported this hypothesis through systematic examination of noncoding regulatory elements of known disease-risk genes in a mutation-negative cardiomyopathy cohort. We identified noncoding variants predicted to disrupt cis-regulatory elements involved in cardiac gene regulation.
By combining inherited and DNV analysis of a clinically well-defined cohort, this study provides novel insights into the complex genetics of cardiomyopathy subtypes, which may lead to improved diagnosis and therapies.
Materials and methods
Study participants
All participants were recruited to the 100,000 Genomes Project (100KGP) (protocol version 7, 2020), with written informed consent. The full protocol is available online at https://doi.org/10.6084/m9.figshare.4530893.v7. Probands of parent–offspring trios (n = 143) and singleton offspring (n = 843) were diagnosed with either ARVC, LVNC, HCM, DCM or DCM and conduction defects. They were included in the study if they had a clear diagnosis under 40 years of age. Patients were excluded if they had an unclear diagnosis or a history suggestive of a non-genetic cause. The control cohort included 19,467 age-, sex-, and ethnicity-matched participants with no known heart disease.
The study adheres to the principles set out in the Declaration of Helsinki. Patients and relatives gave written informed consent for genetic testing. Ethical approval for the 100KGP was granted by the East of England—Cambridge South Research Ethics Committee (REC Ref 14/EE/1112).
Initial data processing and gene selection
Whole-genome sequencing (WGS) were processed by Illumina, and sequencing data were passed through 100KGP's bioinformatics pipeline for alignment, annotation, and variant calling. Variants were prioritised using prespecified virtual gene panels from PanelApp (https://www.genomicsengland.co.uk). To date, 208 genes are listed for the Cardiomyopathies—including childhood onset (v1.37) panel. Based on the Human Phenotype Ontology (HPO) terms entered for these patients, additional gene panels were applied where applicable, including: undiagnosed metabolic disorders (v1.95), mitochondrial disorders (v1.127), intellectual disability (v3.2), and RASopathies (v1.27). Inherited variants were restricted to panel genes, where the allelic state was required to match the curated mode of inheritance.
Identification of candidate variants in coding regions
The variants were categorised according to gene annotation, population allele frequency, functional prediction, and clinical interpretation. The raw list of SNVs and indels was annotated using ANNOVAR [10].
Variants were classified based on their mutational characteristics, position in the genome, allele frequency, and functional role in cardiomyopathy. In silico prediction of pathogenicity was performed using CADD [11] and REVEL [12], and conservation of nucleotides was scored using GERP + + [13]. Population allele frequencies were obtained from 100KGP, 1000Genomes, and gnomAD [14].
Highest priority was given to protein truncating (frameshift, stop gain, stop loss, splice acceptor variant, or splice donor variant) or de novo (protein truncating, missense, or splice region) variants in a gene on the diagnostic-grade list in the virtual gene panel for cardiomyopathy or any additional gene panel relevant to the phenotype to the patient [15].
Inherited protein-altering variants, such as missense and splice region variants, in diagnostic-grade genes were also considered. Variants were retained if they had a minor allele frequency (MAF) < 0.001; the allelic state matches the known mode of inheritance for the gene and disorder and segregates with disease (where applicable). Rare variants were ranked according to a REVEL score above the default threshold of 0.5, a CADD score greater than 20, and GERP + + score greater than 2. Variants with clinical significance as benign or likely benign according to the ClinVar dataset were removed.
In parallel, we used Exomiser (v12.1.0) [16], a phenotype-driven variant prioritisation framework. Exomiser uses computational filters for variant frequency and predicted pathogenicity, protein interaction networks, patient phenotypes, cross-species phenotype comparisons, and pedigree information. A logistic regression model is used to combine the phenotype and variant scores to produce an overall Exomiser score. We considered the top three ranked variants that matched with our candidate-gene discovery analysis.
De novo variant calling and filtering
DNVs were identified by 100KGP's bioinformatics pipeline. Briefly, variants from WGS data were called using Platypus, and filtered for absence of the mutation in both parents, read depth (> 20), allele balance (> 0.3 and 0.7), and no overlap with segmental duplications, simple repeat regions, and patch regions.
To analyse noncoding DNVs, we obtained single-cell ATAC and RNA data of human adult ventricle [14]. DNVs from 143 trios were intersected with the single-cell ATAC-seq peak sets using default parameters of bedtools v2.24.0. Peak sets were tested for an enrichment of DNVs in offspring as compared to a background peak set which contained peaks from all other cell types. We used a chi-squared test to compare the number of peaks with DNVs between the cardiomyocyte-specific peak set and the background peak set.
HiChIP analysis
We used H3K27ac HiChIP to map active chromatin interactions genome wide on iPSC-derived cardiomyocytes (GSM3639703) [17]. HiChIP paired-end reads were aligned to GRCh38 genome using the HiC-Pro pipeline. Duplicate reads were removed and default HiC-pro settings were used to assign reads to MboI restriction fragments, filter for valid interactions, and generate binned interaction matrices. High-confidence contacts (FDR < 0.05) were called using the contact caller FitHiChIP with default settings at 10 kb resolution. These high-confidence contacts were used in visualisation.
Prediction of target genes of enhancers
We used a combination of methods to predict enhancer–gene interactions and interpret the functions of noncoding DNVs.
Candidate enhancers were predicted using the recently developed activity-by-contact (ABC) model [18], which integrates H3K27ac ChIP-seq, HiChIP, and gene expression data with chromatin accessibility to predict enhancers and link them to their target genes. Using this method, we were able to identify sets of high-confidence putative enhancers for cardiomyocytes and their likely target genes.
In addition, publicly available Hi-C data of human left and right ventricle tissue (sample GSM1419085 and GSM2322554, respectively) [19, 20] were analysed in the 3D-genome Interaction Viewer (3DIV) and database (http://kobic.kr/3div/)20.
3DIV was run using distance-normalised interaction frequency ≥ 2 to define significant enhancer–promoter interactions. Topologically associating domains (TADs) were identified using TopDom [22] with a window size of 20. DNVs that were within enhancers predicted by both the ABC model and 3DIV were considered for downstream analysis. Finally, we used a machine learning approach called FATHMM-MKL [23] to predict the functional impact of noncoding SNVs. This tool integrates functional annotations from ENCODE with nucleotide-based sequence conservation measures and provides predictions as p values in the range 0 to 1. We used the default score > 0.5 to indicate putative deleterious variants.
Promoters
Promoters were defined as 2 kb upstream or 1 kb downstream of transcription start sites (TSSs) and determined based on the basic gene annotation file of release 33 from GENCODE [24]. Further, to detect distal promoter-interacting loci we used promoter capture Hi-C data generated from three different human cell/tissue-types, including cardiomyocytes (GSM2297135, GSM2297136, GSM2297137, GSM2297138, GSM2297139), left ventricle (GSM2297192, GSM2297193, GSM2297194, GSM2297195, GSM2297196, GSM2297197, GSM2297198, GSM2297199, GSM2297200, GSM3067218, GSM3067219), and right ventricle (GSM2297289, GSM2297290, GSM2297291, GSM2297292, GSM2297293, GSM2297294, GSM2297295, GSM2297296, GSM2297297) [21].
Network analysis
For noncoding DNVs, a functional enrichment analysis of the candidate genes was performed using the VarElect [25]. This tool uses the deep LifeMap Knowledgebase to infer the “direct” or “indirect” association of biological function between genes and the queried phenotype—i.e. “cardiomyopathy”. A direct association is determined if studies indicate that the gene in question directly affects disease development. An indirect association is based on factors such as shared pathways, protein–protein interaction networks, and mutual publications.
Case-control analysis
Independent to the trio analysis, we analysed 843 probands and 19,467 unrelated controls to identify high-risk noncoding variants in regulatory elements of 12 cardiomyopathy genes with definitive (BAG3, DES, FLNC, LMNA, MYH7, PLN, RBM20, SCN5A, TNNC1, TNNT2, TTN) or strong (DSP) evidence [26]. We focused on regulatory elements of diagnostic genes rather than the entire genome to avoid false-positive results related to genes with an unclear association with the disease. Regulatory elements for each gene were determined using the ABC model (described above).
The functional impact of rare regulatory variants was assessed based on several tools, including RegulomeDB (https://regulomedb.org/), FunMotif (http://bioinf.icm.uu.se:3838/funmotifs/), and FATHMM-MKL. We mapped SNVs to these active regulatory regions of cardiomyopathy genes and defined them as high-risk if they were rare (MAF < 0.0001 in 100KGP and gnomAD population controls), predicted to alter transcription factor (TF) binding, and were enriched in cases versus controls (p < 0.05).
Statistical analysis
To compare variant burden between cases and unrelated controls for high-risk regulatory variants of cardiomyopathy genes, variant calls were required to have an MAF of ≤ 0.0001 in 100KGP controls and gnomAD. Controls were proportionally matched for age, sex and ethnicity. χ2 test, odds ratios (OR), and 95% confidence intervals (95% CIs) were calculated for regulatory regions of all genes by comparing the burden of rare variants.
To evaluate the association between individual noncoding variants and the risk of cardiomyopathy, we performed a Fisher’s exact test as expected value were < 5. Statistical significance was considered at the 5% level (two-tailed). Statistical analyses were undertaken using R 4.2.0 and RStudio 2022.02.2.
Results
Demographics and phenotype data of probands
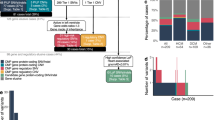
In the trio analysis, 143 probands (85 males, 58 females), with severe or syndromic disease, together with their parents, were analysed. They were of different reported ethnicities across England (Fig. 1A).

Proband demographics and phenotype in trio analysis. A Ethnicities recorded of participants. B, Age distribution of participants at present time and type of diagnosis made. B Top 20 Human Phenotype Ontology (HPO) terms for participants recruited
The age distribution of participants is shown in Fig. 1B. Participants recruited for HCM were more frequent in the 20 to 29 age group, and DCM between ages 20 to 39. LVNC was more common in younger people at enrolment. Of note, participants were enrolled in the 100KGP in their 40 s and 50 s as they still lacked a molecular diagnosis, despite all participants having an age of onset before 40 years of age.
Figure 1C shows the top 20 HPO terms in all participants recruited, including intellectual disability, joint hypermobility, and skeletal myopathy, suggesting syndromic causes.
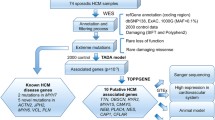
After parent–offspring trio analysis and application of our stringent filtering criteria, each proband had an average of 69.7 DNVs (Fig. 2). Genetic findings and genotype–phenotype correlations are described in Tables 1, 2 and 3. We used Exomiser to help narrow down candidate variants.

Noncoding de novo variant filtering criteria. DCM = dilated cardiomyopathy; ARVC = arrhythmogenic right ventricular cardiomyopathy; LVNC = left ventricular non-compaction cardiomyopathy; and HCM = hypertrophic cardiomyopathy
In addition, 843 exome-negative cardiomyopathy probands and 19,467 controls were incorporated into our case-control study from the 100KGP. The cases are singleton offspring whose parental WGS data were unavailable. Using the PanelApp software, which contains a crowd-sourced curation of genes with diagnostic-grade evidence, only cases lacking a molecular diagnosis were recruited in the case–control analysis. These cases included 61.8% HCM, 26.0% DCM, 8.5% ARVC, and 3.6% LVNC subtypes. Most participants were of European ancestry (70%), and 64% were male.
De novo variants in diagnostic-grade genes.
Using the American College of Medical Genetics (ACMG) guidelines, we identified deleterious DNVs in 11/143 trios (7.7%). These class 4 and 5 variants are defined as likely pathogenic or pathogenic, and are reported as consistent with or confirming a diagnosis, respectively (Table 1). Exomiser ranked the correct diagnosed variants as the top candidate in all these cases, and no parents were affected. Several novel DNVs were identified in syndromic and non-syndromic cases.
In Fam659, the proband had a novel missense DNV c.568C > A (p.Arg190Ser) in the NKX2-5 gene. NKX2-5 (NK2 homeobox 5) encodes for a transcription factor that is important for the development of the myocardium [27]. Mutations in this gene are known to cause congenital heart disease, particularly atrial septal defect (with or without atrioventricular conduction defects), and ventricular septal defect. Consistent with this, the patient was diagnosed with LVNC, including atrial septal defect, atrioventricular block, and abnormal ventricular septum morphology.
In the proband of Fam208, a novel missense DNV c.590 T > C (p.Leu197Pro) was identified in the LMNA gene (lamin A/C). LMNA encodes the A-type lamin proteins, lamin A and C, which are the major components of the nuclear membrane in mammals. Mutations in LMNA have been reported to cause a variety of clinical phenotypes, collectively known as laminopathies. These include cardiac disorders, premature ageing syndromes, and neuropathies [28]. In addition to DCM, the proband had a range of musculoskeletal-related abnormalities (Table 1).
In the proband of Fam520, a missense DNV c.360C > A (p.His120Gln) was found in the LZTR1 gene. Mutations in LZTR1 (leucine-zipper-like transcriptional regulator 1) are associated with Noonan syndrome phenotypes and schwannomatosis [29]. As well as obstructive HCM, the proband had combined disorders of mitral, aortic and tricuspid valves, congestive heart failure, thyrotoxicosis with diffuse goitre, postprocedural hypothyroidism, and rheumatoid arthritis. According to the ACMG guidelines, the variant is classified as a variant of uncertain significance. It is predicated to be deleterious according to in silico algorithms and was not identified in public databases, including gnomAD and 1000G. We require further clinical information to determine a phenotype consistent with Noonan spectrum disorders. Dominant and recessive Noonan syndrome-causing mutations have been described near this variant, all of which within the highly conserved kelch domains [30, 31].
A novel frameshift DNV of high-impact c.6183del (p.Leu2062TrpfsTer25) in the ANKRD11 gene was observed in the proband of Fam451. Mutations in ANKRD11 (ankyrin repeat domain 11) are reported to be associated with KBG syndrome and intellectual disability [32]. The patient presented with LVNC and a variety of other phenotypes, including abnormal vena cava morphology, bicuspid aortic valve, abnormality of the face, delayed fine motor development, intellectual disability, and proportionate short stature (Table 1).
In the proband of Fam478, a known pathogenic missense DNV c.782C > T (p.Pro261Leu) was found in the RAF1 gene. Diseases associated with RAF1 (Raf1 proto-oncogene, serine/threonine kinase) include Noonan and Leopard syndromes. These developmental disorders have overlapping features, including cardiac abnormalities, short stature, and facial dysmorphia [33]. The proband was diagnosed with HCM and had other phenotypes, including congenital malformation of cardiac septum, palpitations, anxiety disorder, and depression. This DNV has previously been reported in an individual affected with Noonan syndrome, including other individuals with clinical features of this disease [33,34,35]. Moreover, functional studies have shown that p.Pro261Leu leads to increased activity of the RAF1 protein [36].
In the proband of Fam828, we found a missense DNV c.836A > G (p.Tyr279Cys) in the PTPN11 gene (protein tyrosine phosphatase non-receptor type 11). Mutations in PTPN11 are well characterised in children with Noonan syndrome and juvenile myelomonocytic leukaemia [37]. In addition to HCM, the proband had other phenotypes, including intellectual disability, failure to thrive, right ventricular cardiomyopathy, and LVNC.
Other known pathogenic DNVs were found consistent with a diagnosis of non-syndromic cardiomyopathy. Three participants with HCM had known missense variants in MYH7. Two variants (c.2156G > A (p.Arg719Gln) and c.1358G > A (p.Arg453His)) are reported as pathogenic on ClinVar, whereas variant c.2420G > C (p.Arg807Pro) is reported as likely pathogenic. The proband harbouring the latter MYH7 variant also had congenital malformations of the heart, congestive heart failure, arrhythmia, and died as an infant with sudden cardiac arrest. Pathogenic variants were found in other known genes, including DES, RYR2, TTN and TNNT2 (Table 1).
De novo variants in other disease-risk genes
We also looked at DNVs in genes not included in PanelApp. In 27/143 probands (18.9%), 30 rare de novo coding variants were identified, which were considered deleterious based on in silico prediction tools (see methods). Using Exomiser, a phenotype-based prioritisation pipeline, 11 DNVs were ranked in the top three as the most likely cause. These include TUBA1B, KIRREL1, DAAM1, DOCK11, and KDM5B (Table 2). It is possible that some of the putative genes identified herein could be novel gene candidates or genetic modifiers.
Interestingly, the proband of Fam231 harbouring a missense DNV c.49 T > C (p.Cys17Arg) in the DAAM1 gene also had a known pathogenic variant in TNNT2 (Table 1). Studies show that DAAM1 is required for cardiomyocyte maturation [38], and deletion of the gene is associated with congenital heart anomalies [39]. Multiple gene mutations occurring in cardiomyopathy families may result in a more severe clinical phenotype because of a compounding effect. Other examples of oligogenic inheritance are described below.
Inherited variants in diagnostic-grade genes
In 10/143 trios (7.0%), 14 rare inherited variants of potential clinical significance were identified in probands and their affected family members based on gene panel testing. These include variants with recessive, dominant, and compound heterozygous segregation patterns (Table 3).
Two participants with HCM had missense variants in MYBPC3. In Fam599, the proband harbouring the c.1504C > T (p.Arg502Trp) variant presented with a range of diagnoses, including abnormal thumb, eye and oral morphology, intellectual disability, and mild microcephaly. The mother, also harbouring the variant, was diagnosed with atrial septal defect, short thumb, polycystic ovaries, and Raynaud syndrome. It is not clear why the proband presented with a more severe phenotype.
In Fam411, the proband inherited the MYBPC3 splice donor variant c.25 + 1G > A from the affected mother. In addition to HCM, the mother presented with hypothyroidism, prolonged QT syndrome, severe depression, anxiety, and schizoid personality disorder.
In Fam484, the proband inherited an autosomal dominant mutation c.2254 T > A in SAMD9 from the affected father. The proband was diagnosed with LVNC, ventricular septal defect, intellectual disability, joint hypermobility, and Wolff–Parkinson–White syndrome. Although the affected father and sibling carrying this variant presented with LVNC, neither had the comorbidities present in the proband. The father, however, presented with gastrointestinal haemorrhage, and diverticular disease of large intestine (without perforation or abscess). Mutations in SAMD9 have been described in patients with MIRAGE syndrome, a severe multisystem disorder [40]. This includes prominent gastrointestinal symptoms and intellectual disability.
In the proband and affected mother of Fam957, a missense variant c.372C > G (p.Ile124Met) was identified in the ACTC1 gene. Mutations in ACTC1 (actin alpha cardiac muscle 1) are associated with atrial septal defect, DCM, and HCM [41, 42]. In addition to DCM, the proband had partial anomalous pulmonary venous return, dyspnoea, myocardial fibrosis, and oligospermia. The mother was diagnosed with DCM, secundum atrial septal defect, and bipolar affective disorder.
Multilocus inheritance may explain the relatively low diagnostic yield for cardiomyopathy cases, or apparent phenotypic expansion. We found evidence for compound heterozygosity, digenic, and oligogenic inheritance in several families (described below). This highlights the importance of screening for additional genes even after a single mutation has been identified.
We observed evidence for oligogenic inheritance in Fam919. The proband, born in 2018 and reported to be deceased, had a recessive mutation in POLR3A (c.1787C > T (p.Thr596Met)) and a compound heterozygous TTN mutation (c.92176C > T (p.Pro30726Ser)). Mutations in POLR3A are associated with a wide array of pathological phenotypes, some of which were present in the proband. In addition to DCM and congestive heart failure, the proband had multiple congenital anomalies, including microcephaly, endocardial fibroelastosis, hydrops fetalis, polymicrogyria, cortical dysplasia, and pedal oedema.
The proband of Fam539 inherited five compound heterozygous TTN variants, one of which from the mother passed our filtering criteria for deleteriousness (c.20335A > T, p.Ser6779Cys) (see methods). The proband also inherited autosomal dominant variants of incomplete penetrance in COL6A1 and LZTR1. The LZTR1 stop gained variant c.1311G > A (p.Trp437Ter) was inherited from the mother, and the COL6A1 missense variant c.1712A > C (p.Lys571Thr) was inherited from the father. The proband was diagnosed with HCM, skeletal myopathy, and increased nuchal translucency. One out of two siblings are also affected with HCM; however, detailed medical notes or WGS data are not available. Although considered unaffected for cardiomyopathy, both parents were diagnosed with primary (essential) hypertension. In addition, the father has a family history of ischaemic heart disease, arrhythmia, syncope and collapse, and other ill-defined heart diseases.
In the proband of Fam992, compound heterozygous mutations c.852_855del (p.Asn284LysfsTer4) and c.1038_1040del (p.Lys346del) in the DSG2 gene were identified. The mother of the proband had the frameshift variant c.852_855del, whereas the father had the inframe deletion c.1038_1040del. In addition to ARVC, the proband was diagnosed with disorders of magnesium metabolism, hypokalaemia, and congenital malformations of cardiac chambers and connections. Of note, a DNV in the enhancer of gene TUSC3 was also identified. TUSC3 constitutes a major component in cellular magnesium transport and homeostasis, and its function in regulation of embryonic development in vertebrates has been suggested [43, 44]. This may explain the disorders of magnesium metabolism and hypokalaemia as a secondary cause in the proband [45].
In Fam180, the proband and mother with HCM harbour a variant of unknown significance (class 3) in the MYH7 gene. In addition, two families with HCM had mutations in mitochondrial genes MT-CO1 and MT-ND6 (Table 3); both previously implicated in heart disease, although heteroplasmy proportions are yet to be determined in multiple tissue samples.
These examples demonstrate that non-Mendelian inheritance may be an important factor in the cardiomyopathy cause-discovery pipeline. Other possibilities exist to help close the diagnostic gap, including noncoding mutations that affect regulatory elements.
Singe-cell chromatin state profiling
We hypothesised that DNVs in human heart regulatory regions are more likely to perturb expression levels of genes that are essential for cardiac function.
Annotation of ventricular cardiomyocyte peak set in genomic features shows enrichment in intronic and distal intergenic regions and in the flanking regions of TSSs, suggesting an enrichment of gene regulatory elements, such as enhancers. We intersected single-cell ATAC-seq peaks with publicly available H3K27ac ChIP-seq data (a marker for active enhancers) of eight healthy adult donors [46] and found significant overlap with our peaks (Permutation test, one-sided, p < 0.001).
Identification of de novo variants in noncoding regulatory regions
To determine selective vulnerabilities across diverse cell types of the human heart, we intersected cell-type-specific ATAC-seq peaks with DNVs identified from parent–offspring trio analysis (see methods). Cardiomyocyte-specific peak sets were not significantly enriched for DNVs in offspring compared to a merged background peak set.
A total of 288 DNVs from 143 trios intersected with a peak signal from ventricular cardiomyocytes. After filtering for parental affected status, H3K27ac overlap, and mapping regulatory regions to genes, 15 DNVs were within promoter regions, and 12 within predicted enhancers linked to their target genes. We used the tool FATHMM-MKL to predict the functional effects of noncoding variants. Additional file 1: Table S1 shows prioritised variants within ventricular cardiomyocyte open chromatin regions.
Predicting the target genes of enhancers
Using the ABC model [18], we predicted likely enhancers by integrating H3K27ac ChIP-seq, HiChIP, and gene expression data with chromatin accessibility. We identified sets of high-confidence putative enhancers for ventricular cardiomyocytes and their likely target genes.
As a complementary approach, histone ChIP-seq experiments on Hi-C samples were analysed to provide epigenetic features using 3DIV. Annotation of enhancer/super-enhancers and histone ChIP-seq signals were provided for the following: H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3. Genes with distance-normalised interaction frequency > 2 were retained. In addition, we used promoter capture Hi-C data to detect interactions with gene promoters. These data are summarised in Additional file 1: Table S1.
Network analysis
We applied another strategy to further prioritise the effect of DNVs on human cardiac regulome. We analysed the 62 genes associated with enhancers and promoters containing prioritised DNVs using VarElect to correlate their functions with different aspects of the clinical phenotype. Results suggest that 20 targets were directly related to cardiomyopathy, whereas 41 were indirectly related (Additional file 2: Table S2). One gene (MIR3143) was unrelated and therefore excluded from the analysis. Among the unified results, the top five genes with the highest score of correlation were JPH2, UTRN, H1-2, RHOD, and SAP30B. This score is an indication of the strength of the connection between the gene and the queried phenotypes. The score helps to rank and prioritise the list of queried genes by relevance to the disease. Interestingly, many of the top scoring genes were associated with the same DNV.
Noncoding de novo variants are associated with cardiomyopathy-risk genes
In the proband of Fam499, we identified a DNV within an enhancer of JPH2 (Fig. 3). This gene exhibited the highest phenotype association (VarElect score 36.89) (Additional file 2: Table S2). The proband, female and of British ethnicity, was diagnosed with HCM and reported to have died due to sudden cardiac arrest in the year 2020, at the age of 19. Both parents and natural sibling recruited in the study were unaffected.

Chromatin interaction map of chr20:44,116,250 locus. A One-to-all interaction plots are shown for the lead variant chr20:44,116,250 A > G shown in blue as bait. Y-axes on the left and the right measure bias-removed interaction frequency (blue bar graph) and distance-normalised interaction frequency (magenta dots), respectively. The arc representation of significant interactions for distance-normalised interaction frequencies ≥ 2 is displayed relative to the Refseq-annotated genes in the locus. The DNV physically interacts with the JPH2 enhancer by long-range chromatin interaction. B Close-up of region containing JPH2. The ABC model independently predicted the DNV (red triangle) is within the JPH2 enhancer. CM = cardiomyocyte
The junctophilin-2 gene (JPH2) is the major structural protein in cardiomyocytes for coupling of transverse tubule-associated L-type Ca2+ channels and type-2 ryanodine receptors on the sarcoplasmic reticulum within junctional membrane complexes (JMC) [47]. Signalling between these two Ca2+ channels is required for normal cardiac contractility. Disruption of the JMC is a common finding in failing hearts. Downregulation of JPH2 gene has been associated with heart failure, and mutations in this gene are associated with HCM [47, 48].
JPH2 was the only high-evidence gene found in our noncoding DNV analysis. We recognise that caution be exercised in the interpretation of variants in cardiomyopathy genes lacking robust evidence; however, the following preliminary results may help to explain the complex genetic architecture of cardiomyopathy.
The proband of Fam126 was diagnosed with DCM and harbours a DNV within an enhancer region that regulates the genes UTRN, STX11, and SF3B5. Diseases associated with UTRN (utrophin) include muscular dystrophy, endothelial dysfunction, and DCM [49, 50].
The proband of Fam313 was diagnosed with DCM, including dysplastic tricuspid valve, right ventricular cardiomyopathy, tricuspid regurgitation, dyspnea, pulmonic stenosis, café au lait spot, and congenital heart disease. Although no DNVs were identified in enhancer regions, a deleterious coding DNV was identified in ADGRV1 (Table 2). In addition, we used pcHi-C to detect distal promoter-interacting regions and found that a DNV in this proband is associated with a large cluster of histone genes on human chromosome 6.
Studies have shown that histone acetylation/deacetylation regulates cardiac morphogenesis, growth, and contractility [51]. Gene expression profiles of DCM patients have also shown that several histone family members are downregulated [52]. We hypothesise that the downregulation of these genes, which are responsible for higher order chromatin structure, may contribute to the clinical presentation in this proband via nucleosome formation blockade [53].
The proband of Fam334 was initially diagnosed with DCM, and later suspected to have aortopathy and early hypertension. Other diagnoses include lethargy, torsion of testis, headaches, and palpitations. The proband died in the accident and emergency department in the year 2018, at the age of 21. Neither parent or three natural siblings were affected. The DNV identified in this proband is (1) within the enhancer region of genes UNC13D, WBP2, SAP30BP and TRIM65; (2) overlapping the promoter regions of genes H3-3B, MIR4738 and UNK; and (3) interacting with distil gene promoters of TRIM56 and TMEM94. Diseases associated with TMEM94 include cardiac defects [54].
In Fam791, the proband was diagnosed with ARVC and DCM, in addition to arrhythmia, varicose veins of lower extremities with ulcer and inflammation, atherosclerotic heart disease, renal failure, gastrointestinal haemorrhage, and anal polyp. A DNV was identified within the enhancer region of genes GRK2 and RHOD, and overlapping the promoter region of RAD9A.
G protein-coupled receptor kinase-2 (GRK2) regulates many cellular and physiological processes, including cardiac contractility, cell proliferation, cell cycle regulation, angiogenesis and vasodilatation. Inhibiting GRK2 can enhance cardiac contractility and protect from adverse heart remodelling in disorders related to cardiac dysfunction [55], suggesting its inhibition as a therapeutic strategy for heart failure. We hypothesise that this DNV may elevate levels and activity of this kinase, thus promoting cardiovascular disease. Moreover, phenome-wide associated loci in the proximity of RHOD is a likely causal gene for cardiomegaly and hematemesis [56], the latter of which may explain the gastrointestinal bleeding observed in this patient.
Case-control analysis reveals high-risk noncoding variants in disease-risk genes
The contribution of disease-causing rare variants in noncoding regulatory regions remains elusive. The identification of candidate noncoding DNVs in our trio analysis led us to investigate high-risk regulatory variants associated with cardiomyopathy genes in a large cohort lacking a pathogenic mutation in gene panel testing.
Overall, combining data from all genes, there was a significant difference in the proportion of cardiomyopathy cases and controls carrying one or more rare variant in regulatory elements of strong or definitive disease-risk genes (p = 0.035, OR = 1.43, 95% Cl = 1.095–1.767).
Of the 843 probands lacking a molecular diagnosis, we performed variant-level analysis and identified 25 noncoding variants that were significantly associated in cases (p< 0.05). Of these, 9 predicted to effect TF binding motifs. Eight of the 12 genes investigated had one or more rare variant in regulatory regions, including DSP, RBM20, LMNA, TNNT1, TNNT2, BAG3, DES, and PLN. The highest-ranking regulatory elements for each gene are listed in Additional file 3: Table S3.
Most of the significant variants (n = 23; 92%) were “private” to a single proband, with only two variants occurring in two unrelated probands, albeit with the same cardiomyopathy subtype. The private variants were not identified in control populations. Most variants (76%) occurred in HCM, 12% occurred in DCM, 8% occurred in ARVC, and 4% were observed in LVNC cases. These data are summarised in Table 4.
Discussion
The pathogenesis of cardiomyopathy is largely unknown, and the diagnosis is challenging due to its clinical heterogeneity, involving incomplete penetrance and variable expression. The analysis of DNVs in clinically well-defined phenotypes is a powerful approach to delineate the aetiological basis of disease as it focuses on a relatively small number of variants that provide strong evidence of pathogenicity [57].
DNVs are responsible for the relatively high prevalence of complex disorders. The estimated rate for human germline de novo SNVs is (1.0 to 2.4) × 10−8 per base per generation [58, 59]. This translates to an average of 32 to 76.8 variants in the human genome, with one or two in exonic regions. We had an average of 69.7 DNVs per trio analysis, giving a mutation rate of 2.2 × 10−8 per base per generation. This is consistent with previous studies, thus showing the high quality of our data.
We combined gene panel testing with Exomiser, a phenotype-based algorithmic framework, to prioritise inherited and DNVs. A definite or probable genetic diagnosis was identified in 21 probands according to the ACMG guidelines. Additional DNVs of potential clinical significance were identified in 30 genes, 11 of which were within the top three ranked by Exomiser, including TUBA1B, KIRREL1, DAAM1, DOCK11, and KDM5B.
In addition, we integrated WGS and single-cell epigenomics to examine the role of regulatory DNVs in cardiomyopathies. Despite the genetic heterogeneity of cardiomyopathy, which stifles efforts to unequivocally demonstrate a causal role for individual noncoding variants, our results provide multiple lines of evidence to indicate the aetiological basis of functional regulatory variants in the human heart regulome. Notably, a DNV was identified within an enhancer of JPH2, a gene associated with HCM and the highest scored in our analysis.
Interestingly, we found that more than one rare variant in different cardiomyopathy genes may be relevant for disease causation. Other studies have shown that cardiomyopathy can arise from co-inheritance of rare genetic variants that are benign on their own but harmful in combination [60]. The assumption that all or most patients will receive a single-gene diagnosis is now relegated to the margins. Investigating additional affected families does not necessarily lead to novel gene discovery, thus necessitating the exploration of non-Mendelian contributors to causation or risk.
To further add weight to the hypothesis that noncoding variants are associated with cardiomyopathy, we performed a case-control analysis in a mutation-negative cohort and found an enrichment of high-impact regulatory SNVs in cases compared to controls. A variant-level association test showed that 25 SNVs were significantly associated with disease, of which 23 were not identified in control populations and nine are predicted to alter TF motifs.
There were several limitations in this study. It is possible that probands in the trio analysis inherited variants within noncoding loci associated with disease, or inherited coding variants in genes beyond those listed in the applied panels. Moreover, in the case–control analysis, we only focused on genes that are strongly associated with cardiomyopathy. We also did not analyse structural variants, such as CNVs, inversions, balanced translocations, or complex rearrangements.
Indeed, functional validation of the novel variants reported herein is warranted, a lack of which is acknowledged as a further limitation. Novel variants should not be considered causal merely because they are rare and predicted to be deleterious in silico [61]. Many of the disease-associated variants identified in this study are noncoding, which are in less-well understood regions of the genome. High-throughput assays with functional readout for putative regulatory elements would enable the identification of functional variants and the biological contexts in which they act. Massively parallel reporter assays (MPRAs) permit the high-throughput functional characterisation of noncoding genetic variation [62]. In Fig. 4, we offer a workflow to identify candidate noncoding variants associated with disease, and to assess the molecular consequences of their disruption experimentally. Adapting MPRAs for use in cardiomyocytes will be critical towards understanding cell-type-specific models of regulatory logic in contexts of greater clinical relevance.

An integrative genomics approach for prioritising noncoding variants. A We performed a rare-variant association study in cardiomyocyte-specific regulatory regions of genes associated with cardiomyopathy. These variants should be tested for deleteriousness or transcriptional activity, and, by inference, for causality. B MPRAs allow for thousands of short DNA sequences to be assayed simultaneously by first synthesising DNA oligos on an array, integrating them into plasmids and inserting into cells. Both input DNA and RNA libraries are sequenced to assess the tag counts associated with the test sequences. Barcode abundance thus scales quantitatively with the regulatory activity of a given tested sequence (figure adapted from Ajore et al. [63]). This technique can be used in future studies to screen all prioritised cardiomyocyte-specific regulatory variants in cardiomyopathy cases
Our work brings together multiple ‘omics datasets to elucidate the role of pathogenic variants in coding and noncoding loci. This study should prompt extensive genetic analyses and variant-specific experimental modelling to elucidate the complex genetic mechanisms underlying cardiomyopathies.
Data availability
All datasets generated for this study are included in the article and supplementary files. Primary data from the 100KGP database are held in a secure Research Environment and available to registered users. Requests to access the primary data should be directed to https://www.genomicsengland.co.uk/about-gecip/for-gecip-members/data-and-data-access. Further information about the data and conditions for access are available from the University of Northampton Research Explorer at: http://doi.org/10.24339/87f60dbe-40e9-4f3e-bcd3-f5d072575431.
References
Brieler J, Breeden MA, Tucker J. Cardiomyopathy: an overview. Am Fam Physician. 2017;96:640–6.
Viswanathan SK, Sanders HK, McNamara JW, Jagadeesan A, Jahangir A, Tajik AJ, et al. Hypertrophic cardiomyopathy clinical phenotype is independent of gene mutation and mutation dosage. PLoS One. 2017;12:e0187948. https://doi.org/10.1371/JOURNAL.PONE.0187948.
Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2010;25:198. https://doi.org/10.1097/HCO.0B013E328337BA52.
Lesurf R, Said A, Akinrinade O, Breckpot J, Delfosse K, Liu T, et al. Whole genome sequencing delineates regulatory and novel genic variants in childhood cardiomyopathy. MedRxiv. 2020. https://doi.org/10.1101/2020.10.12.20211474.
Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. https://doi.org/10.1038/nature11247.
Hrdlickova B, de Almeida RC, Borek Z, Withoff S. Genetic variation in the non-coding genome: Involvement of micro-RNAs and long non-coding RNAs in disease. Biochimica et Biophysica (BBA) Acta Mol Basis Dis. 2014;1842:1910–22. https://doi.org/10.1016/J.BBADIS.2014.03.011.
Rai V, Quang DX, Erdos MR, Cusanovich DA, Daza RM, Narisu N, et al. Single-cell ATAC-Seq in human pancreatic islets and deep learning upscaling of rare cells reveals cell-specific type 2 diabetes regulatory signatures. Mol Metab. 2020;32:109–21. https://doi.org/10.1016/j.molmet.2019.12.006.
Preissl S, Fang R, Huang H, Zhao Y, Raviram R, Gorkin DU, et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci. 2018;21:432–9. https://doi.org/10.1038/s41593-018-0079-3.
Cusanovich DA, Reddington JP, Garfield DA, Daza RM, Aghamirzaie D, Marco-Ferreres R, et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature. 2018;555:538–42. https://doi.org/10.1038/nature25981.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. https://doi.org/10.1093/nar/gkq603.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–94. https://doi.org/10.1093/nar/gky1016.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85. https://doi.org/10.1016/j.ajhg.2016.08.016.
Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP. PLoS Comput Biol. 2010;6:e1001025. https://doi.org/10.1371/journal.pcbi.1001025.
Koch L. Exploring human genomic diversity with gnomAD. Nat Rev Genet. 2020;21:448–448. https://doi.org/10.1038/s41576-020-0255-7.
Vadgama N, Pittman A, Simpson M, Nirmalananthan N, Murray R, Yoshikawa T, et al. De novo single-nucleotide and copy number variation in discordant monozygotic twins reveals disease-related genes. Eur J Hum Genet. 2019;27:1121–33. https://doi.org/10.1038/s41431-019-0376-7.
Smedley D, Jacobsen JOB, Jäger M, Köhler S, Holtgrewe M, Schubach M, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protocols. 2015;10:2004–15. https://doi.org/10.1038/nprot.2015.124.
Wilson KD, Ameen M, Guo H, Abilez OJ, Tian L, Mumbach MR, et al. Endogenous retrovirus-derived lncRNA BANCR promotes cardiomyocyte migration in humans and non-human primates. Dev Cell. 2020;54(6):694-709.e9. https://doi.org/10.1016/j.devcel.2020.07.006.
Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat Genet. 2019;51:1664–9. https://doi.org/10.1038/s41588-019-0538-0.
Leung D, Jung I, Rajagopal N, Schmitt A, Selvaraj S, Lee AY, et al. Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature. 2015;518:350–4. https://doi.org/10.1038/NATURE14217.
Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 2016;17:2042–59. https://doi.org/10.1016/J.CELREP.2016.10.061.
Yang D, Jang I, Choi J, Kim MS, Lee AJ, Kim H, et al. 3DIV: a 3D-genome interaction viewer and database. Nucleic Acids Res. 2018;46:D52–7. https://doi.org/10.1093/NAR/GKX1017.
Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016;44:e70–e70. https://doi.org/10.1093/NAR/GKV1505.
Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day INM, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31:1536–43. https://doi.org/10.1093/BIOINFORMATICS/BTV009.
Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–74. https://doi.org/10.1101/GR.135350.111.
Stelzer G, Plaschkes I, Oz-Levi D, Alkelai A, Olender T, Zimmerman S, et al. VarElect: the phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics. 2016. https://doi.org/10.1186/S12864-016-2722-2.
Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, et al. Evidence-based assessment of genes in dilated cardiomyopathy. Circulation. 2021;144:7–19. https://doi.org/10.1161/CIRCULATIONAHA.120.053033.
Anderson DJ, Kaplan DI, Bell KM, Koutsis K, Haynes JM, Mills RJ, et al. NKX2–5 regulates human cardiomyogenesis via a HEY2 dependent transcriptional network. Nat Commun. 2018;9:1–13. https://doi.org/10.1038/s41467-018-03714-x.
Gonzalo S, Kreienkamp R, Askjaer P. Hutchinson–Gilford progeria syndrome: a premature aging disease caused by LMNA gene mutations. Ageing Res Rev. 2017;33:18–29. https://doi.org/10.1016/J.ARR.2016.06.007.
Schraepen C, Donkersloot P, Duyvendak W, Plazier M, Put E, Roosen G, et al. What to know about schwannomatosis: a literature review. Br J Neurosurg. 2020;10(1080/02688697):1836323.
Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive noonan syndrome associated with Biallelic LZTR1Variants. Genet Med. 2018;20:1175. https://doi.org/10.1038/GIM.2017.249.
Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52:413–21. https://doi.org/10.1136/JMEDGENET-2015-103018.
van Dongen LCM, Wingbermühle E, Oomens W, Bos-Roubos AG, Ockeloen CW, Kleefstra T, et al. Intellectual profiles in kbg-syndrome: A wechsler based case-control study. Front Behav Neurosci. 2017;11:248. https://doi.org/10.3389/FNBEH.2017.00248/BIBTEX.
Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39:1007–12. https://doi.org/10.1038/NG2073.
Kobayashi T, Aoki Y, Niihori T, Cavé H, Verloes A, Okamoto N, et al. Molecular and clinical analysis of RAF1 in Noonan syndrome and related disorders: dephosphorylation of serine 259 as the essential mechanism for mutant activation. Hum Mutat. 2010;31:284–94. https://doi.org/10.1002/HUMU.21187.
Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, Depalma SR, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–601. https://doi.org/10.1038/NG.3970.
Molzan M, Schumacher B, Ottmann C, Baljuls A, Polzien L, Weyand M, et al. Impaired binding of 14–3-3 to C-RAF in Noonan syndrome suggests new approaches in diseases with increased Ras signaling. Mol Cell Biol. 2010;30:4698–711. https://doi.org/10.1128/MCB.01636-09.
Kratz CP, Niemeyer CM, Castleberry RP, Cetin M, Bergsträsser E, Emanuel PD, et al. The mutational spectrum of PTPN11 in juvenile myelomonocytic leukemia and Noonan syndrome/myeloproliferative disease. Blood. 2005;106:2183–5. https://doi.org/10.1182/BLOOD-2005-02-0531.
Ajima R, Bisson JA, Helt JC, Nakaya MA, Habas R, Tessarollo L, et al. DAAM1 and DAAM2 are co-required for myocardial maturation and sarcomere assembly. Dev Biol. 2015;408:126–39. https://doi.org/10.1016/J.YDBIO.2015.10.003.
Bao B, Zhang L, Hu H, Yin S, Liang Z. Deletion of a single-copy DAAM1 gene in congenital heart defect: a case report. BMC Med Genet. 2012;13:1–5. https://doi.org/10.1186/1471-2350-13-63/FIGURES/3.
Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48:792–7. https://doi.org/10.1038/NG.3569.
Frank D, Rangrez AY, Friedrich C, Dittmann S, Stallmeyer B, Yadav P, et al. Cardiac α-actin (ACTC1) gene mutation causes atrial-septal defects associated with late-onset dilated cardiomyopathy. Circ Genom Precis Med. 2019;12:345–56. https://doi.org/10.1161/CIRCGEN.119.002491.
Despond EA, Dawson JF. Classifying cardiac actin mutations associated with hypertrophic cardiomyopathy. Front Physiol. 2018;9:405. https://doi.org/10.3389/FPHYS.2018.00405/BIBTEX.
Zhou H, Clapham DE. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci U S A. 2009;106:15750–5. https://doi.org/10.1073/PNAS.0908332106.
Yu X, Zhai C, Fan Y, Zhang J, Liang N, Liu F, et al. TUSC3: a novel tumour suppressor gene and its functional implications. J Cell Mol Med. 2017;21:1711–8. https://doi.org/10.1111/JCMM.13128.
Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18:2649–52. https://doi.org/10.1681/ASN.2007070792.
Spurrell CH, Barozzi I, Mannion BJ, Blow MJ, Fukuda-Yuzawa Y, Afzal SY, et al. Genome-wide fetalization of enhancer architecture in heart disease. BioRxiv. 2019;1832:591362. https://doi.org/10.1101/591362.
Walsh R, Offerhaus JA, Tadros R, Bezzina CR. Minor hypertrophic cardiomyopathy genes, major insights into the genetics of cardiomyopathies. Nat Rev Cardiol. 2021;2021:1–17. https://doi.org/10.1038/s41569-021-00608-2.
Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, et al. Junctophilin-2 gene therapy rescues heart failure by normalizing RyR2-mediated Ca2+ release. Int J Cardiol. 2016;225:371. https://doi.org/10.1016/J.IJCARD.2016.10.021.
Delfín DA, Zang KE, Schill KE, Patel NT, Janssen PML, Raman SV, et al. Cardiomyopathy in the dystrophin/utrophin-deficient mouse model of severe muscular dystrophy is characterized by dysregulation of matrix metalloproteinases. Neuromuscul Disord. 2012;22:1006. https://doi.org/10.1016/J.NMD.2012.05.002.
Chun JL, O’Brien R, Berry SE. Cardiac dysfunction and pathology in the dystrophin and utrophin-deficient mouse during development of dilated cardiomyopathy. Neuromuscul Disord. 2012;22:368–79. https://doi.org/10.1016/J.NMD.2011.07.003.
Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–802. https://doi.org/10.1101/GAD.1563807.
Zhang H, Yu Z, He J, Hua B, Zhang G. Identification of the molecular mechanisms underlying dilated cardiomyopathy via bioinformatic analysis of gene expression profiles. Exp Ther Med. 2017;13:273–9. https://doi.org/10.3892/ETM.2016.3953.
Bártová E, Kreǰcí J, Harničarová A, Galiová G, Kozubek S. Histone modifications and nuclear architecture: a review. J Histochem Cytochem. 2008;56:711. https://doi.org/10.1369/JHC.2008.951251.
Stephen J, Maddirevula S, Nampoothiri S, Burke JD, Herzog M, Shukla A, et al. Bi-allelic TMEM94 truncating variants are associated with neurodevelopmental delay, congenital heart defects, and distinct facial dysmorphism. Am J Hum Genet. 2018;103:948–67. https://doi.org/10.1016/J.AJHG.2018.11.001.
Sun X, Zhou M, Wen G, Huang Y, Wu J, Peng L, et al. Paroxetine attenuates cardiac hypertrophy via blocking GRK2 and ADRB1 interaction in hypertension. J Am Heart Assoc. 2021;10:1–35. https://doi.org/10.1161/JAHA.120.016364.
Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–11. https://doi.org/10.1038/nbt.2749.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. https://doi.org/10.1038/gim.2015.30.
Pranckėnienė L, Jakaitienė A, Ambrozaitytė L, Kavaliauskienė I, Kučinskas V. Insights Into de novo Mutation Variation in Lithuanian Exome. Front Genet. 2018. https://doi.org/10.3389/FGENE.2018.00315.
Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:1–19. https://doi.org/10.1186/s13059-016-1110-1.
Gifford CA, Ranade SS, Samarakoon R, Salunga HT, Yvanka De Soysa T, Huang Y, et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science. 2019;364:865. https://doi.org/10.1126/SCIENCE.AAT5056.
Li J, te Lei W, Zhang P, Rapaport F, Seeleuthner Y, Lyu B, et al. Biochemically deleterious human NFKB1 variants underlie an autosomal dominant form of common variable immunodeficiency. J Exp Med. 2021;218:18. https://doi.org/10.1084/JEM.20210566/212613.
Abell NS, DeGorter MK, Gloudemans MJ, Greenwald E, Smith KS, He Z, et al. Multiple causal variants underlie genetic associations in humans. Science. 1979;2022(375):1247–54. https://doi.org/10.1126/SCIENCE.ABJ5117/SUPPL_FILE/SCIENCE.ABJ5117_MDAR_REPRODUCIBILITY_CHECKLIST.PDF.
Choi J, Zhang T, Vu A, Ablain J, Makowski MM, Colli LM, et al. Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat Commun. 2020;11:1–16. https://doi.org/10.1038/s41467-020-16590-1.
Acknowledgements
This research was made possible through access to the data and findings generated by the 100,000 Genomes Project (100KGP). The 100KGP is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100KGP is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100KGP uses data provided by patients and collected by the National Health Service as part of their care and support.
Author information
Authors and Affiliations
Consortia
Contributions
IK and NV conceived the project. NV did the literature search, generated the figures, performed the data analysis, and wrote the manuscript. SG helped to generate the HiChIP data. MA and LS provided critical feedback. IK, CG, and JN were involved in planning and supervised the work. All authors edited the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1:
Prioritised noncoding DNVs within the human ventricular cardiomyocyte regulome.
Additional file 2: Table S2:
All gene targets of noncoding DNVs directly or indirectly associated with cardiomyopathy.
Additional file 3: Table S1:
Activity-by-Contact (ABC) model predictions of known cardiomyopathy genes in ventricular cardiomyocytes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Vadgama, N., Ameen, M., Sundaram, L. et al. De novo and inherited variants in coding and regulatory regions in genetic cardiomyopathies. Hum Genomics 16, 55 (2022). https://doi.org/10.1186/s40246-022-00420-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40246-022-00420-0